

Abstract
Phosphorylation of transcription factors is a key link between cell signaling and the control of gene expression. Here we report that phosphorylation regulates DNA binding of the Ets-1 transcription factor by reinforcing an autoinhibitory mechanism. Quantitative DNA-binding assays show that calcium-dependent phosphorylation inhibits Ets-1 DNA binding 50-fold. The four serines that mediate this inhibitory effect are distant from the DNA-binding domain but near structural elements required for autoinhibition. Mutational analyses demonstrate that an intact inhibitory module is required for phosphorylation-dependent regulation. Partial proteolysis studies indicate that phosphorylation stabilizes an inhibitory conformation. These findings provide a structural mechanism for phosphorylation-dependent inhibition of Ets-1 DNA binding and demonstrate a new function for inhibitory modules as structural mediators of negative signaling events.
Keywords: Ets-1, phosphorylation, autoinhibition, transcription factor, DNA binding
Signal transduction pathways modulate gene expression through phosphorylation of regulatory transcription factors. Any of the functions performed by transcription factors, including DNA binding, transcriptional activation, transcriptional repression, subunit association, or nuclear localization, can be targets for this regulation. For example, sequence-specific DNA binding by the p53 protein is stimulated by phosphorylation of the carboxyl terminus (Ko and Prives 1996). Phosphorylation of the NFAT family of factors controls nuclear transport (Rao et al. 1997). Phosphorylation of CREB, NF-κB, and Elk-1 enhances their transcriptional activities (Gonzalez and Montminy 1989; Marais et al. 1993; Treisman 1994; Finco et al. 1997; Montminy 1997; Zhong et al. 1998). Although many examples of phosphorylation-dependent transcriptional regulation have been described, few cases are understood at the structural and mechanistic level. The regulation of Ets-1 DNA binding by phosphorylation provides an opportunity to bring structural data into a mechanistic study of post-translational regulation.
A detailed structural framework is available for studies on the regulation of Ets-1 DNA binding. The ETS domain of Ets-1 displays a winged–helix–turn–helix motif that recognizes a GGAA/T core sequence as a monomer (Nye et al. 1992; Donaldson et al. 1996; Kodandapani et al. 1996; Werner et al. 1997; Batchelor et al. 1998; Mo et al. 1998). More interestingly, an autoinhibitory mechanism regulates Ets-1 DNA binding (Hagman and Grosschedl 1992; Lim et al. 1992; Wasylyk et al. 1992; Graves et al. 1998; Fig. 1). Three α-helices (HI-1, HI-2, and H4) interact with each other and H1 of the ETS domain to produce a metastable inhibitory module (Donaldson et al. 1996; Skalicky et al. 1996). A conformational change of the inhibitory module accompanies DNA binding. This structural transition includes unfolding of helix HI-1 that is detected as DNA-induced protease sensitivity (Petersen et al. 1995; Jonsen et al. 1996). The coupling of helix unfolding to DNA binding is thought to be the basis of Ets-1 autoinhibition. We have proposed that this DNA-induced conformational change provides a structural switch to modulate Ets-1 DNA binding (Graves et al. 1998).
Figure 1.

Model of Ets-1 autoinhibition. (A) Schematic of Ets-1 showing DNA-binding domain (ETS domain, white), regions required for autoinhibition (inhibitory, black) and calcium-dependent phosphorylation (hatched). Expanded: secondary structural elements of the inhibitory module and ETS domain. α-Helices are indicated by barrels; β strands by arrows. (B) Structural model. α-Helices H1, HI-1, HI-2, and H4 form the inhibitory module. DNA binding is coupled to a conformational change that includes loss of structure in helix HI-1 and exposure of proteolytic cleavage sites (asterisks, Petersen et al. 1995; Jonsen et al. 1996). (C) Amino acid sequence spanning sites of calcium-dependent phosphorylation (hatched in A). In vivo calcium-dependent phosphorylation sites are indicated (Rabault and Ghysdael 1994).
A variety of regulatory strategies could use this inhibitory mechanism. It is frequently suggested that autoinhibition could be countered by positive signals to allow high-affinity DNA binding. For example, Ets-1 binds DNA cooperatively with a number of partner proteins (Graves and Petersen 1998). In the case of CBFα2/AML1, the partner protein facilitates Ets-1 DNA binding by counteracting autoinhibition (Goetz et al. 2000; Gu et al. 2000). Phosphorylation of Ets-1 could also rescind autoinhibition. We considered whether Ras/MAP kinase signaling, which stimulates Ets-1 transcriptional activity (Yang et al. 1996), could play this role. However, MAP kinase phosphorylation of Ets-1 does not affect DNA binding (B. Colson and B.J. Graves, unpubl.), ruling out a role for this modification. On the other hand, phosphorylation of Ets-1 by calcium signaling pathways has been reported to inhibit DNA binding (Fisher et al. 1994; Rabault and Ghysdael 1994). Calcium-dependent phosphorylation targets at least four serines amino-terminal of the ETS domain near inhibitory helices HI-1 and HI-2 (Rabault and Ghysdael 1994; Fig. 1C). These observations led us to hypothesize that rather than disrupting autoinhibition, calcium-dependent phosphorylation might use the structural switch to negatively regulate DNA-binding activity. This type of regulation would represent a novel use of autoinhibition in a biological setting.
In this report, we link the autoinhibitory mechanism to phosphorylation-dependent inhibition of DNA binding. We developed an in vitro system that provided a quantitative analysis of phosphorylation-dependent inhibition, establishing both the magnitude of the inhibition and the relative contributions of the multiple phosphorylation sites. Importantly, the in vitro system allowed us to test the role of the inhibitory module directly. We found that mutational disruption of the inhibitory module impairs phosphorylation-dependent repression of DNA binding. Furthermore, phosphorylation of Ets-1 enhances protease resistance within the inhibitory module. Together, these findings indicate that phosphorylation mediates its inhibitory effects via stabilization of the inhibitory module. Thus, this study demonstrates that the inhibitory module of Ets-1 facilitates regulation not only by positive signals that counteract autoinhibition but also by negative signals that reinforce autoinhibition.
Results
In vitro analysis of calcium-dependent Ets-1 phosphorylation
To determine the mechanism by which calcium-dependent signaling pathways regulate Ets-1 DNA binding, we developed an in vitro system suitable for quantitative and structural analysis. T-cell nuclear extract and calmodulin-dependent kinase II (CaMKII) phosphorylated bacterially expressed, purified Ets-1 on approximately six and four sites, respectively (Fig. 2A; Table 1). Approximately one to two sites were modified by nuclear extract without calcium (Fig. 2B). Together, these results indicate that four to five sites are targets of calcium-dependent phosphorylation by either nuclear extract or CaMKII in vitro. In vivo studies with transiently overexpressed Ets-1 suggested that at least four sites are phosphorylated following calcium-ionophore treatment of cells (Rabault and Ghysdael 1994), demonstrating that our in vitro system is suitable for mechanistic studies.
Figure 2.

Calcium-dependent phosphorylation and inhibition of Ets-1 DNA binding. (A) Phosphorylation of Ets-1 by nuclear extract and CaMKII. (Top) PhosphorImage of radiolabeled kinase reaction products separated by SDS-PAGE. (Bottom) Apparent phosphorylation stoichiometry of Ets-1 treated as in lanes 2 and 4 of top panel. Mean (±s.d.) of three experiments. (B) Calcium dependence of Ets-1 phosphorylation by nuclear extract. Ets-1 was treated with nuclear extract and [γ-32P]ATP, with or without calcium and analyzed as in A. (C) Nuclear extract treatment inhibits Ets-1 DNA binding 50-fold. Flag–Ets-1 was incubated with nuclear extract without (mock-treated) or with (phosphorylated) ATP and repurified on α-Flag agarose. (Top) Repurified protein was tested for DNA binding by EMSA. A degradation product with DNA-binding activity is indicated (*). (Bottom) Binding curves were generated by measuring the free DNA signal. Kds (inset) were determined by curve-fitting to [DP]/[Dt] = 1/(1 + (Kd/[P])). (D) CaMKII treatment inhibits Ets-1 DNA binding 50-fold. (Top) EMSA of Ets-1 treated with CaMKII without (mock-treated) or with (phosphorylated) unlabeled ATP. Reaction products were used in DNA-binding assays without repurification. (Bottom) Binding curves were generated from mean (±s.d.) of three EMSA performed as in top panel and analyzed as in C. (E) Ets-1 DNA-binding inhibition is reversed by phosphatase treatment. Phosphorylated, repurified Flag–Ets-1 was mock-treated or incubated with calf intestinal phosphatase (CIP). EMSA analysis was used to derive DNA-binding curves and equilibrium dissociation binding constants as in C.
