

Abstract
Assembly of icosahedral capsids of proper size and symmetry is not understood. Residue F170 in bacteriophage P22 coat protein is critical for conformational switching during assembly. Substitutions at this site cause assembly of tubes of hexamerically arranged coat protein. Intragenic suppressors of the ts phenotype of F170A and F170K coat protein mutants were isolated. Suppressors were repeatedly found in the coat protein telokin-like domain at position 285, which caused coat protein to assemble into petite procapsids and capsids. Petite capsid assembly strongly correlated to the side chain volume of the substituted amino acid. We hypothesize that larger side chains at position 285 torque the telokin-like domain, changing flexibility of the subunit and intercapsomer contacts. Thus, a single amino acid substitution in coat protein is sufficient to change capsid size. In addition, the products of assembly of the variant coat proteins were affected by the size of the internal scaffolding protein.
Keywords: polyheads, virus morphology, suppressor substitutions, size determination, phage
Background
Capsids of many spherical viruses are constructed from multiple copies of the same protein(s), reducing the genetic burden of encoding many different proteins, but then requiring that the single protein be able to assume multiple conformations to produce an icosahedral capsid. A range of icosahedral capsid sizes can be assembled by combining 12 pentamers and a variable number of hexamers into a closed spherical shell (Caspar and Klug, 1962). The total number of subunits is T × 60, where T is the number of quasi-equivalent conformations responsible for the correct product of assembly for isometric capsids. Increasing the T-number, without altering the size of the individual subunits, can change the capsid size by increasing the number of quasi-equivalent conformations. The requirement for conformational switching of identical subunits into the appropriate quasi-equivalent conformation is thus related to capsid size determination. Understanding this self-assembly process is important for many applications from antiviral drug design to nanotechnology.
Bacteriophage P22, which infects Salmonella enterica serovar Typhimurium, is a paradigm for the assembly of tailed bacteriophage and Herpesviridae (Teschke and Parent, 2010). In vivo, assembly of P22 virions involves co-polymerization of 415 coat proteins, 60–300 scaffolding proteins, the dodecameric portal complex, and ejection proteins into a precursor (“procapsid”) structure (King et al., 1976; Prevelige and King, 1993). Procapsids mature into virions as dsDNA is packaged through the portal complex (Bazinet and King, 1988) and scaffolding protein concomitantly exits. Maturation is accompanied by an increase in volume of the head and a change in capsid morphology from one that is nearly spherical to one that is faceted (Earnshaw, Casjens, and Harrison, 1976; Prasad et al., 1993).
Many dsDNA viruses require a scaffolding protein to catalyze proper capsid assembly. Some scaffolding proteins are separately expressed proteins, while some other viruses have the scaffolding protein attached at the end of the capsid protein, called delta domains. P22's scaffolding protein is comprised of 303 amino acids and, like other scaffolding proteins, is highly α-helical (Dokland, 1999). Its dimensions have been shown to be 22 Å in diameter by 247 Å in length (Parker, Stafford, and Prevelige, 1997). The C-terminal helix-turn-helix from amino acids 280–294 is critical for interaction with coat protein during assembly (Sun et al., 2000; Weigele et al., 2005), though other regions are involved as well. Scaffolding protein directs proper assembly of monomeric coat proteins to form a procapsid in which the coat proteins are organized with T=7 quasi-symmetry. In the absence of scaffolding protein in vivo, coat protein assembles into aberrant spiral structures as well as wild-type-sized T=7 and smaller T=4 icosahedral particles (Earnshaw and King, 1978; Lenk et al., 1975; Thuman-Commike et al., 1998). Structural comparisons of the T=7 and T=4 procapsids reveal that each has similar penton and hexon clusters, but with increased curvature of the T=4 hexon (Thuman-Commike et al., 1998). How scaffolding protein inhibits formation of T=4 particles during assembly is not understood.
Much of the information for capsid size determination is intrinsic to the coat protein subunits themselves, which possess natural curvature (Caspar and Klug, 1962). For example, P22 coat protein is capable of assembling into T=7 sized structures even in the absence of all phage proteins, albeit with lower fidelity than when scaffolding protein is present. According to the local-rules theory of Berger et al, the assembly of a capsid is directed by the local interactions of its coat and scaffolding protein subunits (Berger et al., 1994). By this theory, each subunit is switched into the appropriate conformation during assembly based on the conformational state of its neighbors. By changing which conformations bind to each other, the geometric structure of the shell can be altered, producing shells with various T-numbers. A small change in the local rules was modeled to switch the outcome from T=7 to an alternative T=4 structure (Berger et al., 2000).
Cryo-electron microscopy and 3D image reconstruction models of the 47 kDa P22 coat protein revealed that it adopts a fold quite similar to the HK97 capsid protein (Wikoff et al., 1998), but it has an additional telokin-like domain at the outermost surface (magenta in Figure 1) (Chen et al., 2011; Jiang et al., 2003; Parent et al., 2010a). This protrusion, called the “extra-density” domain by Chiu's group (Chen et al., 2011; Jiang et al., 2003), is a domain comprised primarily of β-sheets similar to members of the immunoglobulin-like family of domains. It was originally termed “telokin-like” (Parent et al., 2010a) based on moderate-resolution structural similarity and because telokin was the top match in an unbiased search using FFAS03, a fold and function assignment server (Jaroszewski et al., 2005). The telokin-like domain has been suggested to be important for stabilization of the coat protein monomers (Teschke and Parent, 2010), and the procapsid shell (Chen et al., 2011). The `β-hinge' (orange in Figure 1), a structurally conserved four-stranded β-sheet that links all domains of the protein, is involved in the conformational switching required for assembly and maturation (Parent, Suhanovsky, and Teschke, 2007; Teschke and Parent, 2010). Amino acid substitutions F170L, K and A in the β-hinge cause coat protein to assemble into polyheads (Suhanovsky et al., 2010) comprised entirely of hexons (Parent et al., 2010b). F170 variants decrease the flexibility of the A-domain (cyan in Figure 1) leading to steric crowding at the center of the pentons (which have a smaller axial hole compared to hexons), thereby favoring the hexon configuration during assembly (Suhanovsky et al., 2010). F170A and F170K coat protein variants also cause a temperature-sensitive (ts) phenotype.
