

Abstract
ABCG2 encodes a transporter protein that is associated with multidrug-resistant phenotypes in many cancers, including acute myeloid leukemia (AML); high levels of expression are generally associated with a poor prognosis. To better understand how expression of ABCG2 is controlled in pediatric AML, we performed a detailed analysis of the ABCG2 transcript isoforms from a variety of tissue sources, including 85 pediatric AML samples. These studies revealed a complex 5′ untranslated region (UTR) with 6 novel exons and multiple splice variants. Samples from children with acute megakaryoblastic leukemia (AML FAB-M7) not associated with Down syndrome showed uniformly higher levels of ABCG2 transcripts than samples from children with other AML subtypes. A novel 5′ UTR identified 90 kb upstream of the exon 2 translation initiation site was expressed only in M7 AML subtypes. An associated upstream promoter fragment was shown to be selectively expressed in megakaryoblastic leukemia cells but not in human epithelial cell lines. These findings identify a new tissue-specific ABCG2 promoter that is selectively expressed in pediatric M7 AML. We also show a relatively high incidence of ABCG2 mRNA expression in non-Down associated M7 AML, which may contribute to the relatively poor prognosis of the M7 AML subtype.
Keywords: ABCG2, acute myeloid leukemia, acute megakaryoblastic leukemia, ABC transporters, pediatric leukemia, alternative splicing
Introduction
Despite advances in the treatment of pediatric acute myeloid leukemia (AML), a significant number of cases are refractory to treatment or relapse.1–3 It is important to identify the molecular causes of chemotherapy resistance and design treatment regimens that can overcome resistance. ABCG2 (BCRP, MXR) is a member of the ATP-binding cassette transporter superfamily and reduces exposure to many drugs and environmental toxins through energy-dependent efflux at epithelial barriers.4 Clinical interest in ABCG2 has focused on its role in multidrug resistance in human cancers.5 Many drugs used in oncology, including mitoxantrone, methotrexate, cladribine, topotecan, and imatinib, are ABCG2 substrates, and increased expression of ABCG2 in various cancers has been associated with poor response to chemotherapy.6
Studies of the role of ABCG2 in chemotherapy resistance in AML have produced discordant results, partly due to the use of different assays for ABCG2 expression and function7 and the fact that ABCG2 expression can be limited to small subpopulations of leukemic blasts.8 Despite these confounding factors, the most recent studies suggest that over-expression of ABCG2 portends a poor prognosis.9,10 For instance, high expression of ABCG2 is an independent predictor of poor prognosis in adult AML.11
Children with AML and higher blast-cell expression of ABCG2 had a worse prognosis than children with lower levels of expression.12 Another study found that a CD34+, CD38− subgroup of blast cells enriched for leukemic stem cells expressed both ABCG2 and MDR1 and was associated with decreased response to chemotherapy.9 There is support for a model in which ABCG2 expression in a subset of leukemic stem cells may contribute to the development of resistant disease and relapse, thereby promoting the emergence of a chemoresistant population of rare tumor-initiating cells.8,13
ABCG2 expression is controlled mainly through transcriptional mechanisms. The initial description of the ABCG2 promoter identified a CpG rich promoter that lacked a TATA box and was located about 18 kb upstream of the ATG-containing exon.14 More recent studies have shown ABCG2 transcription to be complex, involving alternative upstream promoters and alternative splicing in the 5′ UTR.15,16 Hypoxia response17 and estrogen response18 elements have also been identified in the promoter regions and likely play a role in tumor cell expression of ABCG2. Epigenetic causes of transcriptional upregulation, including promoter demethylation19,20 and permissive histone modifications on the chromatinized ABCG2 promoter, have been identified.21,22 There is also evidence that ABCG2 can be regulated post-transcriptionally in chronic myelogenous leukemia cell lines via PI3K-AKT signaling.23
This study sought to further characterize ABCG2 expression and transcriptional control in pediatric AML. We identified six novel exons in the 5′ UTR of ABCG2 that were alternatively spliced in various tissue samples and a novel promoter element located approximately 90 kb upstream of the translation initiation codon in exon 2. We found these variant 5′ UTR mRNA isoforms to be expressed in 40% of the bone marrow samples obtained at diagnosis from 85 children with AML. ABCG2 mRNA was expressed in all (8 of 8 cases) of FAB-M7 (megakaryoblastic) AML. Interestingly, expression from the −90kb promoter was detected only in these M7 cases (5 of 8 cases). When tested in reporter gene assays, this upstream promoter was exclusively expressed in an acute megakaryoblastic leukemia (AMKL) cell line but not in non-hematopoietic cell lines, suggesting that this promoter may be specifically active in megakaryocytes and their malignant counterparts.
Patients and Methods
Patients
We evaluated 85 pediatric patients with primary AML who were enrolled on the AML02 clinical trial at St. Jude Children’s Research Hospital between October 31, 2002 and August 18, 2005.3 RNA was isolated from bone marrow samples collected at the time of diagnosis and before the start of chemotherapy. No children were excluded from our analysis, but children with Down syndrome (DS) were not eligible for the treatment protocol. Written informed consent for protocol treatment and procurement of research samples for this study were obtained from patients, parents, or legal guardians. This study was approved by the St. Jude Institutional Review Board.
Computer analysis of the ABCG2 5′ UTR and 5′ flanking genomic region
Searches of human expressed sequence tag (EST) databases (http://blast.ncbi.nlm.nih.gov/Blast.cgi and http://www.genome.ucsc.edu/cgi-bin/hgBlat) were performed to identify transcripts containing a 40 bp sequence from the ABCG2 exon 2 coding region. EST sequences were aligned with human genomic sequence by using Clone Manager software. Putative promoter elements were identified in the 5′ flanking genomic region using the TESS24 and MatInspector25 programs.
RNA isolation and reverse transcription polymerase chain reaction (RT-PCR)
RNA was isolated from bone marrow samples with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. RNA was isolated from cultured cells by using the RNeasy mini kit (Qiagen, Valencia CA). Control RNA from normal human tissues was obtained from Clontech (Mountain View, CA). First-strand cDNA synthesis was performed using Qiagen Omniscript reverse transcriptase with oligo (dT)15 primers (Promega, Madison, WI) and 0.5 μg total RNA. PCR reactions used equal amounts of the RT reaction products (1/20 of total RT reaction volume) mixed with the PCR master mix. Reactions contained 5 pmol of each primer, 0.2 mM each dNTP, and Qiagen Tag DNA polymerase and buffers. PCR products were electrophoresed on 2% NuSieve 3:1 agarose gels (Cambrex, East Rutherford, NJ), subcloned using TOPO TA cloning kits (Invitrogen), and sequenced using both universal vector-specific primers and internal primers.