Table 1.
Phosphate incorporation and phosphorylation-dependent inhibition of Ets-1 and mutants
| Ets-1 protein
|
Treatmenta
|
mol phosphate/mol Ets-1 (mean ± s.d.)
|
Fold inhibitionb (mean ± s.d.)
|
|---|---|---|---|
| Ets-1 | extract | 5.7 ± 0.9 | 16.6 ± 1.3c |
| Flag–Ets-1 | extract, repurified | N.D. | 62.6 ± 8.7d |
| Flag–Ets-1 | extract + ATP, repurified, ± phosphatase | N.D. | 41.9e |
| Ets-1 | CaMKII | 4.4 ± 0.3 | 55.7 ± 9.5 |
| 4S-A | extract | 0.73 ± 0.2 | 1.3 ± 0.2 |
| 4S-A | CaMKII | 1.4e | 3.0d |
| S251A, S257A | extract | 2.0 ± 0.4 | 3.7 ± 1.6 |
| S282A, S285A | extract | 3.3 ± 0.6 | 5.7 ± 1 |
| L429A | extract | 4.3 ± 0.5 | 2.1 ± 0.4 |
| Flag–L429A | extract + ATP, repurified, ± phosphatase | N.D. | 6.2e |
| L429A | CaMKII | 4.9 ± 0.4 | 5.8 ± 2.0 |
| Y307P | extract | 4.2 ± 2.3 | 7.0 ± 3.0 |
Values represent mean of three experiments except where noted otherwise. (N.D.) Not determined
Ets-1 proteins were treated with indicated agent ± ATP except where noted otherwise.
Fold inhibition = Kd phosphorylated/Kd mock-treated.
Represents a minimum estimate (see Materials and Methods).
Mean of two experiments.
Value from a single experiment.
Phosphorylation inhibits Ets-1 DNA binding 50-fold
To measure inhibitory effects of phosphorylation on Ets-1 DNA binding, we used a quantitative electrophoretic mobility shift assay (EMSA) to determine equilibrium dissociation binding constants (Kd) of mock-treated or phosphorylated Ets-1. In the case of nuclear extract, competing binding activities in the extract interfered with the ability to accurately measure the affinity of phosphorylated Ets-1 (see Materials and Methods; Table 1). Therefore, Flag-tagged Ets-1 was treated and subsequently repurified prior to analysis of DNA binding. Phosphorylation of Ets-1 by either nuclear extract or CaMKII reduced its affinity for DNA ∼50-fold (Fig. 2C,D; Table 1). Alkaline phosphatase treatment of extract-phosphorylated, repurified Flag–Ets-1 resulted in a 42-fold increase in affinity (Fig. 2E; Table 1), demonstrating that the effect was reversible and was not a result of Ets-1 degradation in the nuclear extract. Together, these results establish that calcium-dependent phosphorylation inhibits Ets-1 DNA binding ∼50-fold. These quantitative analyses set the stage to investigate the mechanism of phosphorylation-dependent Ets-1 inhibition.
Four phosphoserines cooperatively inhibit Ets-1 DNA binding
We first explored the contributions of various phosphorylation sites to inhibition. A previous study identified four serines as targets of calcium-dependent phosphorylation in vivo (Rabault and Ghysdael 1994; Fig. 1C). We mutated all four serines together or in pairs to alanine and tested the phosphorylation and inhibition of these mutants. Mutation of all four serines to alanine (4S-A) reduced phosphorylation by either nuclear extract or CaMKII to a level of approximately one mole of phosphate per mole of Ets-1 (Table 1). The residual phosphorylation of this mutant had very little effect on its DNA-binding affinity (Table 1), indicating that at least one of these serines is required to mediate dramatic effects on Ets-1 DNA binding. Mutation of the serines in pairs resulted in intermediate phosphorylation levels of two to three phosphates per Ets-1 molecule by nuclear extract (S251A;S257A, S282A;S285A, Table 1). However, phosphorylation of these double mutants decreased their DNA-binding affinities only four- and sixfold, respectively (Table 1). This result suggests that the 50-fold inhibition of wild-type Ets-1 is due to a synergistic effect of the two pairs of phosphorylated residues.
The cooperative requirement for multiple phosphoserines in inhibition suggests that structural elements might coordinate the interdependent activity of each phosphate. The phosphorylation sites are near the amino-terminal inhibitory helices (Fig. 1), suggesting that phosphorylation might inhibit DNA binding by regulating the function of the inhibitory module. Specifically, our model of autoinhibition predicts that the conformational equilibrium of the inhibitory module determines the affinity of Ets-1 for DNA. Phosphorylation could inhibit Ets-1 DNA binding by affecting this equilibrium. This model makes two principal predictions. First, the structure and function of the inhibitory module should be critical for phosphorylation-dependent inhibition. Second, an increase in the stability of the inhibitory module should be detectable following phosphorylation.
Disruption of the inhibitory module blocks phosphorylation-dependent inhibition
To determine whether the structure and function of the inhibitory module are important for phosphorylation-dependent regulation, we first generated single amino acid substitutions that disrupt Ets-1 autoinhibition. Tyrosine 307 in helix HI-1 was mutated to proline, which is expected to constitutively unfold this metastable helix (Fig. 3A, Y307P). Leucine 429 in helix H4 was mutated to alanine to disrupt packing of the inhibitory module (Fig. 3A, L429A; Jonsen 1999). The Y307P and L429A proteins displayed enhanced DNA binding relative to wild-type Ets-1 (Fig. 3B), consistent with a loss of autoinhibition. Partial proteolysis was used to determine if the mutations disrupted the inhibitory module (Fig. 3C). Multiple sites within inhibitory helix HI-1 become accessible to trypsin cleavage after the unfolding of the inhibitory module (Petersen et al. 1995; Jonsen et al. 1996; Fig. 1). When compared with wild-type Ets-1, cleavage at these sites was dramatically enhanced in both the L429A and Y307P proteins as evidenced by the appearance of T5 (Fig. 3C). Other cleavage sites such as the lysine at the amino terminus of T3 were unaffected by the mutations, indicating that the proteins were not globally misfolded. The enhanced DNA binding and protease sensitivity of the Y307P and L429A mutants indicate that the mutations cause constitutive unfolding of HI-1 and a loss of autoinhibition.
Figure 3.