Figure 1. P22 coat protein subunit.

Cα backbone model of P22 coat protein subunit color-coded by domain (N-arm, red; E-loop, yellow; P-domain, green; A-domain, cyan; telokin-like domain, magenta) from the WT procapsid shell (PDB accession number 2XYY) (Chen et al., 2011). The A-domain points towards the center of the capsomer hole and is involved in intracapsomer interactions. The P-domain is at the periphery of the capsomer and is involved in intercapsomer interactions. The β-hinge is highlighted in orange. Amino acids F170 and A285 are labeled.
Here, we describe intragenic second site suppressors of the ts phenotype caused by substitutions at position F170 (orange sphere in Figure 1). In particular we investigated the suppressors found in the telokin-like domain, and determined that this domain is important for proper capsid size determination. In addition, we demonstrate that scaffolding protein can correct improper size determination caused by coat variants.
Results
Suppressors of F170 substitutions highlight the telokin-like domain
F170A and F170K variants showed temperature-sensitive (ts) defects at 39 °C and 41 °C, respectively (data not shown), in addition to an altered ability to assemble (Suhanovsky et al., 2010). Therefore, we searched for intragenic second site suppressors by plating phage carrying these substitutions at high temperature and isolating individual plaques. A total of 17 independent suppressors were sequenced. Of these, substitutions in the β-hinge were isolated 7 times and substitutions at position A285 were separately isolated 9 times (Table 1). WT revertants were not found because reverting to the WT phenylalanine required two or three base changes. The substitutions at A285 (magenta sphere in Figure 1), which is located in the telokin-like domain, are intriguing as this is residue is distant both in sequence and in space from the F170 site. For this reason, and because of the high frequency of their isolation, the suppressors at position A285 were chosen for further investigation.
Table 1.
Suppressors of F170 variants.
| Parent ts coat protein mutant | Suppressor substitutiona |
|---|---|
| F170A | D163G (1) |
| T166I (2) | |
| A170V (1) | |
| A285S (1) | |
| G424A (1) | |
| F170K | T166I (2) |
| G403V (1) | |
| A285T (8) |
Number in parentheses is the number of times the substitution was independently isolated from the ts parent phage.
F170K:A285T coat protein forms both T=7 and petite particles
F170A and F170K coat proteins cause polyhead formation in vivo at 30 °C (Suhanovsky et al., 2010). We analyzed cell lysates from complementation experiments with the F170A/K:suppressor coat proteins to determine if the second site substitutions affected polyhead formation. In these complementation experiments, the phage carried an amber mutation in gene 5 (5− am) so that coat protein was not expressed from the phage genome. The cells carried a plasmid that coded for either WT or mutated gene 5, from which coat protein was expressed. The soluble proteins from a clarified lysate of such phage-infected cells were applied to 5–20% linear sucrose gradients. Procapsids, comprised of both coat (c) and scaffolding (s) proteins, run in the lower middle of these gradients. Phage and polyheads sediment to the bottom upon centrifugation. The sucrose gradient fractions were evaluated by SDS-PAGE (Figure 2A) and negative stain electron microscopy (Figure 2B,C). Micrographs of the WT sample (data not shown) showed correctly sized procapsids in fraction 16 and mature phage in fractions 22–23. Analysis of SDS gels of the F170A:A285S sucrose gradient fractions showed that the particles sedimented similarly to that of the WT coat protein sample. However, micrographs of fraction 23 revealed that F170A:A285S coat protein still produced some polyheads (arrow in Figure 2B), but fewer than previously shown for F170A coat protein alone (Suhanovsky et al., 2010). Surprisingly, F170K:A285T assembly products were distributed throughout the gradient with a significant fraction sedimenting to positions higher on the gradient, indicating smaller products. Micrographs of fractions 10, 16, and 23 from the F170K:A285T coat protein lysate are shown in Figure 2C. Fraction 10 contained procapsids with a smaller diameter (`petites') than procapsids generated from WT coat protein. F170K:A285T coat protein also produced some correctly sized procapsids seen in fraction 16. Both petite and correctly sized procapsids were competent for maturation. The white arrows in Figure 2C (fraction 23) point to petite angular particles with tailspikes present. The diameter of the mature petite phage, ~ 45 nm, is consistent with T=4 sized particles. There were no polyheads in bottom fractions of the F170K:A285T lysate. These results are the first report of a P22 coat protein variant that produces petite heads.
Figure 2. F170K:A285T coat protein assembles into petite phage in vivo.

(A) Sucrose gradient sedimentation of lysates from 5− am phage-infected cells that express WT, F170A:A285S, or F170K:A285T coat proteins. Samples were run on a 5–20% linear sucrose gradient and analyzed by SDS-PAGE. Coat protein (c) and scaffolding protein (s) are labeled. The F170K:A285T particles migrate higher in the gradient. Both (B) and (C) show representative micrographs of negatively stained samples from the sucrose gradients shown in (A). (B) Fraction 23 from the sucrose gradient of F170A:A285S coat protein cell lysate shows some polyhead structures (black arrow) and T=7 mature phage. (C) The micrographs from fractions of the sucrose gradient of the F170K:A285T coat protein cell lysate show petite procapsids in fraction 10, T=7 procapsids in fraction 16, and both petite (white arrows) and T=7 mature phage in fraction 23. The bright white appearance and presence of tailspike indicates that the petite particles had packaged dsDNA.
A285F and A285Y coat proteins form only petite procapsids
F170K:A285T coat protein produced petite procapsids in vivo, while F170A:A285S did not. Since threonine is larger than serine, we hypothesized that the size of the side chain might affect formation of the petite procapsids. We also investigated the role of the side chain hydroxyl. The side chain at the A285 position was investigated in the absence of the F170 substitutions in order to understand why altering this amino acid produces petite procapsids in vivo.
Site-directed mutagenesis was used to introduce serine, threonine, valine, phenylalanine, and tyrosine at position 285. The ability of the A285 variant coat proteins to assemble into infectious virions was compared to that of WT coat protein using complementation as described above. A285S, A285T, and A285V coat protein variants all showed similar cold-sensitive (cs) and temperature-sensitive (ts) phenotypes, while A285F and A285Y variants each caused a lethal phenotype at all temperatures (Figure 3A).