Forward primers used for PCR amplification of the human ABCG2 cDNA isoforms were: exon 1U, 5′-TTTAGTAGGCGTGGGTCCTG-3′; exon 1A, 5′-ACCAAACCCAGCTAGGTCAG-3′; exon 1C, 5′-CTGTGGAGGAACTGGGTAGG-3′; and exon 2, 5-TCCCAGTGTCACAAGGAAAC-3′. The common reverse primer for human ABCG2 was located in exon 4 and was 5′-GTCGCGGTGCTCCATTTATC-3′. GAPDH control primers were as follows: forward, 5′-AGGTCATCCCTGAGCTGAAC-3′; reverse, 5′-TACTCCTTGGAGGCCATGTG-3′. Conditions for the ABCG2 PCR reactions were: denaturation at 94°C for 3 min; 35 cycles of amplification (94°C for 60 s, 60°C for 60 s, 72°C for 60 s); and final extension at 72°C for 10 min. GAPDH control reactions used the same conditions but only 24 amplification cycles.
5′ Rapid Amplification of cDNA Ends (5′ RACE)
5′ RACE was performed using the SMART RACE cDNA amplification kit (Clontech) according to the manufacturer’s instructions. 5′ RACE-ready cDNA was generated from 1 μg total RNA using PowerScript reverse transcriptase, SMART IIA oligonucleotides and the 5′ CDS primer. Two gene-specific primers derived from ABCG2 exons 3 and 4 were used in the RACE reactions: exon 3, 5′-CCAGGATGGCGTTGAGACCAGGTTTC-3′; exon 4, 5′-GGCAGGTCGCGGTGCTCCATTTATC-3′. Touch-down PCR was performed using Advantage 2 DNA polymerase with the following conditions: denaturation for 1 min at 94°C; 5 cycles of (94°C for 30 s, 72°C for 30 s); 5 cycles of (94°C for 30 s, 70°C for 30 s, 72°C for 30 s); and 25 cycles of (94°C for 30 s, 68°C for 30 s, 72°C for 30 s). RACE products were checked by electrophoresis on a 1% agarose gel prior to TA cloning into the pCR4-TOPO vector (Invitrogen). RACE clones were then sequenced using vector-specific universal primers.
Construction of ABCG2 1U promoter GFP expression vectors
The flanking genomic DNA upstream of the ABCG2 exon 1U transcription start site was amplified by PCR using genomic DNA isolated from K562 cells as a template. The following primers containing XmaI and MluI or NcoI restriction enzyme sites were used for PCR: forward, 5′-CTAGCCCGGGACGCGTAGAAACAGTGACGGTGACACAAC-3′; reverse, 5′-GTAGCCATGGGCAGCTACCAGGGAACATTGAG-3′. The PCR fragment, which also contained the 5′ end of exon 1A1, was digested with XmaI and NcoI and ligated into a shuttle vector containing the eGFP reporter cDNA. Subclones were selected and verified by sequence analysis.
A DNA fragment consisting of the full ABCG2 5′ UTR sequence contained in exons 1U1, 1U2, 1U3, 1U4, 1U5 and exon 2 was generated by PCR amplification of the sequence from a 5′ RACE clone. The primers used contained either EcoRI and SacII restriction enzyme sites or an NcoI site for subsequent subcloning: forward, 5′-GAATTCCGCGGCACAAAGAG-3′; reverse, 5′-GAAGCCATGGGGAGAGTTTTTATCTTTCTGTAATCCC-3′. The PCR product and the exon 1U promoter GFP shuttle vector were digested with SacII and NcoI and ligated to create an exon 1U promoter 5′UTR eGFP shuttle vector. Subclones were selected and sequenced for verification.
The exon 1A promoter-GFP and exon 1A promoter 5′UTR GFP DNA fragments were excised from the shuttle vectors with MluI and NotI and inserted into the pCL20cw INS1R MpGFP lentiviral expression vector26 to generate the exon 1A promoter GFP and exon 1A promoter 5′UTR GFP lentiviral vectors. A promoter-less lentiviral vector was generated as previously described.26
Preparation of lentiviral stocks
Lentiviral vector stocks were prepared as previously described.27 Briefly, 293T cells growing on 10 cm plates were transiently transfected with 10 μg of the vector plasmid, 6 μg of pCAGkGP1.1R (Gag/Pol), 2 μg of pCAG4-RTR2 (Rev/Tat), and 2 μg of pCAG-VSV-G (VSV-G envelope) by the calcium phosphate precipitation method. After 18 h, the cells were washed twice with phosphate-buffered saline (PBS) and cultured for an additional 24 h in fresh medium. The supernatant was then harvested, cleared by centrifugation, and filtered through a 0.45 μm filter. The viral particles were concentrated by ultracentrifugation, resuspended in 1:100 of the initial supernatant volume, and quick-frozen on dry ice for storage at −80°C.
Lentiviral transduction and GFP reporter assays
Lentiviral transduction of adherent 293T or CMK cells was done as previously described.27 Briefly, cells were seeded into 6-well plates coated with RetroNectin (Takara, Madison, WI). Concentrated vector particles were added either once or twice (after a delay of 24 h). For transduction, 6 μg/ml polybrene was added to the medium. 24 h after the final transduction, the cells were washed with PBS and cultured for 5 to 7 days. Expression of the GFP reporter gene in the transduced cells was analyzed by flow cytometry after addition of propidium iodide to label dead cells.
Southern blot analysis
Southern blot analysis of genomic DNA isolated from transduced cells used standard techniques. Briefly, cells were grown to near confluence on 10 cm plates, then rinsed and lysed. Genomic DNA was isolated using the Gentra Puregene DNA extraction kit (Qiagen). 10 mg of DNA was digested with BstEII and electrophoresed on a 0.8% agarose gel, then transferred to a nitrocellulose membrane. Blots were probed with a 750 bp restriction fragment of the RRE sequence contained in the proviral vector genome. Hybridization was carried out in Hybrisol I hybridization buffer (Chemicon, Billerica, MA) at 42°C overnight, and blots were analyzed using the Storm PhosphorImager system and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Transcript stability studies
CMK cells transduced with the MSCV GFP or the MSCV 1-5 GFP vector were treated with 5uM Actinomycin D (SIGMA). At various time points, total RNA were extracted using RNA easy mini kit (Qiagen), and EGFP transcript levels were measured using quantitative RT-PCR. cDNA were synthesized using the SuperScript vilo cDNA Synthesis kit (Invitrogen). Gapdh was used as internal control and were measured using primers/probes from Applied Biosystems. EGFP half lives were determined by linear regression of the time points using GraphPad Prism 4 program. Student’s t test was used to assess the difference between mRNA half lives.
Statistical analysis
Because of the small sample size, ABCG2 expression based on semi-quantitative PCR was categorized as either transcript-detectable or transcript-undetectable. The exact Cochran-Mantel-Haenszel test was used to evaluate the association between ABCG2 transcript expression (yes/no) and day 22 MRD, stratified by treatment arm, for each of the 4 transcripts. Event-free survival (EFS) was defined as the time elapsed from study enrollment to study removal for any cause or to last follow-up. EFS was estimated by the Kaplan-Meier method,28 and estimates were compared by using the exact log-rank test.