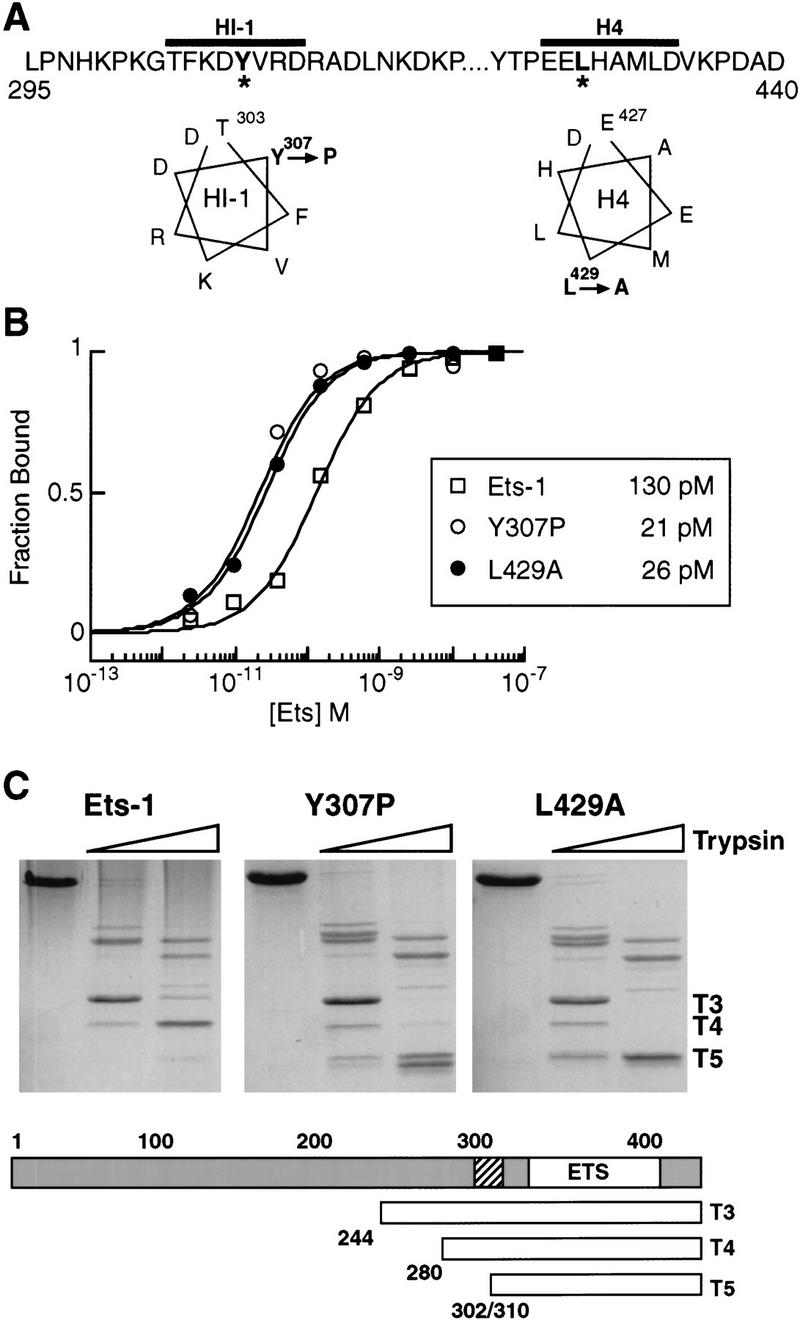
Mutational disruption of the Ets-1 inhibitory module. (A) Sequence and secondary structure of inhibitory helices HI-1 and H4. Y307P and L429A mutations are marked with asterisks and designated on helical wheel diagrams. (B) DNA-binding affinities of Ets-1, Y307P, and L429A. Binding curves and Kd values (inset) were derived from EMSA of bacterially expressed, purified proteins. (C) Trypsin proteolysis of Ets-1, Y307P, and L429A. (Top) Coomassie blue-stained SDS-PAGE of protease digestion products; (bottom) cleavage positions of T3, T4, and T5 fragments are indicated relative to inhibitory helix HI-1 (hatched) and the ETS domain (ETS, white). Note that T5 represents a doublet with products cleaved at position 302 or 310.
We analyzed phosphorylation and DNA binding of L429A and Y307P to determine the effects of inhibitory module disruption on phosphorylation-dependent inhibition. L429A and Y307P were phosphorylated by nuclear extract at levels similar to wild-type Ets-1 (Fig. 4A; Table 1). Additionally, CaMKII phosphorylated L429A to the same extent as wild-type Ets-1 (Fig. 4A; Table 1). However, L429A and Y307P were inhibited only three- to sevenfold by treatment with either nuclear extract or CaMKII (Fig. 4B–D; Table 1). The same result also was found when Flag-tagged L429A was phosphorylated by nuclear extract, repurified, and treated with phosphatase (Table 1). These findings contrast with the 50-fold inhibition of wild-type Ets-1. The data suggest that the two mutant forms of Ets-1, although fully phosphorylated, cannot mediate the inhibitory effects of this phosphorylation because of the constitutive disruption of the inhibitory module. We conclude that a functional inhibitory module is essential for the phosphorylation-dependent inhibition of Ets-1 DNA binding.
Figure 4.
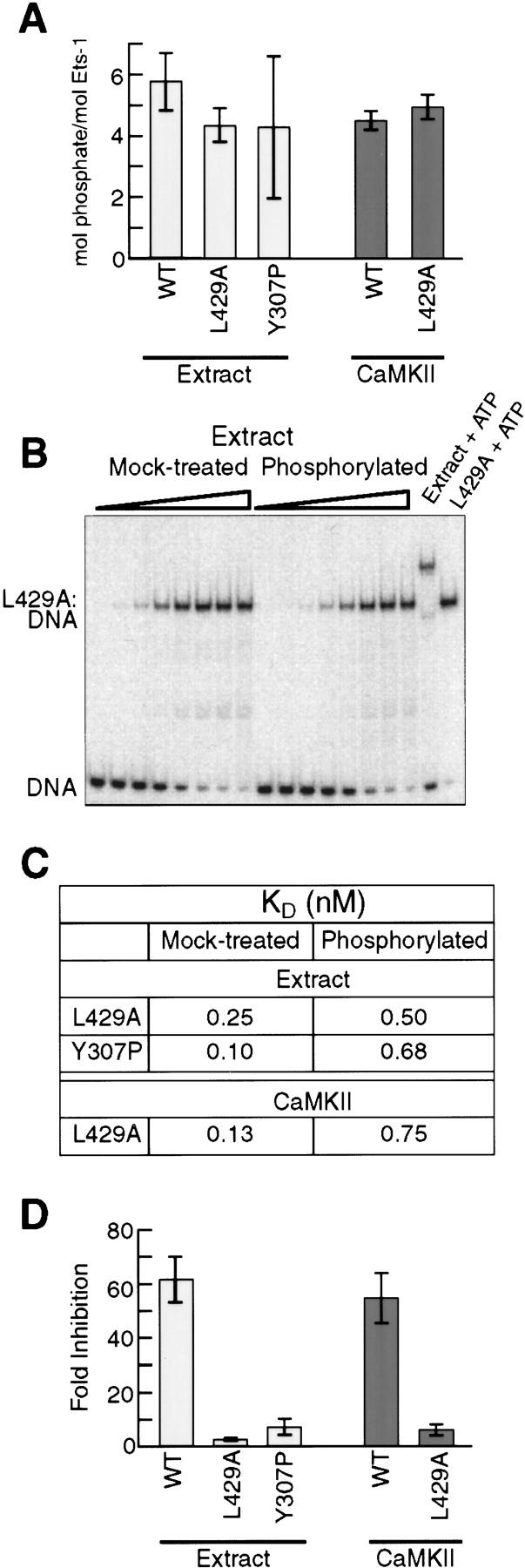
A disrupted inhibitory module impairs phosphorylation-dependent repression of DNA binding. (A) Phosphorylation of wild-type Ets-1 and inhibitory-module mutants by nuclear extract or CaMKII. Bars indicate mean phosphate incorporation (±s.d.) from three experiments. (B) Equilibrium DNA-binding studies of L429A. L429A was treated with nuclear extract without (mock-treated) or with (phosphorylated) ATP. Reaction mixtures were diluted into DNA-binding buffer and analyzed by EMSA. (C) Kd of mock-treated and phosphorylated L429A and Y307P. L429A and Flag–Y307P were treated with nuclear extract and analyzed as in B; L429A was treated with CaMKII and analyzed as in Fig. 2D. Kd values were obtained from mean data points of three experiments. (D) Phosphorylation-dependent inhibition of wild-type Ets-1, L429A, and Y307P. Mean (±s.d.) inhibition values were determined from Kd values of two [extract-treated wild-type (WT)] or three (all others) individual EMSA experiments. Extract-treated wild-type represents inhibition of Flag–Ets-1 repurified from nuclear extract as in Fig. 2C.
Phosphorylation stabilizes the inhibitory module
We next sought to test the second prediction of our model, that the repressive effects of phosphorylation are due to stabilization of the inhibitory module. As presented earlier, the loss of structure of the inhibitory module can be monitored by partial trypsin proteolysis. We reasoned that protease sensitivity could also detect inhibitory module stabilization. However, trypsin was not suitable for this purpose because a major cleavage site of trypsin lies at position 280, between the two pairs of phosphoacceptor sites (Fig. 5A). To solve this problem, we chose endoproteinase Lys-C, which generates two proteolytic products from the carboxyl terminus of Ets-1 (Fig. 5A). The ΔN246 fragment is produced first by cleavage carboxy-terminal of lysine 245 and includes all of the calcium-dependent phosphorylation sites. This fragment is then cleaved within the inhibitory module to yield ΔN302. The inhibitory module of the L429A mutant displays dramatically enhanced production of ΔN302 by Lys-C (data not shown). Thus, this Lys-C sensitivity is similar to the trypsin sensitivity in helix HI-1, which has been used previously to gauge the conformation of the inhibitory module (Fig. 3; Petersen et al. 1995; Jonsen et al. 1996).