Figure 3. A285F and A285Y coat proteins exclusively produce petite particles.

(A) Efficiency of plating experiments of the A285 variants by complementation. WT and the A285 variant coat proteins were expressed from a plasmid and the cells were infected with phage that cannot synthesize coat protein. The substitutions A285F and A285Y cause a lethal phenotype. (B) Sucrose gradient sedimentation of assembly products from phage-infected cell lysates of WT and the A285 variants. (C) Representative micrographs of negatively stained samples of the assembly products in fractions 6, 10, 16, and 23 for A285T and A285Y coat proteins. The micrograph of fraction 23 from the A285T cell lysate shows both T=4 mature phage (white arrow) T=7 mature phage (black arrow).
We analyzed the assembly products produced in cell lysates from complementation experiments at 30 °C by sucrose gradient sedimentation and SDS-PAGE (Figure 3B). Assembly products of A285S coat protein migrated to a similar position on the sucrose gradient as that of WT coat protein products. A285T and A285V assembly products were slightly shifted up the gradients. A285F and A285Y particles migrated much higher on the gradients, with very little coat protein present beyond fraction 16, indicating smaller products. Negative stain electron microscopy was used to visualize the assembly products. Representative micrographs taken of sucrose gradient fractions from lysates generated with A285T and A285Y coat proteins are shown in Figure 3C. A285V and A285T assembly products were similar to the F170K:A285T particles. A285F and A285Y coat proteins produced petite particles present as high as fraction 6 in the sucrose gradient. The particles in fraction 6 had a smaller diameter (~ 33 nm) than the T=4 sized particles present in fraction 10. There were no mature phage particles in the bottom fractions of either A285F or A285Y coat protein sucrose gradients. Thus, the formation of petite assembly products appears to correlate to the volume of the side chain at position A285, but did not require the presence of a side chain hydroxyl since phenyalanine and tyrosine had similar affects on assembly.
We investigated the in vivo products of assembly of WT and the A285 variant coat proteins in the absence of all other phage proteins to determine the inherent propensity of the proteins to form shells with increased curvature. Clarified cell lysates generated from cells expressing only the coat protein variants were examined by negative stain electron microscopy. The fraction of spirals, T=7, and petite particles were determined by counting over 500 particles for each variant (Table 2). WT coat protein produced ~22% petite shells, consistent with previous studies (Earnshaw and King, 1978). As the size of the amino acid at position A285 was increased, so did the fraction of petite shells. Therefore, the larger side chain at position 285 must be affecting the curvature of the subunits and/or the interaction interfaces.
Table 2.
Petite formation of A285 variants.
| Amino Acid | Volume (Å3) | Number counted | Petite (%) | T=7 (%) | Spiral (%) |
|---|---|---|---|---|---|
| Alanine (WT) | 88.6 | 561 | 22 | 65 | 13 |
| Serine | 89.0 | 543 | 25 | 62 | 13 |
| Threonine | 116.1 | 502 | 56 | 27 | 17 |
| Valine | 140.0 | 587 | 59 | 25 | 16 |
| Phenylalanine | 189.9 | 521 | 89 | 5 | 6 |
| Tyrosine | 193.6 | 548 | 90 | 4 | 6 |
Assembly of the coat proteins was also examined in the absence of any phage proteins in vitro. WT, A285T, and A285Y coat proteins were used in all the subsequent experiments as representatives for not defective, mildly defective, and severely defective coat proteins, respectively. Refolded coat protein monomers of these representatives were concentrated above 2 mg/mL to promote uncontrolled assembly (Teschke, King, and Prevelige, 1993). The products were again visualized by negative stain electron microscopy (Figure 4). WT coat protein produced mainly T=7 sized products, A285T coat protein produced both T=7 and petite products, while A285Y coat protein produced only petite products, consistent with the in vivo assembly in the absence of all phage proteins.
Figure 4. Petite formation is intrinsic to the coat protein variants.

Representative micrographs of negatively stained samples of WT, A285T, and A285Y coat proteins assembled in vitro in the absence of scaffolding protein.
Interactions between the A285 variant coat protein monomers and scaffolding protein
We observed that A285T and A285V coat protein lysates had scaffolding protein associated with the T=7 procapsids but less was observed in the petite procapsids. A285F and A285Y coat proteins make only petite procapsids that have just a fraction of normal scaffolding protein levels (Figure 3B). Since WT coat protein will assemble into petite particles in the absence of scaffolding protein, we hypothesized that the variant coat proteins may be less able to interact with scaffolding protein during assembly.
Weak-affinity chromatography (Zopf, 1990), which evaluates easily reversible but specific interactions, was used to qualitatively assess the binding of coat protein monomers to scaffolding protein. Refolded coat protein monomers were applied to a WT scaffolding protein-affinity column, where retention of coat protein monomers is directly related to the strength of their binding to scaffolding protein (Suhanovsky et al., 2010; Teschke and Fong, 1996; Zopf, 1990). The intrinsic tryptophan fluorescence of the coat protein monomers was used to monitor their elution from the column (Figure 3), with ovalbumin serving as a negative control (elutes at 1.5 mL). WT monomers eluted in a broad peak centered at 2.25 mL. A285T and A285Y coat protein monomers also eluted from the column at 2.25 mL, indicating that they bind full-length scaffolding protein with similar affinity as WT coat protein. Therefore, a change in affinity between the variant coat proteins and scaffolding protein is not likely to be the cause of the assembly defect.