Results
Characterization of ABCG2 5′ UTR isoforms
To characterize all potential isoforms of ABCG2 transcripts in human leukemia samples, we first performed database searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi and http://www.genome.ucsc.edu/cgi-bin/hgBlat) to identify all human ESTs homologous to the coding region and known 5′ UTR exons of human ABCG2. The previously identified transcripts and several novel transcripts were identified. Each of three upstream exons (1U, 1A, and 1C) was directly spliced to the same site in exon 2, which contains the translation initiation site (Fig. 1A). Alignment of these sequences with the ABCG2 genomic region showed transcription start sites for exons 1C, 1A, and 1U approximately 18.4 kb, 19.3 kb, and 91.5 kb, respectively, upstream of the exon 2 splice-acceptor sequence (Figure 1A). These results suggest that there are at least 3 distinct upstream promoters in the human ABCG2 locus, generally similar to what has been reported for the murine Abcg2 gene 26.
Figure 1.
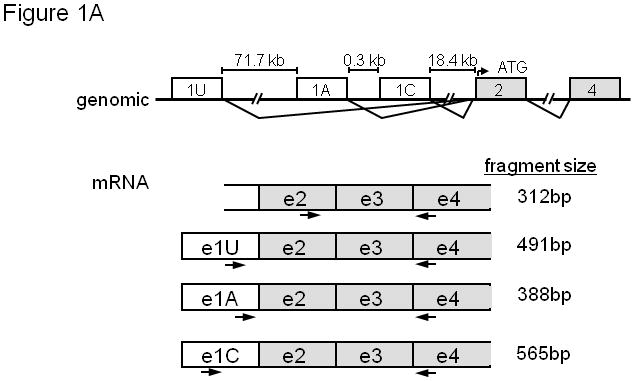
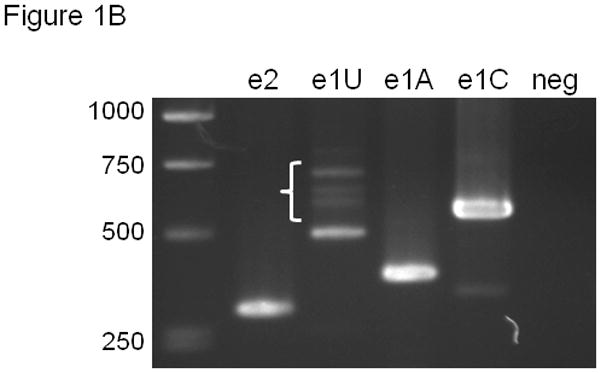
(A) Organization of the 5′ portion of the human ABCG2 gene showing splicing pattern of exons. mRNA isoforms are shown with PCR primers. A common reverse orientation exon 4 primer was used with forward primers specific to exons 1U, 1A, 1C, and 2. (B) RT-PCR of ABCG2 5′ UTR isoforms. The PCR primers in (A) were used after RT of human placental RNA with Oligo dT primers. Negative control reactions used exon 2 forward primer and no RT. Brace identifies higher molecular weight exon 1U PCR products.
EST database sequences were used to design PCR primers specific for the various 5′UTR isoforms and a common region from ABCG2 exons 2 through 4 (Figure 1A). Placental RNA template was chosen for cDNA synthesis because ABCG2 expression is relatively high in this tissue.29 RT-PCR amplification products of the predicted size were detected for each 5′ UTR isoform (Figure 1B). The exon 1U RT-PCR product reproducibly showed not only the 491 bp fragment but numerous larger products (Figure 1B, brace), demonstrating further complexity in exon1U-containing transcripts.
We cloned and sequenced these RT-PCR products. While exon 1A and 1C sequences were identical to those in the EST database, the exon 1U clones revealed a significantly more complicated 5′UTR structure. Four additional, smaller exons (designated 1U2 – 1U5 “subexons”) were identified between exons 1U and exon 1A (Figure 2). All of the exon 1U-containing transcripts were spliced directly to exon 2 and contained various combinations of these subexons.
Figure 2.
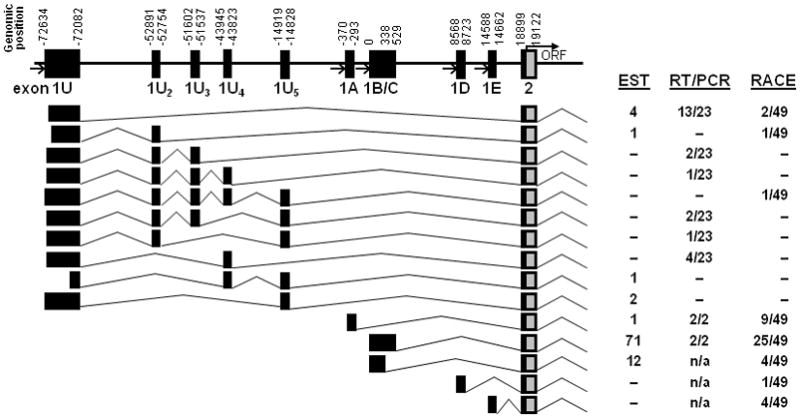
Splice patterns of all ABCG2 transcripts identified. All 5′ UTR variants use the same exon 2 splice acceptor and are predicted to initiate translation at the same amino-terminal ATG in exon 2. Figure is not to scale but shows the relative genomic position of each exon. Variable transcription start sites were identified for exons 1U, 1B, and 1C. Boxes indicate untranslated (black) and translated (gray) regions. Open arrows indicate putative promoter regions. The full ORF and 3′UTR of each isoform was not sequenced. The relative frequency of each isoform in the EST database and by RT-PCR and 5′ RACE is indicated at right.
We also performed 5′ rapid amplification of cDNA ends (5′ RACE) on human placental RNA using reverse primers specific for ABCG2 exon 3 or exon 4. Sequencing of the products confirmed the RT-PCR results and revealed additional variability (Figure 2). Approximately half of the RACE clones (25 of 49) contained the 5′ 1C exon identified in the ABCG2 reference sequence (NM_004827). Four of 49 clones isolated contained the previously described exon 1C splice variant (exon1B),16 and 9 of 49 contained exon 1A. Two previously unreported exon 1 isoforms identified by RACE (designated 1D and 1E) were located downstream of exon 1C (Figure 2).
Expression pattern of ABCG2 5′UTR variants in normal tissues
We characterized the expression pattern of the variant ABCG2 mRNA isoforms by RT-PCR of RNA from normal human tissue (Figure 3). Expression of the exon 1C isoform was detected at variable levels in all tissues examined, while isoforms containing exons 1A and 1U had a more limited distribution (e.g., exon 1A mRNA was detected only in placenta and the MCF-7 breast cancer cell line). Exon 1U-mRNAs were detected in placenta, prostate, small intestine, liver, kidney, and bone marrow. All reactions showed the multiple E1U variants in the typical step-ladder pattern (Figure 3). These results show that transcription of ABCG2 involves multiple mRNA isoforms with varying degrees of tissue specificity.