Figure 5.
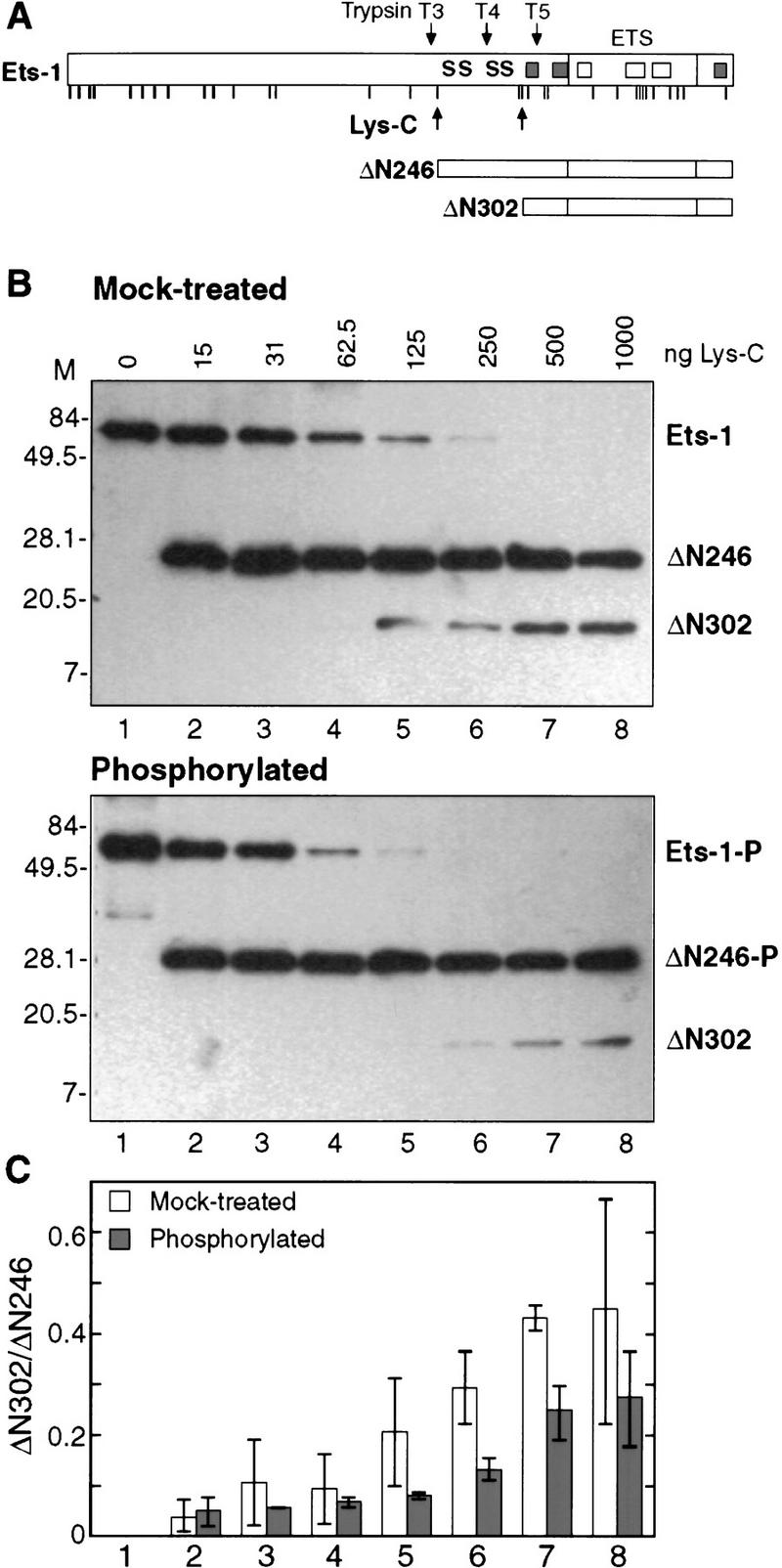
CaMKII phosphorylation stabilizes the Ets-1 inhibitory module. (A) Schematic of full-length Ets-1 and carboxy-terminal proteolytic fragments from endoproteinase Lys-C digestion. Potential Lys-C cleavage sites are indicated by vertical lines (see Fig. 3A for sequence). Previously characterized trypsin cleavage sites are labeled as in Fig. 3C. Calcium-dependent phosphorylation sites are indicated by S. α-Helices of the ETS domain and inhibitory module are indicated by rectangles. (B) Western analysis of Lys-C partial proteolysis. Mock-treated (top) or phosphorylated (bottom) Ets-1 was digested with indicated amounts of endoproteinase Lys-C and analyzed by SDS-PAGE and Western blotting with antiserum specific to the Ets-1 carboxyl terminus. (C) Quantitation of Western blots. The ratio of ΔN302/ΔN246 was obtained by densitometry of Western blots from B. Data show mean (±s.d.) of two experiments.
The digestion of ΔN246 to yield ΔN302 provides an indicator of the stability of the inhibitory module. To test the effect of phosphorylation on the stability of the inhibitory module, the Lys-C sensitivity of Ets-1 was analyzed after mock treatment or phosphorylation by CaMKII (Fig. 5B,C). The amount of ΔN246 produced at each concentration of Lys-C was approximately equal for unmodified or phosphorylated Ets-1. However, the ratio of ΔN302 to ΔN246 was significantly higher in mock-treated Ets-1 than in phosphorylated Ets-1. Thus, phosphorylated ΔN246 is less sensitive to Lys-C at residues near helix HI-1. These findings demonstrate that the inhibitory module is shifted toward the folded conformation by phosphorylation and strongly indicate that phosphorylation inhibits Ets-1 DNA binding by stabilizing the inhibitory module.
Discussion
Model for phosphorylation-dependent inhibition of Ets-1 DNA binding
Our studies of Ets-1 DNA binding have established a link between the phenomena of autoinhibition and phosphorylation-dependent regulation. The Ets-1 inhibitory module is modeled as a four-helix bundle created by packing of three inhibitory α-helices with helix H1 of the ETS domain (Skalicky et al. 1996; Fig. 1). Ets-1 DNA binding is accompanied by a conformational change from the folded to the unfolded state. Specifically, helix HI-1 unfolds on DNA binding (Petersen et al. 1995; Jonsen et al. 1996). The relatively low DNA-binding affinity of Ets-1 is proposed to be due to this required conformational change that couples the stability of the inhibitory module to DNA binding. These findings lead to a model of Ets-1 in which the inhibitory module exists in equilibrium between folded and unfolded states. Three lines of evidence in this report suggest that phosphorylation inhibits Ets-1 DNA binding by shifting this equilibrium toward the folded state (Fig. 6). First, phosphorylation of serines 251, 257, 282, and 285 synergistically inhibits Ets-1 DNA binding, suggesting the involvement of structural elements. Next, Ets-1 mutants that display constitutively disrupted inhibitory modules are resistant to inhibition by phosphorylation, indicating that the folding of the inhibitory module is critical for this inhibition. Finally, the protease experiments indicate that phosphorylation reduces cleavage within the inhibitory module, consistent with a shift of the conformational equilibrium towards the folded state. Together, these three observations support the model that phosphorylation inhibits Ets-1 DNA binding by stabilizing the inhibitory module.
Figure 6.

Model of phosphorylation-dependent inhibition of Ets-1 DNA binding. Electrostatic interactions between phosphoserines (P) and basic residues of the inhibitory module stabilize the inhibitory conformation, shifting the equilibrium toward the folded state. DNA binding is repressed by the higher energetic cost of the conformational change that accompanies DNA binding.
How might phosphorylation stabilize the inhibitory module? We hypothesize that negatively charged phosphate groups promote electrostatic interactions between the phosphorylated region and the inhibitory module (Fig. 6). The calcium-dependent phosphorylation sites are found in primary sequence environments punctuated by regularly spaced acidic residues (Fig. 1C). Phosphorylation of the four serines would increase the local negative charge substantially, facilitating ion-pairing interactions with positively charged residues of the inhibitory module. Inhibitory helix HI-1 is within a 21-residue segment of Ets-1 that contains eight basic amino acids (Fig. 3A). Interactions between these basic residues and the phosphorylated serines could reduce local unfolding, thus stabilizing the entire inhibitory module. Consistent with this proposal, substitution of the four phosphoacceptor serines with glutamic acid partially mimics the effects of phosphorylation (data not shown).