Interactions between scaffolding protein and assembled forms of A285 variant coat proteins
We reasoned that petite procapsids may not have a normal complement of scaffolding protein because it might simply not fit inside. We used a scaffolding protein re-entry assay with full-length WT scaffolding protein and a shorter N-terminal deletion mutant (Δ1-140), which has been shown to retain function (Parker, Casjens, and Prevelige, 1998; Weigele et al., 2005) to determine if scaffolding protein is not found at normal levels in petite procapsids because it does not fit or because it cannot bind the inside of the procapsid lattice. Scaffolding protein can re-enter empty procapsid shells (procapsids stripped of scaffolding protein), through capsomer holes and bind to the inner wall of the shell (Greene and King, 1994; Prasad et al., 1993; Thuman-Commike et al., 2000). WT (T=7), A285T (mixture of T=7 and petite), and A285Y (only petite) empty procapsid shells were mixed with either WT or Δ1-140 scaffolding protein. The equilibrated samples were sedimented through a linear 5–40% sucrose gradient, and the coat and scaffolding proteins in the fractions were quantified (Figure 6). WT scaffolding protein (open circles in Figure 6A) readily entered the WT coat protein shells. Some WT scaffolding protein was able to enter and bind the A285T coat protein shells, yet the scaffolding protein peak was offset from the coat protein peak suggesting that the WT scaffolding protein was able to enter the T=7 sized shells, but not the petite particles. The WT scaffolding protein peak with the A285Y coat protein shells looked similar to the control sample of WT scaffolding protein alone, indicating that it is unable to bind the A285Y coat protein shells. On the other hand, the Δ1-140 scaffolding protein was able to enter and bind all three procapsid shells (Figure 6B). These data indicate that the petite shells are capable of binding scaffolding protein via its C-terminal coat-binding domain; however, WT scaffolding protein likely does not fit.
Figure 6. Δ1-140 scaffolding protein but not WT scaffolding protein re-enters and binds to petite procapsid shells.
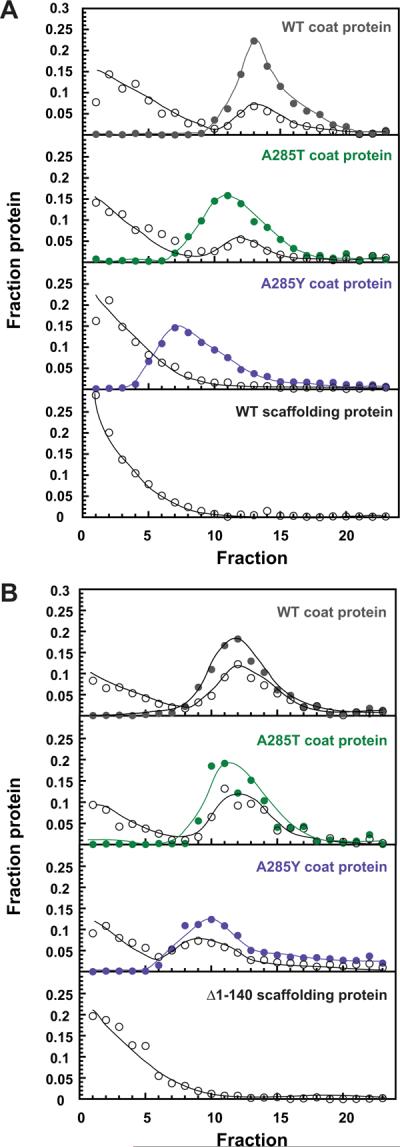
Both panels show sucrose gradient profiles of coat protein (closed circles) and scaffolding protein (open circles) from shell re-entry experiments. Panel (A) shows re-entry of WT scaffolding protein. Panel (B) shows re-entry of the N-terminally deleted, Δ1-140 scaffolding protein. The lines are drawn to aid the eye. The A285T and A285Y peaks were shifted up the gradient compared to WT due to the presence of petite shells in these samples.
In vitro assembly of the A285 variant coat proteins
Since the coat protein monomers showed normal affinity for scaffolding protein, we determined the ability of WT, A285T, and A285Y coat proteins to assemble in vitro with WT and Δ1–140 scaffolding proteins. Coat protein monomers and scaffolding protein were mixed at final concentrations of 0.5 mg/mL and monitored by following the increase in light scattering with time (Figure 7). Scaffolding protein is needed to promote rapid and robust assembly of coat protein monomers below 1 mg/mL (Suhanovsky and Teschke, unpublished data). WT coat protein assembled regardless of the scaffolding protein used in the assay. Assembly of A285T coat protein was more efficient with WT scaffolding protein than with Δ1–140 scaffolding protein, as measured by the final light scattering intensity. Surprisingly, A285Y coat protein assembly was more efficient with Δ1–140 scaffolding protein. We used negative stain electron microscopy to visualize the assembly products (Figure 8). WT coat protein assembled into procapsid-like particles with the correct size and shape with both WT and Δ1–140 scaffolding protein, though with slightly better fidelity with the WT scaffolding protein. A285T coat protein assembled predominately into T=7 sized procapsids with WT scaffolding protein (black arrows in Figure 8), but assembly with the Δ1–140 scaffolding protein resulted in a preponderance of petite procapsids (white arrows in Figure 8). Assembly with A285Y coat protein resulted in petite procapsids with both WT and Δ1–140 scaffolding protein; however, assembly was more efficient with the Δ1–140 scaffolding protein, consistent with the light scattering results (Figure 7). In addition, assembly of A285Y coat protein with WT scaffolding protein resulted in petite procapsids that were not able to close (see insets in bottom panel of Figure 8). When the Δ1–140 scaffolding protein was used, the assembly products had no gaps, indicating that they made closed petite procapsids. These data are consistent with the idea that the A285 coat protein variants are able to interact properly with scaffolding protein, but WT scaffolding protein cannot fit inside petite procapsids. We hypothesize that when the effect of the coat protein substitution is not too severe, as with A285T, the larger size of WT scaffolding protein sterically hinders assembly of petite particles, indicating that scaffolding protein plays a direct role in size determination.
Figure 7. In vitro assembly of A285Y coat is more efficient with Δ1-140 scaffolding protein than WT scaffolding protein.

In vitro assembly reactions of WT, A285T, and A285Y coat proteins with WT scaffolding protein (top) or the N-terminally deleted, Δ1-140 scaffolding protein (bottom). The assembly kinetics were followed by monitoring the increase in light scattering at 500 nm. The light scattering was normalized to the final intensity of assembly of WT coat protein. Open circles (WT), diamonds (A285T), and squares (A285Y) were added to the data for identification.
Figure 8. Δ1-140 scaffolding protein is not sufficient for assembly of A285T T=7 procapsids.