Figure 3.
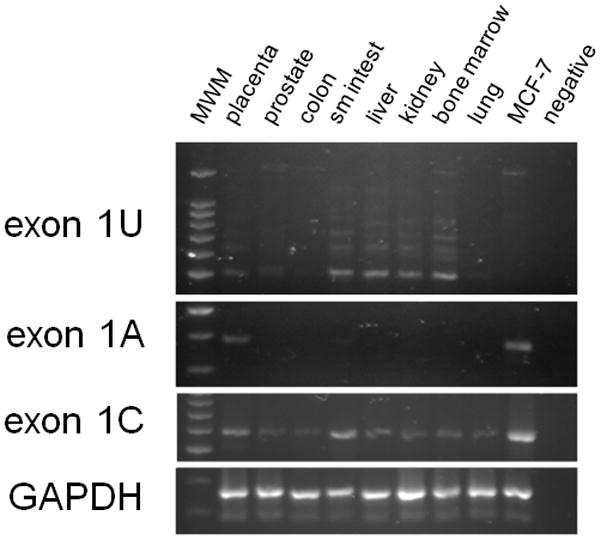
Evaluation of of ABCG2 mRNA isoform expression in normal human tissues. PCR primers are listed in Fig. 1A; GAPDH was added as positive control.
Expression pattern of ABCG2 isoforms in human pediatric AML samples
Because mounting evidence suggests that expression of ABCG2 is associated with worse prognosis in adult AML,10,11, we analyzed the transcript expression patterns in primary pediatric AML samples. RNA was extracted from whole bone marrow samples obtained at diagnosis from 85 patients enrolled on the St. Jude AML02 protocol. RT-PCR analysis revealed transcripts containing the ABCG2 coding region (exons 2-4) in 36 of the 85 patients (Table 1), demonstrating a relatively high prevalence of ABCG2 mRNA expression at diagnosis. The 1C isoform was expressed in 33 of 43 cases and was the predominant species based on this semiquantitative analysis (Figure 4).
Table 1.
Expression of ABCG2 mRNA isoforms in pediatric AML blasts by RT-PCR analysis (number positive/total)
| Exons 2-4 | Exon 1U | Exon 1A | Exon 1C | |
|---|---|---|---|---|
| All samples | 36/85 | 5/43 | 2/43 | 33/43 |
| Non-M7 AML | 28/77 | 0/35 | 1/35 | 25/35 |
| M7 AML | 8/8 | 5/8 | 1/8 | 8/8 |
Figure 4.
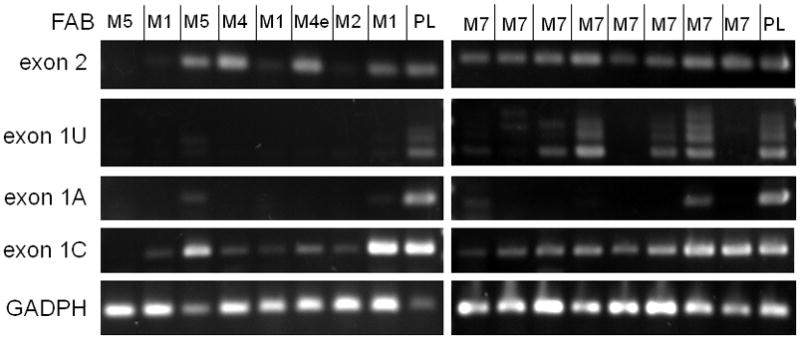
Evaluation of ABCG2 mRNA isoform expression in pediatric AML samples. Left: a variety of selected non-AMKL subtypes. Right: AMKL (AML M7) samples. PCR primers are listed in Fig. 1A; GAPDH was added as positive control. Placental RNA (PL) served as a positive control template.
Exon 1U-containing transcripts were detected in only 5 of 43 patients evaluated. Notably, all five of these patients had AML M7; and all cases showed various patterns of e1U sub-exon splicing (Figure 4). All eight of the AML M7 patients showed expression of the ABCG2 coding region (exons 2-4) and the 1C isoform. In contrast, in other AML subtypes, expression of ABCG2 coding sequences was detected in a smaller proportion of cases (28/77 non-M7 versus 8/8 M7) (Table 1). In these non-M7 cases, exon 1C was the most commonly expressed isotype. The clinical parameters of this pediatric cohort are shown in Table 2.
Table 2.
Characteristics of patients
| Total | |
|---|---|
| Age | 8.49 (0.01, 21.2) |
| Gender | |
| Male | 45 |
| Female | 40 |
| Race | |
| White | 56 |
| Black | 14 |
| Unknown | 1 |
| Other | 14 |
| Risk group | |
| Low | 26 |
| Standard | 33 |
| High | 26 |
| Cytogenetics | |
| Normal | 23 |
| t(8;21) | 12 |
| inv(16) | 10 |
| t(9;11) | 4 |
| any 11q23 (except t(9;11)) | 11 |
| Insufficient sample | 2 |
| Miscellaneous | 23 |
| Arm | |
| Low-dose cytarabine | 47 |
| High-dose cytarabine | 36 |
| Not randomized | 2 |
| WBC | 27.5 (1.1, 351) |
| FAB | |
| M0 | 2 |
| M1 | 12 |
| M2 | 16 |
| M4 | 15 |
| M4eo | 8 |
| M5 | 18 |
| M6 | 1 |
| M7 | 8 |
| N/A | 5 |
| FLT3 status | |
| ITD | 12 |
| Point mutation | 5 |
| Wild type | 68 |
Data are numbers of patients or median (range). There are total 85 patients in each category
Statistical comparison of ABCG2 expression and EFS
We analyzed the relation between expression of four ABCG2 transcripts as determined by semi-quantitative PCR, minimal residual disease (MRD) at day 22 of therapy, EFS, risk stratification, FAB classification, and cytogenetic findings. We detected no statistically significant association between expression of ABCG2 transcripts and MRD on day 22. Expression of exon 1U-containing transcripts was significantly associated with high-risk AML (p=0.013) and FAB M7 AML (p=0.003) but not with EFS. This probably reflects the small sample size and heterogeneity of this group.
Analysis of the exon 1U promoter region
Our RT-PCR results suggested the presence of a novel promoter immediately upstream of exon 1U of ABCG2. We computationally analyzed 1100 bp upstream of the transcription initiation site in exon 1U to identify potential promoter elements (Figure 5). Like the previously characterized ABCG2 promoter region upstream of exon 1C,16 the region upstream of exon 1U contains no TATA box. Multiple potential transcription factor binding sites, such as AP-1, Sp1, and c-Ets1 binding motifs, were identified, as well as potential binding sites for hematopoietic transcription factors, including GATA1, Pbx-1, and Fli1 (Figure 5). Interestingly, Fli1 is known to be important in regulating the development and differentiation of megakaryocytes.30,31
Figure 5.