The synergistic inhibitory effects of the four phosphoserines suggest that structural elements may exist within the phosphorylated region. Arginine 263, a potential trypsin site between the pairs of phosphoacceptor residues, is not cleaved in proteolysis experiments (Jonsen et al. 1996), suggesting that residues in this region may be constrained. Secondary or tertiary structure could cluster the pairs of phosphorylated serines, enhancing electrostatic interactions with the positively charged surface of the inhibitory module. Experiments are underway to test the predictions of this model.
A new biological role for autoinhibition
The finding that phosphorylation enhances Ets-1 autoinhibition provides a new perspective on the role of autoinhibition in a biological setting. Autoinhibition has been viewed as a mechanism to repress protein activity in the absence of biological inputs that counteract the inhibition. By this view, autoinhibitory mechanisms function as simple switches to keep the activity of a protein off until a signal turns the activity on. For example, cooperative DNA binding between heterologous transcription factors can disrupt autoinhibition of DNA binding, allowing one or both factors to bind with high affinity in the presence of the partner. Examples of this phenomenon include cooperative binding partnerships between a1 and α2 (Stark et al. 1999), PU.1 and Pip (Brass et al. 1996), Elk-1 or Sap-1 and SRF (Dalton and Treisman 1992; Ling et al. 1997), and others. Phosphorylation also has been described as a biological input that can counteract transcription factor autoinhibition. For example, phosphorylation derepresses the DNA-binding activity of p53 (Ko and Prives 1996) and the transcriptional activation capabilities of C/EBPβ (Kowenz-Leutz et al. 1994) and NF-κB p65 (Zhong et al. 1998). Thus, autoinhibition can restrict transcriptional activities to cellular contexts in which an appropriate partner protein or signal counteracts the repression. Our results expand this traditional view of autoinhibition by demonstrating that an inhibitory module can provide a structural switch to repress activity in response to a signaling event.
In concert with other studies of Ets-1 DNA binding and autoinhibition, our results suggest that the Ets-1 inhibitory module acts as a rheostat that regulates DNA binding in response to biological signals. Our data demonstrate that calcium-dependent phosphorylation uses the inhibitory module to repress Ets-1 DNA binding. On the other hand, Ets-1 binds DNA cooperatively with several binding partners that could counteract autoinhibition. Ets-1 partners include NF-κB (Bassuk et al. 1997; Dickinson et al. 1999), Jun/Fos (Logan et al. 1996), USF and TFE3 (Sieweke et al. 1998; Tian et al. 1999), and CBFα2 (Wotton et al. 1994; Kim et al. 1999; Goetz et al. 2000; Gu et al. 2000). Whereas the mechanism of cooperative binding with most of these proteins has not been characterized, in the case of CBFα2, cooperative binding has been shown to occur via CBF-mediated disruption of Ets-1 autoinhibition (Goetz et al. 2000; Gu et al. 2000). Thus, the Ets-1 inhibitory module maintains the DNA-binding activity in a partially repressed state, allowing the binding to be enhanced by protein partners or repressed further by calcium-dependent phosphorylation in response to cellular activation.
Biological roles for calcium-dependent regulation of Ets-1 DNA binding
Functional data indicate that Ets-1 regulates genes important for lymphocyte biology. Gene inactivation studies in mice have demonstrated that Ets-1 is required for the development of natural killer cells and for normal differentiation, homeostasis, and activation of T and B lymphocytes (Bories et al. 1995; Muthusamy et al. 1995; Barton et al. 1998). Although the critical target genes regulated by Ets-1 are not known, these phenotypes support a role for Ets-1 in lymphocyte activation. Endogenous Ets-1 is phosphorylated by calcium signals during lymphocyte activation (Pognonec et al. 1988; Fisher et al. 1991), suggesting that the phenomenon regulates activation-dependent transcriptional responses. The results presented here confirm that these calcium signals inhibit Ets-1 DNA binding dramatically. These observations are compatible with two distinct models for the role of calcium-signaling in Ets-1 function.
First, the calcium-dependent inhibition of Ets-1 DNA binding could be part of a general mechanism to remove Ets-1 function from the cell during activation. Expression of ets-1 is down-regulated after cellular activation (Bhat et al. 1990), suggesting that Ets-1 may need to be eliminated from the activated cell. Thus, calcium-dependent phosphorylation could be a dominant, rapid mechanism to inactivate Ets-1 DNA binding as a part of this clearance mechanism. In this model, Ets-1 function would be restricted to resting lymphocytes.
Our new mechanistic insights suggest an alternative role for calcium signaling in the regulation of Ets-1. As discussed above, the inhibitory module mediates both negative regulation of DNA binding in response to phosphorylation and cooperative DNA binding with protein partners. We propose that certain protein partners are able to override the inhibitory effects of phosphorylation. In this model, cellular activation and phosphorylation would specifically direct Ets-1 to target sites with partner proteins that could overcome the phosphorylation-induced inhibition of DNA binding. In support of this model, Ets-1 can mediate activation-dependent transcriptional responses from certain promoter elements in transient overexpression assays in association with other DNA binding proteins (Lambert et al. 1997; Thomas et al. 1997; Blumenthal et al. 1999). The ets-1 expression data could be accommodated in this model if such transcriptional events were restricted to a relatively short interval after cellular activation but prior to elimination of Ets-1. The mechanistic modeling from this study will guide future biological and biochemical investigation of these proposed models.
In summary, our results highlight the critical role of the Ets-1 inhibitory module as an effector of both positive and negative DNA-binding regulation. Autoinhibition is a common feature of kinases, phosphatases, and transcription factors (Soderling 1990; Kemp et al. 1996; Graves et al. 1998). These classes of proteins are the key players in the chain of signaling events that control cell growth, differentiation, and homeostasis. This essential role necessitates tight control of these protein activities as well as mechanisms to modulate the activities in response to appropriate stimuli. Our results demonstrate that autoinhibitory mechanisms can provide this control by acting as structural switches that integrate various biological inputs to generate appropriate responses.
Materials and methods
Site-directed mutagenesis and construction of bacterial expression constructs
Mutant forms of murine Ets-1 were generated by site-directed mutagenesis. Primers for phosphorylation site mutants include serines 251 and 257 to alanine: 5′-CTATCGTAGGCCTCTACGCTCTCAAACGCGTCCTGGCCCCCG-3′; serines 282 and 285 to alanine: 5′-CGTAGTCGAAGGCGTCATATGCGGGGACCCGCTGCAGGC-3′. L429A substitution was generated using the primer 5′-CCAGCATGGCGTGCGCCTCTTCAGGG-3′. Mutations were placed into the context of full-length ets-1 in the pAED-4 expression vector, a derivative of pET3 that bears a high-copy origin of replication (Doering 1992). Flag–Ets-1 was generated by cloning a FLAG/HMK (Blanar and Rutter 1992) cassette into the NdeI site of the pET3a–Ets-1 construct (Nye et al. 1992). The Flag–L429A and Flag–Y307P mutants were generated with the following primers L429A, 5′-CAGCATGGCGTGCGCCTCTTCAGGGGTG-3′ and Y307P, 5′-CACCTTCAAGGACCCTGTGCGTGACCGTGC-3′.
Protein expression and purification
All mutant Ets-1 proteins were expressed in Escherichia coli BL21 (DE3). In the case of wild-type Ets-1, a previously described strain bearing pET3 with a full-length open reading frame of murine Ets-1 and the pLysS plasmid was available (Nye et al. 1992). Cultures (500 ml) were grown at 37°C in Luria broth and protein expression was induced at mid-log phase for 2 hr by addition of 1 mm IPTG. Harvested cells were resuspended in 25 ml of lysis buffer (25 mm Tris-Cl at pH 7.9, 0.1 m EDTA, 1 m KCl) with 1 mm phenylmethylsulfonyl fluoride (PMSF) and 1 mm dithiothreitol (DTT) and lysed by sonication (Heat Systems, Inc). The insoluble fraction was collected by centrifugation at 27,000g for 15 min. To wash the pellet, sonication and centrifugation were repeated. The insoluble pellet was solubilized by sonication in 25 ml of 10 mm Tris-Cl (pH 7.6), 750 mm NaCl, 5 m urea, 0.1% (vol/vol) Triton X-100, 1 mm DTT, and 1 mm PMSF, then centrifuged at 27,000g for 15 min at 4°C. The supernatant was loaded on a 5-ml DEAE–cellulose column (Pierce) at 4°C. The flowthrough fraction was subjected to dialysis at 4°C, two times for 2 hr each against 10 volumes of TGEK50 (25 mm Tris-Cl at pH 7.9, 10% glycerol, 0.1 mm EDTA, 50 mm KCl) with 1 mm DTT and 0.2 mm PMSF. After dialysis, soluble protein was loaded onto a 26/10 Hi Load S-Sepharose column (Pharmacia) in TGEK with 1 mm DTT at 4°C and eluted with a linear gradient of KCl from 50 to 500 mm over five column volumes. All Ets-1 species eluted between ∼200 and 300 mm KCl. Protein aliquots were snap frozen and stored at −80°C.