Representative micrographs of negatively stained samples of in vitro assembly products from reactions with WT scaffolding protein (A) and the N-terminally deleted, Δ1-140 scaffolding protein (B). The bright white appearance of the A285Y procapsids assembled with Δ1-140 scaffolding protein suggests that there is a substantially greater number of these scaffolding proteins associated with the procapsids compared to the full-length scaffolding protein, as a very full head resists staining. The insets in the bottom panel show enlarged images of A285Y assembly products (magnification × 2.5).
Substitutions at A285 increase flexibility of the A-domain
Limited proteolysis and difference map analysis of 3D reconstructions of WT and the F170 variants revealed a decrease in flexibility of the A-domain of F170K procapsid shells, which resulted in polyhead assembly (Suhanovsky et al., 2010). Since a decrease in flexibility of the A-domain diminishes the ability of F170K coat protein to assemble into pentons, we reasoned that the A285 variants might have increased flexibility. We used limited proteolysis of empty procapsid shells to probe for changes in the F170 and A285 variant coat proteins compared to WT coat protein. WT and the variant empty procapsid shells were digested for four hours using trypsin, chymotrypsin, and elastase. The cleavage sites have been mapped to a flexible region (residues 157–207, blue in Figure 10) of the coat protein A-domain (Kang et al., 2006; Lanman, Tuma, and Prevelige, 1999; Suhanovsky et al., 2010). The cleavage patterns were similar for each of the variant coat protein shells, with the major peptides having masses of 20–25 kDa. However, there were dramatic differences in the amount of these peptides (Figure 9A). F170K coat protein shells were significantly more resistant to protease than WT shells indicating a decrease in A-domain flexibility, as shown previously (Suhanovsky et al., 2010). The A285T substitution slightly increased the protease sensitivity of the procapsid shells compared to F170K substitution alone. A285Y procapsid shells showed a substantial increase in digestion, with complete digestion by trypsin of the full-length protein in four hours (Figure 9B). To confirm that the A285Y procapsid shells remained intact during the digestion, the samples were run on a 1.0% agarose gel (Figure 9D). The band corresponding to A285Y shells shifts slightly after incubation with trypsin; however, the particles remain intact.
Figure 10. Substitutions at position A285 torque the telokin-like domain, affecting both intercapsomer interactions and A-domain flexibility.

(A) Modeled asymmetric unit from the WT procapsid shell (PDB accession number 2XYY) viewed from the outside of the capsid with the telokin-like domain facing the reader. The seven quasi-equivalent subunits are labeled; subunits a – f comprise a hexamer and subunit g is from the neighboring pentamer. The subunits a and g, showing the capsomer interface, are color-coded to highlight important features; β-hinge (orange), flexible region of the A-domain (blue), D-loop (black), telokin-like domain (magenta), the α-helix containing A285 (yellow), the A285 position (green), and the N78 position (cyan). Subunits b – f have only the flexible region of the A-domain colored (blue) to emphasize the requirement for A-domain flexibility in pentamer and hexamer formation, as previously shown (Suhanovsky et al., 2010). (B) An enlarged view of the region highlighted by a dashed circle in (a), subunits a and g. (C) Same as (B), but with the telokin-like domain removed except for the α-helix containing A285 and the D-loop. The green box shows a zoom-in view of two-fold interactions between capsomers. The magenta box shows the connection of the telokin-like domain (first and last several amino acids in the telokin-like domain are colored in magenta) to the HK97-like body of the protein through two strands of the β-hinge.
Figure 9. Substitutions at A285 increase flexibility of the A-domain.

(A) Tricine-SDS-PAGE of WT and variant procapsid shells digested for four hours with trypsin, chymotrypsin, and elastase. The major proteolytic cleavage sites, which result in peptides that are ~ 20 – 25 kDa, are located in the A-domain. Trypsin digestion of empty procapsid shells (B) and refolded monomers (C) was analyzed over time. The amount of full-length coat protein remaining at each time point was quantified by SDS-PAGE and densitometry. (D) Agarose gel electrophoresis of WT and A285Y procapsid shells before and after trypsin digestion showing that the shells remain intact. Negative stain electron microscopy confirmed the presence of intact particles in these samples (data not shown). (E) Circular dichroism spectra of A285T and A285Y coat proteins, in mean residue ellipticity, show WT-like secondary structure. Amino acid substitutions at position 285 in coat protein cause an increase in flexibility of both monomers and subunits in shells.
The enhanced protease sensitivity observed in the shell samples could be due to either an overall increase in flexibility of the variant proteins or to a change in conformation of the assembled subunits due to the increased curvature in petite sized shells. To differentiate between these possibilities, a time course of trypsin proteolysis of WT, A285T, and A285Y in vitro refolded monomers was done. Both A285T and A285Y monomers were dramatically more sensitive to protease than WT coat protein monomers (Figure 9C). Circular dichroism spectra of A285T and A285Y coat protein monomers revealed WT-like secondary structure (Figure 9E), and taken with the ability of the proteins to assemble in vitro, suggests that the monomers are properly folded despite the increase in protease sensitivity. These results suggest that the increase in protease sensitivity of A285T and A285Y shells was due to greater flexibility of the A-domain and not due to the increased curvature of the petite shells. Together these data indicate that increasing the size of the side chain at A285 results in greater flexibility of the A-domain, which likely counteracts the observed decrease in A-domain flexibility of the original F170A/K variants. Thus, we hypothesize that flexibility of the A-domain is important for the intracapsomer interactions responsible for pentamer and hexamer formation.
Discussion
The HK97-like fold found in most, if not all, major capsid proteins of tailed phages and Herpesviridae points to a common ancestry and highlights the ability of this fold to serve as building blocks to form shells of highly variable sizes (Fokine et al., 2005). Capsids using the HK97-like fold range in size from ~45 nm to ~200 nm in diameter (Bamford, Grimes, and Stuart, 2005). How this structure evolved to accommodate drastic differences in diameters and how conformational switching of these subunits is regulated is not understood.