Genomic nucleotide sequence of the 5′ flanking region of ABCG2 exon 1U. Transcription start site (base 1) is labeled. The exon 1U sequence (the 5′-most transcription start site identified by 5′ RACE) is italicized. Putative transcription factor binding sites identified by MatInspector and TESS are underlined. The putative promoter region cloned into the lentiviral expression vectors is bracketed.
Tissue specificity of the e1U promoter
To confirm promoter activity in the genomic region immediately upstream of the e1U exon, we cloned a 1050-bp fragment of the putative promoter region and inserted it upstream of a GFP reporter gene in a self-inactivating lentiviral vector (Figure 6A). A 1.2 kb fragment of the chicken β-globin locus hypersensitive site insulator (HS4)32,33 flanked the expression cassette to shield from inadvertent interactions with the host cell genome. We also generated vectors containing all 5 e1U sub-exons to assess their possible role in regulating expression (Figure 6A).
Figure 6.
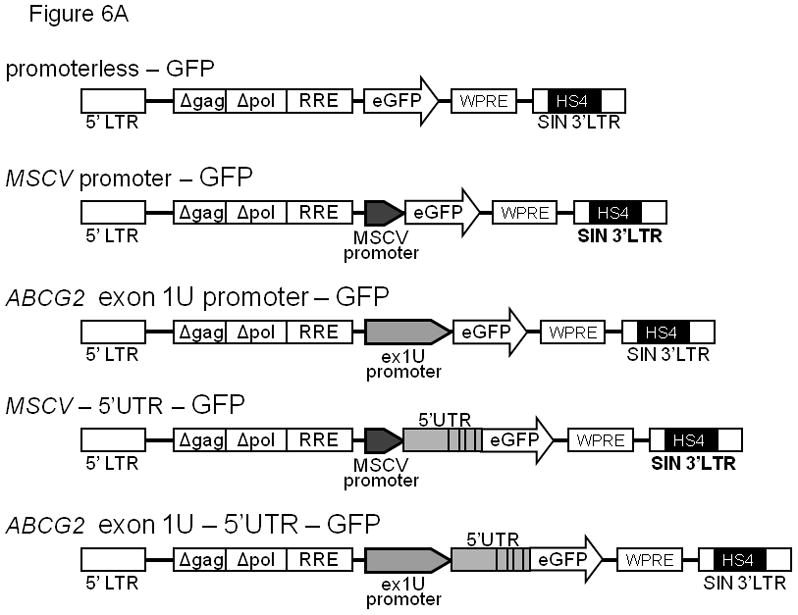
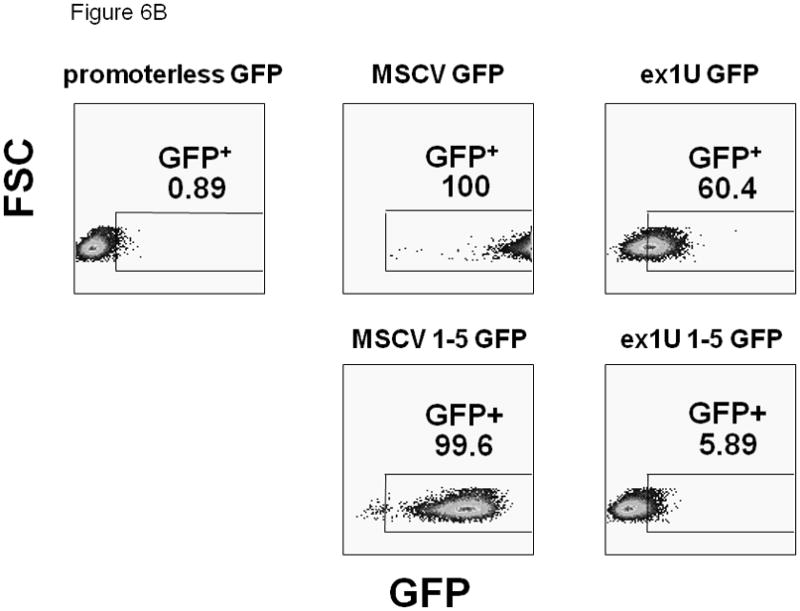
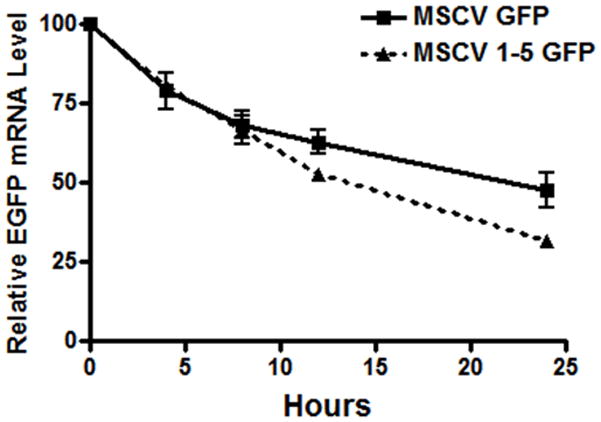
(A) Diagram of the self-inactivated (SIN) lentiviral ABCG2 expression constructs. All constructs contain an HS4 insulator and woodchuck hepatitis virus posttranscriptional regulatory element (WPRE). The putative exon 1U promoter region, a 1050 bp fragment of genomic DNA directly upstream of the 5′-most exon 1U transcriptional start site, was placed adjacent to an EGFP reporter gene. Positive (containing an MSCV promoter) and negative (promoterless) control vectors were used. (B) Flow cytometric analysis of GFP activity in transduced CMK cells. GFP is plotted on the x-axis and forward scatter on the y-axis. Number represents percentage of GFP+ cells. (C) The stability of GFP transcripts were studied using actinomycin D treated CMK cells and quantitative RT-PCR. CMK cells containing either the MSCV 1-5 GFP vector (dashed line) or the MSCV GFP control vector (solid line) were cultured with actinomycin D and RNA was analyzed at the various time points shown. GFP transcript levels were then normalized to GAPDH and expressed as a ratio relative to untreated cells. This experiment was performed 3 times and error bars show the standard deviation for each measurement.
To test whether the e1U promoter might be megakaryocyte-specific, we transduced the megakaryoblastic leukemia cell line CMK 34 to test whether the promoter could drive GFP expression. Flow cytometry showed significant but modest levels of GFP expression in cells with the exon 1U promoter construct, indicating that this promoter fragment was transcriptionally active in these cells (Figure 6B). Overall expression from the 1U promoter was significantly less than that obtained using the strong MSCV LTR promoter. Inclusion of the five upstream e1U subexons attenuated GFP expression, with both the e1U promoter and the internal MSCV LTR promoter (Figure 6B). Therefore, the multiple 5′UTR sequences in the e1U region may be a target for mRNA degradation or may inhibit translation.