Purity of Ets-1 species was estimated to be >90% by Coomassie brilliant blue staining of SDS–polyacrylamide gels. Protein concentrations were determined by spectrophotometry after denaturing in 6 m guanidine–HCl (Pierce), using extinction coefficients calculated based on the method of Gill and von Hippel (1989). The following extinction coefficients were used: Ets-1 wild-type, L429A, 4S-A, S251A;S257A, and S282A;S285A—81200/(m × cm) at 278 nm; Flag–Ets-1, Flag–L429A—80650/m × cm) at 280 nm; Flag–Y307P—79370 m x cm at 280 nm. The concentration of the Ets-1 4S-A protein was corrected to reflect active protein concentration as determined experimentally by DNA titration (Jonsen et al. 1996). The active concentrations of other serine mutant Ets-1 proteins were not determined because, unlike 4S-A, the binding affinities were within the range of 2 × 10−10 to 4 × 10−10 m expected from previous analyses in which preparations were determined to be 100% active (Jonsen et al. 1996).
Kinase assays and quantitation of Ets-1 phosphorylation
Nuclear extract was prepared from the murine T cell hybridoma Hd11.2 (Meikle et al. 1992, a generous gift of Dr. Ray Daynes, University of Utah) as described previously (Gunther and Graves 1994) except that buffer A (lysis buffer) was supplemented with 1 mm EGTA. Total protein concentration of nuclear extract preparations ranged from 4 to 6 mg/ml as determined by Bradford assay (Bio-Rad). Nuclear extract kinase assays were performed at 30°C for 30 min in kinase buffer (30 mm HEPES at pH 7.5, 7 mm MgCl2, 100 μm CaCl2, and 1 mm DTT) with 1 mg/ml nuclear extract protein, 1.25 μm Ets-1 and 1 mm unlabeled or [γ-32P]ATP (∼150 mCi/mmole). Products of reactions with unlabeled ATP were diluted into DNA-binding buffer for EMSA analysis. Reactions with [γ-32P]ATP were stopped by adding Laemmli SDS-PAGE sample buffer and heating to 95°C for 5 min, then separated by SDS-PAGE. Gels were stained with Coomassie brilliant blue, dried, and analyzed by PhosphorImager with Image Quant (v. 1.1, Molecular Dynamics).
CaMKIIα, purified from a baculovirus expression system, was generously provided by Dr. Thomas Soderling (Oregon Health Sciences University, Portland). CaMKII kinase assays were performed at 30°C for 30 min with 50 mm HEPES (pH 7.5), 10 mm magnesium acetate, 0.5 mm CaCl2, 2 mm DTT, 1 μm calmodulin (Calbiochem), 2 μm Ets-1, and 100 nm CaMKII. Unlabeled or [γ-32P]ATP was added at 0.4 mm to appropriate reactions. CaMKII was diluted appropriately in 50 mm HEPES (pH 7.5), 10% ethylene glycol, 2 mg/ml bovine serum albumin (BSA) before adding to reactions.
The apparent stoichiometry of Ets-1 phosphorylation (moles of phosphate per mole of Ets-1) was estimated from kinase reactions that included [γ-32P]ATP as follows. Precise quantities of [γ-32P]ATP (10–500 pmoles) were spotted on filter paper. Phosphorimaging of these known quantities of isotope concurrently with SDS gels containing known quantities of phosphorylated Ets-1 provided data to convert the signal from Ets-1 gel bands into moles of phosphate per mole of Ets-1.
Repurification and phosphatase treatment of phosphorylated Flag–Ets-1 proteins
Kinase assays were performed as described above with 2 mg/ml nuclear extract protein, 2.5 μm Flag–Ets-1 or Flag–L429A, and 1 mm unlabeled ATP in appropriate reactions. Flag-tagged proteins were repurified with anti-Flag M2–Agarose (Sigma) and eluted with Flag peptide (Sigma) at 500 μg/ml in 25 mm Tris-Cl (pH 7.9), 0.1 mm EDTA, 50 mm KCl, 1 mm DTT. Concentrations of repurified proteins were determined by densitometry of Coomassie-blue-stained SDS–polyacrylamide gels with a standard curve prepared from known amounts of pure Flag–Ets-1. Phosphatase experiments were performed with phosphorylated, repurified Flag–Ets-1, or Flag–L429A. Phosphorylated proteins were incubated with 10 units of calf intestinal phosphatase (New England Biolabs) at 30°C for 30 min, then the reaction was stopped by addition of phosphatase inhibitors (50 mm NaF, 20 mm β-glycerol phosphatase, 2 mm Na3VO4, and 5 mm Na4P2O7). An equivalent amount of control protein was incubated at 4°C with phosphatase inhibitors. Reaction mixtures were stored at 4°C until dilution into binding buffer for DNA-binding assays.
DNA-binding assays
Kds were measured by EMSA with the high-affinity ets-binding site duplex SC1 (Nye et al. 1992) as described previously (Jonsen et al. 1996). In brief, binding reaction mixtures were prepared on ice in 20 μl with 25 mm Tris-Cl (pH 7.9), 10% glycerol, 0.1 mm EDTA, 60 mm KCl, 6 mm MgCl2, 0.2 mg/ml BSA, 10 mm DTT, and 50 ng of poly[d(I-C)]. EMSA was performed at 4°C on 5% native polyacrylamide gels in 0.5× TBE buffer. Kds were determined by plotting the fraction of bound DNA ([DP]/[Dt]) versus the concentration of free protein ([P]) and fitting the mean data points to the rearranged mass action equation [DP]/[Dt] = 1/(1 + (Kd/[P])) using nonlinear least-squares analyses (KaleidaGraph, v. 3.0, Synergy Software) (Jonsen et al. 1996). The total concentration of DNA ([Dt]) was at least 10-fold below the expected Kd of the Ets-1 species, allowing the assumption that free protein concentration was approximately equal to total protein concentration. The fraction of bound DNA was calculated from PhosphorImager analysis of the free DNA signal as this is the most accurately measured parameter in EMSA (Carey 1991). The fraction of bound DNA at each Ets-1 concentration was calculated from the fraction of free DNA, [DP]/[Dt] = 1 − ([D]/[Dt]). Data points are mean values from 3 experiments except where noted. Error bars represent standard deviation of the mean for each point. Fold inhibition reported in Fig. 4D and Table 1 was calculated as the ratio Kd phosphorylated/Kd mock treated.
The phosphorylation-dependent inhibition measured for nonrepurified, extract-treated Ets-1 was only ∼17-fold (Table 1), owing to the fact that binding curves were generated by measuring the free DNA fraction. Upon inhibition of Ets-1 DNA binding, the background DNA-binding activities in nuclear extract contributed significantly to the disappearance of the free DNA signal (data not shown). Thus, the presence of nuclear extract prohibited accurate measurement of binding constants for phosphorylated wild-type Ets-1. DNA-binding experiments reported for mutant Ets-1 proteins were performed without repurification because, unlike wild-type Ets-1, the binding activities of mutant proteins were above the background of the nuclear extract regardless of their phosphorylation state (e.g., Fig. 4B).
L429A and Y307P display 5- to 10-fold higher affinity than wild-type Ets-1 only in the absence of MgCl2 (data not shown). Therefore, in experiments with these proteins, MgCl2 was omitted from DNA-binding buffer and EDTA was added to a final concentration of 5 mm. This change had very little effect (2×) on the binding affinity of wild-type Ets-1 (data not shown). Nevertheless, DNA-binding experiments of wild-type Ets-1 treated with CaMKII or Flag–Ets-1 repurified from nuclear extracts were performed without MgCl2 to allow direct comparison to L429A. Flag–L429A, Flag–Y307P, and wild-type Ets-1 experienced a loss of DNA-binding activity during incubation at 30°C (cf. Figs. 3B and 4C). Our experimental design controlled for this phenomenon by directly comparing the effect of phosphorylation only to mock-treated controls.