The role of coat protein amino acid substitutions in size determination
Amino acid substitutions in bacteriophage P22 coat protein at position F170 result in polyhead formation (Suhanovsky et al., 2010). Here we take advantage of the fact that the substitutions at position F170 also cause a ts defect to search for other regions that might be involved in conformational switching. A search for suppressors highlighted position A285, which is located in the telokin-like domain of coat protein (Parent et al., 2010a). The telokin-like domain is linked to the HK97 body of the coat protein through two strands of the β-hinge (Figure 10C). We hypothesize that increasing the size of the side chain at position 285 may torque the telokin-like domain, thereby affecting the conformation of the β-hinge and consequently the flexibility of the A-domain. Proper A-domain flexibility is essential for the balance of penton and hexon incorporation during assembly to achieve the correct products (Suhanovsky et al., 2010). The A285T coat protein suppressor may act by increasing the F170K coat protein A-domain flexibility and thereby alleviating polyhead formation by allowing proper penton incorporation during assembly.
Our work provides evidence that the telokin-like domain is important for size determination. The telokin-like domain was recently modeled to have a large loop (D-loop, black in Figure 10) that was suggested to make intercapsomer interactions in the procapsid (Chen et al., 2011). Position 285 is in an α-helix directly following the D-loop. Twisting of the telokin-like domain due to the increased size of the side chain at position 285 may also affect intercapsomer contacts through the D-loop (Figure 10C). Interestingly, in ongoing work, the substitution N78S was independently isolated multiple times from a preliminary suppressor search of the cs phenotype of A285T. Position 78 (cyan in Figure 10) is located in the β-sheet portion of the P-domain, which has been suggested to interact with the D-loop from the neighboring subunit in the adjacent capsomer. We hypothesize that this suppressor may act by correcting the intercapsomer contacts to achieve a T=7 sized capsid and are currently testing this idea.
The role of scaffolding protein in size determination
Auxiliary proteins (such as scaffolding or core proteins) are necessary to constrain the conformational possibilities of the capsid protein (Dokland, 2000). P22 scaffolding protein is 303 amino acids with an axial ratio of 11 and estimated dimensions of 247 Å by 22 Å (Parker, Stafford, and Prevelige, 1997). The Δ1–140 scaffolding protein is also a prolate ellipsoid with an axial ratio of 10 (Tuma et al., 1998). P22 scaffolding protein has a helix-turn-helix domain at the C-terminus that is necessary for coat protein binding (Weigele et al., 2005), but the amino-terminus has been shown to also be involved in coat protein binding (Cortines et al., personal communication).
The Δ1–140 scaffolding protein is sufficient for assembly of WT coat protein into T=7 sized particles (Parker, Casjens, and Prevelige, 1998). A285T coat protein assembly in the absence of scaffolding protein resulted in heterogeneously sized and shaped particles. While the Δ1–140 scaffolding protein was able to promote assembly of A285T coat protein into more homogeneous products, there were mostly T=4 sized particles. WT scaffolding protein was able to shift A285T coat protein assembly to T=7 sized heads, with very few petite particles. The C-terminus of scaffolding protein has been identified as the coat protein-binding domain (Weigele et al., 2005). However, our results highlight that the N-terminal 140 amino acids of scaffolding protein must also be important for size determination. WT coat protein, which prefers to assemble into a T=7 sized particles even in the absence of scaffolding protein, does not require the N-terminal 140 amino acids of scaffolding protein for size regulation. However, the N-terminal 140 amino acids of scaffolding protein are important for assembly of A285T coat protein into a T=7 sized procapsid. A285T coat protein must have a propensity to form a capsid with increased curvature in the absence of scaffolding protein, thereby making petite particles. We propose that the size of WT scaffolding protein may sterically inhibit the formation of petite procapsids. Alternatively, the N-terminal region of scaffolding protein may interact directly with the coat protein, influencing the conformation of the coat protein subunits and capsid size.
Interestingly, WT scaffolding protein was not able to shift assembly of A285Y coat protein into T=7 sized procapsids. The Δ1–140 scaffolding protein was better in directing the formation of closed particles, most likely because it was able to fit inside the smaller particles. We hypothesize that the A285Y coat protein has a greater inherent curvature than A285T coat protein; therefore, WT scaffolding protein may not be able to cause formation of a larger shell.
Size determination in other viruses
Unlike P22, bacteriophage T4 uses different proteins for its hexons and pentons (gp23 and gp24, respectively). These capsid proteins are 21% identical in sequence and both have the HK97-like fold (Fokine et al., 2005). Single amino acid substitutions in the A-domain of gp24 allow it to occupy the pentameric vertices, thereby abolishing the requirement for gp23. This supports our hypothesis that flexibility of the A-domain of the HK97-like fold is important for association of monomers into pentamers or hexamers. Other point mutations in gp23 have been isolated that affect the size and shape of the head. In particular, two mutations in gene 23 produce high levels of isometric phage, which possess greater curvature than the WT-shaped prolate phage. These substitutions are localized to the P-domain, possibly affecting intercapsomer interactions and capsid size in a similar manner to our amino acid substitutions at A285.
The ability to form different sized capsids from identical coat proteins with the HK97-like fold has been demonstrated with bacteriophages P2, 80α, and λ. These studies have provided evidence that assembly of T=7 and T=4 shells is indeed closely related. Bacteriophage P2 normally forms a T=7 capsid from its major capsid protein, gpN, with the help of its scaffolding protein, gpO. The satellite bacteriophage P4 codes for an external scaffolding protein, Sid, which has the ability to cause gpN to form a smaller T=4 capsid (Christie and Calendar, 1990; Dokland, Lindqvist, and Fuller, 1992). This indicates that the major capsid protein has a high degree of conformational flexibility and the scaffolding proteins are responsible for providing the constraints necessary to produce capsids of either size with high fidelity. A similar example involves the Staphylococcus aureus pathogenicity island SaPI1, which can be mobilized by infection with S. aureus helper phage 80α. The capsid protein of 80α can be compelled to assemble into smaller SaPI1-transducing particles. In this case, evidence suggests a SaPI1-encoded internal scaffolding protein directs the 80α capsid protein to assemble into small capsids (Poliakov et al., 2008). Finally, single amino acid substitutions in bacteriophage λ coat protein can cause it to assemble into smaller T=4 shells rather than T=7 (Katsura and Kobayashi, 1990).