To determine whether the decreased GFP expression seen with the exon 1U construct could be due to decreased stability of the transcript, actinomycin D inhibition experiments were performed (Figure 6C). These studies showed a modest but statistically significant decrease in transcript stability due to the 5′ UTR sequences, with the GFP half life in MSCV GFP cells 20.4 +/− 1.35 hours versus 15.5 in the MSCV 1-5 GFP cells. These results indicate that decreased transcript stability contributes to the decreased protein expression seen with the 5 UTR, but given the large difference in protein expression seen comparing the two constructs, it is likely that other mechanisms such as translational inhibition may also play a role.
We also transduced HeLa cells to test the tissue specificity of the exon 1U promoter construct. No GFP expression was detected in HeLa cells transduced with the exon 1U promoter construct (Figure 7A). Genomic integration of the vector (retroviral integration >80%) was confirmed by Southern blot analysis with a probe for the RRE sequence within the lentiviral DNA (Figure 7B), showing that the lack of expression was not due to lack of gene transfer. In contrast, significant GFP expression was seen using the positive control MSCV LTR promoter in HeLa cells. Together, these data indicate that the 1050 kb fragment immediately upstream of e1U contains a promoter with relative specificity for the megakaryocytic lineage, potentially explaining the exclusive presence of this transcript in M7 AML.
Figure 7.
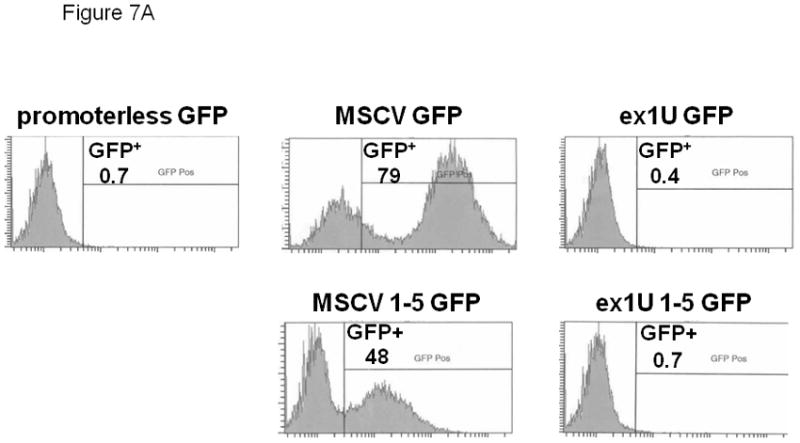
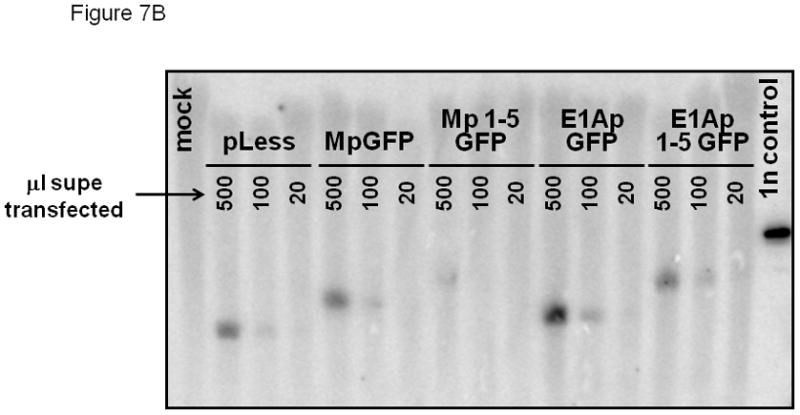
(A) Flow cytometric analysis of GFP expression in HeLa cells transduced with exon 1A expression construct and controls. GFP intensity is plotted on the x-axis and cell count on the y-axis. Number represents percentage of GFP+ cells. (B) Southern blot confirmation of genomic proviral integration. 10 mg genomic DNA was loaded in each lane. Blot was probed with RRE fragment specific for proviral DNA. Three lanes are above each lane. Negative control: supernatant from mock-transfected HeLa cells. Positive control: lentiviral plasmid vector DNA.
Discussion
We have identified a novel group of ABCG2 mRNA isoforms with variable 5′UTRs that originate from a promoter approximately 90 kb upstream of the translation initiation site in exon 2. Transcription of these isoforms initiates from a promoter region immediately upstream of exon 1U. We showed the activity of this promoter to be relatively tissue specific, as shown by reporter gene expression in a megakaryocytic, but not in an epithelial, cell line. While expression of the exon 1U isoforms were noted in other whole tissue, it is currently not clear if this could be due to hematopoietic cells contained within these tissues. Inclusion of the e1U “subexons” in the lentiviral reporter constructs led to decreased expression with either the e1U promoter or the MSCV promoter, and transcript stability studies showed that the 5′UTR region contains elements that destabilize the transcripts and may also inhibit their translation. This demonstration of additional transcription control mechanisms for human ABCG2 parallel previous findings in mouse Abcg2.26
A significant number of human genes contain multiple upstream promoters with alternative 5′ untranslated regions. Alternative splicing is particularly prevalent in genes of the ABC transporter family15. The use of alternative promoters enhances regulatory control,35,36 and alternative UTR sequences can affect transcript stability and translational efficiency in a tissue-specific manner37,38. For example, the human arylamine N-acetyltransferase type 1 gene contains eight 5′ UTR exons expressed from three alternative promoters, and several of the isoforms are expressed in a tissue-specific manner.39 Although the function of the extensive variability in the ABCG2 5′UTR is not fully understood, it may facilitate the multiple roles and the tissue- and cell-type–specific expression of ABCG2.
Increased ABCG2 expression in M7 AML has not previously been reported. Our RNA samples from untreated pediatric AML showed greater expression of ABCG2 transcripts in the M7 subtype than in other subtypes. All eight M7 cases showed increased expression of the exon 1C 5′UTR isoform, and five of the eight samples showed significant expression of the e1U isoforms, which were not detected in other AML subtypes. These results indicate that ABCG2 transcripts are present in AML cells in approximately 40% of children at diagnosis, including most if not all M7 cases.