Proteolysis assays
For Lys-C experiments, Ets-1 was mock-treated or phosphorylated by CaMKII as described above except that 50-μl reactions contained Ets-1 and CaMKII concentrations of 10 μm and 250 nm, respectively, and CaMKII was diluted in 50 mm HEPES (pH 7.5) and 10% ethylene glycol without BSA. In control reactions, these conditions resulted in full phosphorylation of Ets-1 in <30 min at 30°C (data not shown). After 30 min at 30°C, reactions were stopped by addition of EDTA to 20 mm final concentration. Sequencing grade endoproteinase Lys-C (Promega) was diluted in 25 mm sodium phosphate (pH 7.7), 1 mm EDTA. Aliquots of kinase reaction mix (5 μl, ∼2.5 μg of Ets-1) were added to 5 μl appropriately diluted Lys-C and incubated 1 hr at 30°C. Reactions were stopped by addition of SDS-PAGE sample buffer and 5 min incubation at 95°C. Approximately 200 ng of Ets-1 digestion products were resolved by 15% SDS-PAGE and transferred to PVDF membrane (Bio-Rad) in 25 mm Tris (pH 8.3), 192 mm glycine, 20% methanol, 0.1% SDS. Western analysis used rabbit polyclonal antiserum directed against the carboxyl terminus of Ets-1 (Gunther and Graves 1994) and ECL Plus detection system (Amersham). Quantitation was performed by autoradiography and densitometry using Image Quant (v. 1.1 Molecular Dynamics).
Trypsin digestions were performed in 20 μl of buffer (25 mm Tris at pH 7.9, 10 mm CaCl2, 1 mm DTT) with 5.5 μm Flag–Ets-1, Flag–L429A, or Flag–Y307P and 0, 20, or 200 ng of trypsin (Sigma). Reaction mixtures were incubated 2 min at 25°C, then stopped as described above. Products were separated by 15% SDS-PAGE and visualized by staining with Coomassie blue. Amino termini of trypsin and Lys-C digestion products were determined by Edman sequencing on an ABI 477A protein sequencer (Applied Biosystems).
Acknowledgments
We thank Nathan Heaps and Drs. Thomas Soderling and Raymond Daynes for providing reagents used in these studies. We are grateful to Dr. Brad Cairns and members of the Graves laboratory for helpful suggestions. This work was supported by the National Institutes of Health (GM-38663 to B.J.G., predoctoral fellowship HD-07491 to D.O.C., and CA-24014 to core facilities of the Huntsman Cancer Institute). Support from the Center for Excellence in Molecular Hematology DK49219 is also acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL barbara.graves@hci.utah.edu; FAX (801) 585-1980.
References
- Barton K, Muthusamy N, Fischer C, Ting C-N, Walunas T, Lanier L, Leiden J. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity. 1998;9:555–563. doi: 10.1016/s1074-7613(00)80638-x. [DOI] [PubMed] [Google Scholar]
- Bassuk AG, Anandappa RT, Leiden JM. Physical interactions between Ets and NF-kappaB/NFAT proteins play an important role in their cooperative activation of the human immunodeficiency virus enhancer in T cells. J Virol. 1997;71:3563–3573. doi: 10.1128/jvi.71.5.3563-3573.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor A, Piper D, Charles de la Brousse F, Mcknight S, Wolberger C. The structure of GABPα/β: An ETS domain-ankryin repeat heterodimer bound to DNA. Science. 1998;279:1037–1041. doi: 10.1126/science.279.5353.1037. [DOI] [PubMed] [Google Scholar]
- Bhat NK, Thompson CB, Lindsten T, June CH, Fujiwara S, Koizumi S, Fisher RJ, Papas TS. Reciprocal expression of human ETS1 and ETS2 genes during T-cell activation: Regulatory role for the protooncogene ETS1. Proc Natl Acad Sci. 1990;87:3723–3727. doi: 10.1073/pnas.87.10.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanar MA, Rutter WJ. Interaction cloning: Identification of a helix-loop-helix zipper protein that interacts with c-Fos. Science. 1992;256:1014–1018. doi: 10.1126/science.1589769. [DOI] [PubMed] [Google Scholar]
- Blumenthal SG, Aichele G, Wirth T, Czernilofsky AP, Nordheim A, Dittmer J. Regulation of the human interleukin-5 promoter by Ets transcription factors. Ets1 and Ets2, but not Elf-1, cooperate with GATA3 and HTLV-I Tax1. J Biol Chem. 1999;274:12910–12916. doi: 10.1074/jbc.274.18.12910. [DOI] [PubMed] [Google Scholar]
- Bories J-C, Willerford DM, Grevin D, Davidson L, Camus A, Martin P, Stehelin D, Alt FW. Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature. 1995;377:635–638. doi: 10.1038/377635a0. [DOI] [PubMed] [Google Scholar]
- Brass AL, Kehrli E, Eisenbeis CF, Storb U, Singh H. Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the ets factor PU.1. Genes & Dev. 1996;10:2335–2347. doi: 10.1101/gad.10.18.2335. [DOI] [PubMed] [Google Scholar]
- Carey J. Gel retardation. Methods Enzymol. 1991;208:103–117. doi: 10.1016/0076-6879(91)08010-f. [DOI] [PubMed] [Google Scholar]
- Dalton S, Treisman R. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell. 1992;68:597–612. doi: 10.1016/0092-8674(92)90194-h. [DOI] [PubMed] [Google Scholar]
- Dickinson LA, Trauger JW, Baird EE, Dervan PB, Graves BJ, Gottesfeld JM. Inhibition of Ets-1 DNA binding and ternary complex formation between Ets-1, NF-κB and DNA by a designed DNA-binding ligand. J Biol Chem. 1999;274:12765–12773. doi: 10.1074/jbc.274.18.12765. [DOI] [PubMed] [Google Scholar]
- Doering DS. “Functional and structural studies of a small F-actin binding domain.” Ph.D. Thesis. Cambridge, MA: Massachusetts Institute of Technology; 1992. [Google Scholar]
- Donaldson LW, Petersen JM, Graves BJ, McIntosh LP. Solution structure of the ETS domain from murine Ets-1: A winged helix-turn-helix DNA binding motif. EMBO J. 1996;15:125–134. [PMC free article] [PubMed] [Google Scholar]
- Finco TS, Westwick JK, Norris JL, Beg AA, Der CJ, Baldwin AS., Jr Oncogenic Ha-Ras-induced signaling activates NF-κB transcriptional activity, which is required for cellular transformation. J Biol Chem. 1997;272:24113–24116. doi: 10.1074/jbc.272.39.24113. [DOI] [PubMed] [Google Scholar]
- Fisher CL, Ghysdael J, Cambier JC. Ligation of membrane Ig leads to calcium-mediated phosphorylation of the proto-oncogene product, Ets-1. J Immunol. 1991;146:1743–1749. [PubMed] [Google Scholar]
- Fisher RJ, Favash M, Casas-Finet J, Erickson JW, Kondoh A, Bladen SV, Fisher C, Watson DK, Papas T. Real-time DNA binding measurements of the ETS1 recombinant oncoproteins reveal significant kinetic differences between the p42 and p51 isoforms. Protein Sci. 1994;3:257–266. doi: 10.1002/pro.5560030210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Goetz TL, Gu TL, Speck NA, Graves BJ. Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor α2. Mol Cell Biol. 2000;20:81–90. doi: 10.1128/mcb.20.1.81-90.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Graves BJ, Petersen JM. Specificity within the ets family of transcription factors. In: Woude GV, Klein G, editors. Advances in cancer research. San Diego, CA: Academic Press; 1998. pp. 1–55. [DOI] [PubMed] [Google Scholar]
- Graves BJ, Cowley DO, Goetz TL, Petersen JM, Jonsen MD, Gillespie ME. Autoinhibition as a transcriptional regulatory mechanism. Cold Spring Harbor Symp Quant Biol. 1998;63:621–629. doi: 10.1101/sqb.1998.63.621. [DOI] [PubMed] [Google Scholar]