Hendrix has suggested that like capsid proteins, the capsid size of tailed bacteriophages has also evolved over time (Hendrix, 2009). It is much more plausible for this evolution to be in the direction of larger capsids due to the constraints imposed on genome size by capsid size (Hendrix, 2009). Our study shows how a single mutation in a coat protein gene could increase the size of a capsid allowing for a larger genome to be packaged. We suggest that this may be a mechanism by which evolution has transformed viruses to assume diverse capsid morphologies. A detailed structural study of the petite procapsids and phage is underway to help understand the differences at the molecular level that affect size determination.
Material and methods
Phage and bacteria
The P22 bacteriophage used in this study all carried the c1–7 allele to prevent lysogeny. The phage carried an amber mutation in gene 5 (5− am N114), or point mutations in gene 5 resulting in F170K or F170A. The host strain Salmonella enterica serovar Typhimurium strain DB7136 (leuA414am, hisC525am, su0) has been previously described (Gordon et al., 1994; Winston, Botstein, and Miller, 1979). For certain experiments, DB7136 was transformed with a plasmid (pHBW1, ampR) (Parent, Suhanovsky, and Teschke, 2007) that codes for WT coat protein or a coat protein variant.
Isolation of revertants ofts phage
Second site suppressors of the ts F170A and F170K phage were isolated as previously described (Aramli and Teschke, 1999). Briefly, the gene 5 mutant phage, F170A and F170K, were plated at non-permissive temperatures. Each plaque is an independent revertant of the ts phage. Plaques were isolated, purified, and gene 5 was sequenced.
Generation of plasmid-encoded coat variants
Site-directed mutagenesis was used to alter pHBW1 to generate mutations in the coat protein gene at the codons for residues 170 and 285. The codon changes were: the F170 TTC codon was changed to AAG for F170K and to GCC for F170A. The A285 GCT codon was changed to TCT for A285S, ACT for A285T, GTT for A285V, TTT for A285F, and TAT for A285Y.
In vivo complementation
Salmonella DB7136 was transformed with a plasmid (pHBW1) that codes for WT coat protein or a coat protein variant (Parent, Suhanovsky, and Teschke, 2007). The gene 5 amber phage was complemented by expression of coat protein from the various plasmids.
Analysis of in vivo phage production
The methods for analysis of in vivo phage production were previously described (Suhanovsky et al., 2010). Briefly, Salmonella DB7136, transformed with the various plasmids that would express coat protein, was infected with phage carrying an amber mutation in gene 5. The phage also had an amber mutation in gene 13 (13− am H101) to block cell lysis. The phage-infected cells were concentrated, lysed by cycles of freezing and thawing with lysozyme and EDTA, and the clarified samples were applied to 2.2 mL linear 5–20% (w/w) sucrose gradients. The gradients were centrifuged in a Sorvall RC M120EX centrifuge with a RP55S rotor for 35 minutes at 104,813 × g at 20 °C and then fractionated from the top into 100 μL aliquots. The fractions were analyzed by SDS-PAGE and negative stain electron microscopy.
Negative stain electron microscopy
Aliquots (3 μL) of the sucrose gradient fractions and the in vitro assembly reactions were applied to carbon-coated, 300-mesh copper grids, allowed to absorb for 1 minute, and the grids were then washed with 2–3 drops of water followed by staining with 1% aqueous uranyl acetate for 30 seconds. Excess stain was blotted off with filter paper and the grids were air-dried and viewed in an FEI Technai Biotwin TEM (nominal magnification of 68,000) operated at 80 kV.
Determination of phage relative titer
Gene 5 amber phage were plated on Salmonella DB7136 at temperatures from 16 – 41 °C. Coat protein was supplied via complementation from plasmids (described above). LB plates were supplemented with 100 μg/mL ampicillin and 2 mM IPTG. The relative titer was calculated by dividing the titer at the experimental temperature by the titer of phage complemented with WT gene 5 at 30 °C.
Scaffolding protein re-entry into empty coat shells
Scaffolding protein was extracted from procapsids by treatment with 0.5 M GuHCl to generate empty procapsid shells (Greene and King, 1994). Empty procapsid shells at final concentrations of 4.3 μM were mixed with full-length or Δ1–140 scaffolding protein at a final concentration of 9.0 μM in 20 mM sodium phosphate (pH 7.6) with 50 mM NaCl and allowed to equilibrate at 20 °C for >20 h. Samples were applied to linear 5–40% sucrose gradients, as described above. Samples from each gradient fraction were analyzed by SDS-PAGE and the bands quantified by densitometry using BioRad Quantity One software.
Scaffolding protein column
Scaffolding protein affinity column experiments were previously described (Suhanovsky et al., 2010). Briefly, scaffolding protein with an N-terminal hexa-histidine tag was applied to a 1 mL TALON immobilized metal affinity column (Clontech) in 20 mM sodium phosphate buffer (pH 7.6) with 50 mM NaCl. Ovalbumin and coat protein monomers (100 μL each) were applied to the column and 250 μL fractions were collected. The elution profile of each protein was determined by tryptophan fluorescence, as described previously.
Analysis of coat protein monomers
Empty procapsid shells containing the coat protein substitutions were prepared as previously described (Anderson and Teschke, 2003; Parent, Suhanovsky, and Teschke, 2007). Briefly, urea-unfolded coat protein monomers were refolded by extensive dialysis against 20 mM phosphate buffer, pH 7.6 at 4 °C, and clarified by centrifugation at 175,000 × g at 4 °C for 20 min. Circular dichroism experiments were done with an Applied Photophysics (Leatherhead, Surrey, UK) Pi-Star 180 circular dichroism spectrapolarimeter with the 0.1 cm cuvette maintained at 20 °C. Refolded coat protein monomers were at a final concentration of 10.7 μM in 20 mM sodium phosphate buffer (pH 7.6). Wavelength scans were done over 200 – 250 nm with the following settings: step resolution of 0.5 nm, bandwidth of 3.0 nm, and data averaging for 15 sec/point, leading to scan time of 25 minutes.