M7 AML carries a relatively good prognosis in children with DS but not in other children.1,40,41 No patients in our study had DS, and the ABCG2 expression patterns in DS-associated M7 AML have yet to be determined. Although we found no association between ABCG2 transcript expression and prognosis, this may be due to the relatively small sample size of our cohort. The possibility that drug resistance in non-DS M7 AML cases may be related to the relatively high prevalence and/or extent of ABCG2 expression will require further study but is consistent with several previous findings. In vitro, leukemic blasts from children without DS showed greater resistance to common AML chemotherapy agents than did blasts from children with DS; the tested agents included daunorubicin and mitoxantrone,42, both of which are substrates of ABCG2.43 In DS AML M7, a common GATA-1 mutation produces a truncated, dominant-negative form of the transcription factor (GATA-1s).44 Because GATA-1s lacks the N-terminal transactivation domain, it is thought to be unable to regulate megakaryocytic target genes.45 The presence of potential GATA-1 binding sites in the e1U promoter raises the possibility that transcription of ABCG2 is lessened in DS AML M7 as a direct consequence of the GATA-1s mutation, although direct studies in DS AML M7 blasts will be needed to address this issue.
Recent data suggest that in adult AML, expression of ABCG2 and the MDR1 transporter correlate with clinical response to chemotherapy.9 While more work must be done to clarify the clinical significance of ABCG2 expression in non-DS M7 AML, our results raise questions about current therapy for this disorder, especially the customary use of anthracyclines during induction. Our data suggest that known ABCG2 substrates may not be optimal and that addition of ABCG2 inhibitors to current regimens for non-DS AML M7 should be explored. Imatinib and nilotinib are powerful inhibitors of ABCG2 and MDR1,46–48 and their inhibition of oncogenic tyrosine kinase activities may have other anti-leukemic effects. Another possibility is the multikinase inhibitor sorafenib. Sorafenib not only inhibits ABCG2 transport but also enters cells passively, and its effectiveness is not likely to be reduced by high expression of ABC transporters.49
Acknowledgments
We thank Charles Mulligan for providing the CMK cell line, the St. Jude flow cytometry core facility for analysis of the lentiviral reporter constructs, Stanley Pounds for assistance with the statistical analyses, and Sharon Naron for editing assistance. Supported in part by National Institutes of Health grants R01-HL67366 (to B. P. S.), T32-CA070089 (to P.K.C.), and P30 CA021765 and by the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Authors’ Contributions
P.K.C., and Y.Z. performed experiments; P.K.C., S.Y. and B.P.S. analyzed results; P.K.C. made the figures and P.K.C., J.E.R. and B.P.S. designed the research and wrote the paper.
Conflict of Interest
The authors declare no competing financial interests or any other real or apparent conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rubnitz JE, Razzouk BI, Lensing S, Pounds S, Pui CH, Ribeiro RC. Prognostic factors and outcome of recurrence in childhood acute myeloid leukemia. Cancer. 2007;109:157–63. doi: 10.1002/cncr.22385. [DOI] [PubMed] [Google Scholar]
- 2.Rubnitz JE, Gibson B, Smith FO. Acute myeloid leukemia. Hematol Oncol Clin North Am. 2010;24:35–63. doi: 10.1016/j.hoc.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010;11:543–52. doi: 10.1016/S1470-2045(10)70090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Herwaarden AE, Schinkel AH. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol Sci. 2006;27:10–6. doi: 10.1016/j.tips.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007;26:39–57. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- 6.Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M, et al. ABCG2: a perspective. Adv Drug Deliv Rev. 2009;61:3–13. doi: 10.1016/j.addr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suvannasankha A, Minderman H, O’Loughlin KL, Nakanishi T, Greco WR, Ross DD, et al. Breast cancer resistance protein (BCRP/MXR/ABCG2) in acute myeloid leukemia: discordance between expression and function. Leukemia. 2004;18:1252–7. doi: 10.1038/sj.leu.2403395. [DOI] [PubMed] [Google Scholar]
- 8.Abbott BL, Colapietro AM, Barnes Y, Marini F, Andreeff M, Sorrentino BP. Low levels of ABCG2 expression in adult AML blast samples. Blood. 2002;100:4594–601. doi: 10.1182/blood-2002-01-0271. [DOI] [PubMed] [Google Scholar]
- 9.Ho MM, Hogge DE, Ling V. MDR1 and BCRP1 expression in leukemic progenitors correlates with chemotherapy response in acute myeloid leukemia. Exp Hematol. 2008;36:433–42. doi: 10.1016/j.exphem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 10.van den Heuvel-Eibrink MM, van der Holt B, Burnett AK, Knauf WU, Fey MF, Verhoef GE, et al. CD34-related coexpression of MDR1 and BCRP indicates a clinically resistant phenotype in patients with acute myeloid leukemia (AML) of older age. Ann Hematol. 2007;86:329–37. doi: 10.1007/s00277-007-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damiani D, Tiribelli M, Michelutti A, Geromin A, Cavallin M, Fabbro D, et al. Fludarabine-based induction therapy does not overcome the negative effect of ABCG2 (BCRP) over-expression in adult acute myeloid leukemia patients. Leukemia research. 2010;34:942–5. doi: 10.1016/j.leukres.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Steinbach D, Sell W, Voigt A, Hermann J, Zintl F, Sauerbrey A. BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia. Leukemia. 2002;16:1443–7. doi: 10.1038/sj.leu.2402541. [DOI] [PubMed] [Google Scholar]
- 13.Raaijmakers MH, de Grouw EP, Heuver LH, van der Reijden BA, Jansen JH, Scheper RJ, et al. Breast cancer resistance protein in drug resistance of primitive CD34+38− cells in acute myeloid leukemia. Clin Cancer Res. 2005;11:2436–44. doi: 10.1158/1078-0432.CCR-04-0212. [DOI] [PubMed] [Google Scholar]
- 14.Bailey-Dell KJ, Hassel B, Doyle LA, Ross DD. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP-binding cassette transporter G2) gene. Biochim Biophys Acta. 2001;1520:234–41. doi: 10.1016/s0167-4781(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 15.Srinivasan S, Bingham JL, Johnson D. The ABCs of human alternative splicing: a review of ATP-binding cassette transporter splicing. Curr Opin Drug Discov Devel. 2009;12:149–58. [PubMed] [Google Scholar]
- 16.Nakanishi T, Bailey-Dell KJ, Hassel BA, Shiozawa K, Sullivan DM, Turner J, et al. Novel 5′ untranslated region variants of BCRP mRNA are differentially expressed in drug-selected cancer cells and in normal human tissues: implications for drug resistance, tissue-specific expression, and alternative promoter usage. Cancer Res. 2006;66:5007–11. doi: 10.1158/0008-5472.CAN-05-4572. [DOI] [PubMed] [Google Scholar]
- 17.Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–25. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]