- Gu TL, Goetz TL, Graves BJ, Speck NA. Auto-inhibition and partner proteins, core-binding factor β (CBFβ) and Ets-1, modulate DNA binding by CBFβ2 (AML1) Mol Cell Biol. 2000;20:91–103. doi: 10.1128/mcb.20.1.91-103.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther CV, Graves BJ. Identification of ETS domain proteins in murine T lymphocytes that interact with the Moloney murine leukemia virus enhancer. Mol Cell Biol. 1994;14:7569–7580. doi: 10.1128/mcb.14.11.7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagman J, Grosschedl R. An inhibitory carboxyl terminal domain in Ets-1 and Ets-2 mediates differential binding of ETS family factors to promoter sequences of the mb-1 gene. Proc Natl Acad Sci. 1992;89:8889–8893. doi: 10.1073/pnas.89.19.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsen M. “Auto-inhibition of DNA binding in the transcription factor Ets-1: A mechanistic and structural study.” Ph.D. Thesis. Salt Lake City, UT: University of Utah; 1999. [Google Scholar]
- Jonsen MD, Petersen JM, Xu Q, Graves BJ. Characterization of the cooperative function of inhibitory sequences of Ets-1. Mol Cell Biol. 1996;16:2065–2073. doi: 10.1128/mcb.16.5.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp BE, Barden JA, Kobe B, House C, Parker MW. Intrasteric regulation of calmodulin-dependent protein kinases. Adv Pharmacol. 1996;36:221–249. doi: 10.1016/s1054-3589(08)60584-0. [DOI] [PubMed] [Google Scholar]
- Kim W-Y, Sieweke M, Ogawa D, Wee H-J, Englemeier U, Graf T, Ito Y. Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J. 1999;18:1609–1620. doi: 10.1093/emboj/18.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes & Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Kodandapani R, Pio F, Ni C-Z, Piccialli G, Klemsz M, McKercher S, Maki RA, Ely KR. A new pattern for helix-turn-helix recognition revealed by the PU.1 ETS-domain-DNA complex. Nature. 1996;380:456–460. doi: 10.1038/380456a0. [DOI] [PubMed] [Google Scholar]
- Kowenz-Leutz E, Twamley G, Ansieau S, Leutz A. Novel mechanism of C/EBP (NF-M) transcriptional control: Activation through derepression. Genes & Dev. 1994;8:2781–2791. doi: 10.1101/gad.8.22.2781. [DOI] [PubMed] [Google Scholar]
- Lambert PF, Ludford-Menting MJ, Deacon NJ, Kola I, Doherty RR. The nfkb1 promoter is controlled by proteins of the Ets family. Mol Biol Cell. 1997;8:313–323. doi: 10.1091/mbc.8.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim F, Kraut N, Frampton J, Graf T. DNA binding by c-Ets-1, but not v-Ets, is repressed by an intramolecular mechanism. EMBO J. 1992;11:643–652. doi: 10.1002/j.1460-2075.1992.tb05096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y, Lakey JH, Roberts CE, Sharrocks AD. Molecular characterization of the B-box protein-protein interaction motif of the ETS-domain transcription factor Elk-1. EMBO J. 1997;16:2431–2440. doi: 10.1093/emboj/16.9.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan SK, Garabedian MJ, Campbell CE, Werb Z. Synergistic transcriptional activation of the tissue inhibitor of metaloproteinases-1 promoter via functional interaction of AP-1 and Ets-1 transcription factors. J Biol Chem. 1996;271:774–776. doi: 10.1074/jbc.271.2.774. [DOI] [PubMed] [Google Scholar]
- Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- Meikle AW, Dorchuck RW, Araneo BA, Stringham JD, Evans TG, Spruance SL, Daynes RA. The presence of a dehydroepiandrosterone-specific receptor binding complex in murine T cells. J Steroid Biochem Mol Biol. 1992;42:293–304. doi: 10.1016/0960-0760(92)90132-3. [DOI] [PubMed] [Google Scholar]
- Mo Y, Vaessen B, Johnston K, Marmorstein R. Structures of SAP-1 bound to DNA targets from the E74 and c-fos promoters: Insights into DNA sequence discrimination by Ets proteins. Mol Cell. 1998;2:201–212. doi: 10.1016/s1097-2765(00)80130-6. [DOI] [PubMed] [Google Scholar]
- Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- Muthusamy N, Barton K, Leiden JM. Defective activation and survival of T-cells lacking the Ets-1 transcription factor. Nature. 1995;377:639–642. doi: 10.1038/377639a0. [DOI] [PubMed] [Google Scholar]
- Nye JA, Petersen JM, Gunther CV, Jonsen MD, Graves BJ. Interaction of murine Ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes & Dev. 1992;6:975–990. doi: 10.1101/gad.6.6.975. [DOI] [PubMed] [Google Scholar]
- Petersen JM, Skalicky JJ, Donaldson LW, McIntosh LP, Alber T, Graves BJ. Modulation of transcription factor Ets-1 DNA binding: DNA-induced unfolding of an alpha helix. Science. 1995;269:1866–1869. doi: 10.1126/science.7569926. [DOI] [PubMed] [Google Scholar]
- Pognonec P, Boulukos KE, Gesquiere JC, Stehelin D, Ghysdael J. Mitogenic stimulation of thymocytes results in the calcium-dependent phosphorylation of c-ets-1 proteins. EMBO J. 1988;7:977–983. doi: 10.1002/j.1460-2075.1988.tb02904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabault B, Ghysdael J. Calcium-induced phosphorylation of ETS1 inhibits its specific DNA binding activity. J Biol Chem. 1994;269:28143–28151. [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: Regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Sieweke MH, Tekotte H, Jarosch U, Graf T. Cooperative interaction of Ets-1 with USF-1 required for HIV-1 enhancer activity in T cells. EMBO J. 1998;17:1728–1739. doi: 10.1093/emboj/17.6.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalicky JJ, Donaldson LW, Petersen JM, Graves BJ, McIntosh LP. Structural coupling of the inhibitory regions flanking the ETS domain of murine Ets-1. Prot Sci. 1996;5:296–309. doi: 10.1002/pro.5560050214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR. Protein kinases: Regulation by auto-inhibitory domains. J Biol Chem. 1990;265:1823–1826. [PubMed] [Google Scholar]
- Stark MR, Escher D, Johnson AD. A trans-acting peptide activates the yeast a1 repressor by raising its DNA-binding affinity. EMBO J. 1999;18:1621–1629. doi: 10.1093/emboj/18.6.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Tymms MJ, McKinlay LH, Shannon MF, Seth A, Kola I. ETS1, NFκB and AP1 synergistically transactivate the human GM-CSF promoter. Oncogene. 1997;14:2845–2855. doi: 10.1038/sj.onc.1201125. [DOI] [PubMed] [Google Scholar]
- Tian G, Erman B, Ishii H, Gangopadhyay SS, Sen R. Transcriptional activation by ETS and leucine zipper-containing basic helix-loop-helix proteins. Mol Cell Biol. 1999;4:2946–2957. doi: 10.1128/mcb.19.4.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R. Ternary complex factors: Growth regulated transcriptional activators. Curr Opin Genet Dev. 1994;4:96–101. doi: 10.1016/0959-437x(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Wasylyk C, Kerckaert J-P, Wasylyk B. A novel modulator domain of Ets transcription factors. Genes & Dev. 1992;6:965–974. doi: 10.1101/gad.6.6.965. [DOI] [PubMed] [Google Scholar]
- Werner MH, Clore GM, Fisher CL, Fisher RJ, Trinh L, Shiloach J, Gronenborn AM. Correction of the NMR structure of the ETS1/DNA complex. J Biomol NMR. 1997;10:317–328. doi: 10.1023/a:1018399711996. [DOI] [PubMed] [Google Scholar]
- Wotton D, Ghysdael J, Wang S, Speck NA, Owen MJ. Cooperative binding of Ets-1 and core binding factor to DNA. Mol Cell Biol. 1994;14:840–850. doi: 10.1128/mcb.14.1.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B-S, Hauser CA, Henkel G, Colman MS, Van Beveren C, Stacey KJ, Hume DA, Maki RA, Ostrowski MC. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets-1 and c-Ets-2. Mol Cell Biol. 1996;16:538–547. doi: 10.1128/mcb.16.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-κ B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]