- 18.Ee PL, Kamalakaran S, Tonetti D, He X, Ross DD, Beck WT. Identification of a novel estrogen response element in the breast cancer resistance protein (ABCG2) gene. Cancer Res. 2004;64:1247–51. doi: 10.1158/0008-5472.can-03-3583. [DOI] [PubMed] [Google Scholar]
- 19.Turner JG, Gump JL, Zhang C, Cook JM, Marchion D, Hazlehurst L, et al. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood. 2006;108:3881–9. doi: 10.1182/blood-2005-10-009084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.To KK, Zhan Z, Bates SE. Aberrant promoter methylation of the ABCG2 gene in renal carcinoma. Mol Cell Biol. 2006;26:8572–85. doi: 10.1128/MCB.00650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.To KK, Polgar O, Huff LM, Morisaki K, Bates SE. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol Cancer Res. 2008;6:151–64. doi: 10.1158/1541-7786.MCR-07-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calcagno AM, Fostel JM, To KK, Salcido CD, Martin SE, Chewning KJ, et al. Single-step doxorubicin-selected cancer cells overexpress the ABCG2 drug transporter through epigenetic changes. Br J Cancer. 2008;98:1515–24. doi: 10.1038/sj.bjc.6604334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108:678–84. doi: 10.1182/blood-2005-10-4020. [DOI] [PubMed] [Google Scholar]
- 24.Schug J. Curr Protoc Bioinform. Vol. 21. John Wiley & Sons; 2008. Using TESS to Predict Transcription Factor Binding Sites in DNA Sequence; pp. 2.6.1–2.6.15. [DOI] [PubMed] [Google Scholar]
- 25.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 26.Zong Y, Zhou S, Fatima S, Sorrentino BP. Expression of mouse Abcg2 mRNA during hematopoiesis is regulated by alternative use of multiple leader exons and promoters. J Biol Chem. 2006;281:29625–32. doi: 10.1074/jbc.M606314200. [DOI] [PubMed] [Google Scholar]
- 27.Hanawa H, Persons DA, Nienhuis AW. Mobilization and mechanism of transcription of integrated self-inactivating lentiviral vectors. J Virol. 2005;79:8410–21. doi: 10.1128/JVI.79.13.8410-8421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. J Am Stat Assoc. 2006;53:457–81. [Google Scholar]
- 29.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–9. [PubMed] [Google Scholar]
- 30.Hart A, Melet F, Grossfeld P, Chien K, Jones C, Tunnacliffe A, et al. Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity. 2000;13:167–77. doi: 10.1016/s1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- 31.Kruse EA, Loughran SJ, Baldwin TM, Josefsson EC, Ellis S, Watson DK, et al. Dual requirement for the ETS transcription factors Fli-1 and Erg in hematopoietic stem cells and the megakaryocyte lineage. Proc Natl Acad Sci U S A. 2009;106:13814–9. doi: 10.1073/pnas.0906556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aker M, Tubb J, Groth AC, Bukovsky AA, Bell AC, Felsenfeld G, et al. Extended core sequences from the cHS4 insulator are necessary for protecting retroviral vectors from silencing position effects. Hum Gene Ther. 2007;18:333–43. doi: 10.1089/hum.2007.021. [DOI] [PubMed] [Google Scholar]
- 33.Chung JH, Bell AC, Felsenfeld G. Characterization of the chicken beta-globin insulator. Proc Natl Acad Sci U S A. 1997;94:575–80. doi: 10.1073/pnas.94.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melemed AS, Ryder JW, Vik TA. Activation of the mitogen-activated protein kinase pathway is involved in and sufficient for megakaryocytic differentiation of CMK cells. Blood. 1997;90:3462–70. [PubMed] [Google Scholar]
- 35.Miyazaki K, Inoue S, Yamada K, Watanabe M, Liu Q, Watanabe T, et al. Differential usage of alternate promoters of the human stress response gene ATF3 in stress response and cancer cells. Nucleic Acids Res. 2009;37:1438–51. doi: 10.1093/nar/gkn1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singer GA, Wu J, Yan P, Plass C, Huang TH, Davuluri RV. Genome-wide analysis of alternative promoters of human genes using a custom promoter tiling array. BMC Genomics. 2008;9:349. doi: 10.1186/1471-2164-9-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkie GS, Dickson KS, Gray NK. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem Sci. 2003;28:182–8. doi: 10.1016/S0968-0004(03)00051-3. [DOI] [PubMed] [Google Scholar]
- 38.Hughes TA. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006;22:119–22. doi: 10.1016/j.tig.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Butcher NJ, Arulpragasam A, Goh HL, Davey T, Minchin RF. Genomic organization of human arylamine N-acetyltransferase Type I reveals alternative promoters that generate different 5′-UTR splice variants with altered translational activities. Biochem J. 2005;387:119–27. doi: 10.1042/BJ20040903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnard DR, Alonzo TA, Gerbing RB, Lange B, Woods WG. Comparison of childhood myelodysplastic syndrome, AML FAB M6 or M7, CCG 2891: report from the Children’s Oncology Group. Pediatr Blood Cancer. 2007;49:17–22. doi: 10.1002/pbc.20951. [DOI] [PubMed] [Google Scholar]
- 41.Athale UH, Razzouk BI, Raimondi SC, Tong X, Behm FG, Head DR, et al. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution’s experience. Blood. 2001;97:3727–32. doi: 10.1182/blood.v97.12.3727. [DOI] [PubMed] [Google Scholar]
- 42.Polgar O, Robey RW, Bates SE. ABCG2: structure, function and role in drug response. Expert Opin Drug Metab Toxicol. 2008;4:1–15. doi: 10.1517/17425255.4.1.1. [DOI] [PubMed] [Google Scholar]
- 43.Yamada S, Hongo T, Okada S, Watanabe C, Fujii Y, Hori H, et al. Distinctive multidrug sensitivity and outcome of acute erythroblastic and megakaryoblastic leukemia in children with Down syndrome. Int J Hematol. 2001;74:428–36. doi: 10.1007/BF02982087. [DOI] [PubMed] [Google Scholar]
- 44.Taub JW, Mundschau G, Ge Y, Poulik JM, Qureshi F, Jensen T, et al. Prenatal origin of GATA1 mutations may be an initiating step in the development of megakaryocytic leukemia in Down syndrome. Blood. 2004;104:1588–9. doi: 10.1182/blood-2004-04-1563. [DOI] [PubMed] [Google Scholar]
- 45.Muntean AG, Ge Y, Taub JW, Crispino JD. Transcription factor GATA-1 and Down syndrome leukemogenesis. Leuk Lymphoma. 2006;47:986–97. doi: 10.1080/10428190500485810. [DOI] [PubMed] [Google Scholar]
- 46.Dohse M, Scharenberg C, Shukla S, Robey RW, Volkmann T, Deeken JF, et al. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib and dasatinib. Drug Metab Dispos. 2010 doi: 10.1124/dmd.109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brendel C, Scharenberg C, Dohse M, Robey RW, Bates SE, Shukla S, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21:1267–75. doi: 10.1038/sj.leu.2404638. [DOI] [PubMed] [Google Scholar]
- 48.Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004;64:2333–7. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- 49.Hu S, Chen Z, Franke R, Orwick S, Zhao M, Rudek MA, et al. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin Cancer Res. 2009;15:6062–9. doi: 10.1158/1078-0432.CCR-09-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]