For procapsid assembly experiments, coat protein monomers at a final concentration of 10.7 μM were mixed with WT or Δ1–140 scaffolding protein at final concentration of 14.9 μM. The reactions were monitored for 33 min at 20 °C. Assembly reactions with WT scaffolding protein were performed in 20 mM sodium phosphate (pH 7.6) with 50 mM NaCl. Assembly reactions with N-terminal hexa-histidine tagged Δ1–140 scaffolding protein were performed in 20 mM sodium phosphate (pH 7.6) without NaCl, to accommodate for the effect the hexa-histidine tag has on coat protein affinity (Cortines et al., personal communication). Light scattering intensity was normalized to the WT coat protein assembly with each of the scaffolding proteins. Coat protein monomers were also assembled in the absence of scaffolding protein by centrifugation in a 10K MWCO Microcon (Millipore) at 14,000 × g, 4 °C for 15 minutes until the coat protein concentration exceeded 42.8 μM.
Protease Digestion
WT and A285 variant procapsid shells were digested using trypsin, chymotrypsin, and elastase with an enzyme:substrate ratio of 1:100 at 20 °C. Refolded monomers were digested using trypsin at an enzyme:substrate ratio of 1:1000 at 4 °C. Samples were quenched with reducing sample buffer, heated for 5 min at 95 °C, and analyzed by 16% Tricine-SDS-PAGE (Schägger and von Jagow, 1987). Samples for native agarose gels (Parent et al., 2010b; Serwer and Pichler, 1978) were quenched with 1 mM phenylmethylsulfonyl fluoride.
Figure 5. The A285 variant coat protein monomers can bind scaffolding protein.

Elution profiles of WT, A285T, and A285Y coat protein monomers from a metal affinity matrix bound with histidine-tagged scaffolding protein and monitored by intrinsic tryptophan fluorescence. The A285 variants are able to bind to scaffolding protein with an affinity similar to that of WT coat protein.
Acknowledgements
We thank Dr. Marie Cantino for use of the University of Connecticut Electron Microscopy Center and her expert guidance in electron microscopy. We also thank Dr. Kristin Parent for helpful discussions and for reading of this manuscript. This work was supported by NIH grant to CMT (GM076661).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson E, Teschke CM. Folding of Phage P22 Coat Protein Monomers: Kinetic and Thermodynamic Properties. Virology. 2003;313:184–197. doi: 10.1016/s0042-6822(03)00240-x. [DOI] [PubMed] [Google Scholar]
- Aramli LA, Teschke CM. Single amino acid substitutions globally suppress the folding defects of temperature-sensitive folding mutants of phage P22 coat protein. J. Biol. Chem. 1999;274(32):22217–22224. doi: 10.1074/jbc.274.32.22217. [DOI] [PubMed] [Google Scholar]
- Bamford DH, Grimes JM, Stuart DI. What does structure tell us about virus evolution? Curr Opin Struct Biol. 2005;15:655–63. doi: 10.1016/j.sbi.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Bazinet C, King J. Initiation of P22 procapsid assembly in vivo. J. Mol. Biol. 1988;202(1):77–86. doi: 10.1016/0022-2836(88)90520-7. [DOI] [PubMed] [Google Scholar]
- Berger B, King J, Schwartz R, Shor PW. Local rule mechanism for selecting icosahedral shell geometry. Discreet Applied Mathematics. 2000;104:97–111. [Google Scholar]
- Berger B, Shor PW, Tucker-Kellogg L, King J. Local rule-based theory of virus shell assembly. Proc. Natl. Acad. Sci. USA. 1994;91(16):7732–6. doi: 10.1073/pnas.91.16.7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspar DLD, Klug A. Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol. 1962;27:1–24. doi: 10.1101/sqb.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- Chen D-H, Baker ML, Hyrc CF, DiMaio F, Jakana J, Weimin W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King J, Chiu W. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. PNAS. 2011;108(4):1355–60. doi: 10.1073/pnas.1015739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie GE, Calendar R. Interactions between satellite bacteriophage P4 and its helpers. Annu. Rev. Genet. 1990;24:465–90. doi: 10.1146/annurev.ge.24.120190.002341. [DOI] [PubMed] [Google Scholar]
- Dokland T. Scaffolding proteins and their role in viral assembly. Cellular & Molecular Life Sciences. 1999;56(7–8):580–603. doi: 10.1007/s000180050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokland T. Freedom and restraint: themes in virus capsid assembly. Structure. 2000;8(8):R157–62. doi: 10.1016/s0969-2126(00)00181-7. [DOI] [PubMed] [Google Scholar]
- Dokland T, Lindqvist BH, Fuller SD. Image reconstruction from cryo-electron micrographs reveals the morphopoietic mechanism in the P2-P4 bacteriophage system. EMBO Journal. 1992;11(3):839–46. doi: 10.1002/j.1460-2075.1992.tb05121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw W, Casjens S, Harrison SC. Assembly of the head of bacteriophage P22: x-ray diffraction from heads, proheads and related structures. J. Mol. Biol. 1976;104(2):387–410. doi: 10.1016/0022-2836(76)90278-3. [DOI] [PubMed] [Google Scholar]
- Earnshaw W, King J. Structure of phage P22 coat protein aggregates formed in the absence of the scaffolding protein. J. Mol. Biol. 1978;126:721–747. doi: 10.1016/0022-2836(78)90017-7. [DOI] [PubMed] [Google Scholar]
- Fokine A, Leiman PG, Shneider MM, Ahvazi B, Boeshans KM, Steven AC, Black LW, Mesyanzhinov VV, Rossmann MG. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. USA. 2005;102:7163–68. doi: 10.1073/pnas.0502164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CL, Sather SK, Casjens S, King J. Selective in vivo rescue by GroEL/ES of thermolabile folding intermediates to phage P22 structural proteins. J. Biol. Chem. 1994;269(45):27941–27951. [PubMed] [Google Scholar]
- Greene B, King J. Binding of scaffolding subunits within the P22 procapsid lattice. Virology. 1994;205:188–197. doi: 10.1006/viro.1994.1634. [DOI] [PubMed] [Google Scholar]
- Hendrix RW. Jumbo Bacteriophages. In: Van Etten JL, editor. Lesser Known Large dsDNA Viruses. Vol. 328. 2009. [PubMed] [Google Scholar]
- Jaroszewski L, Rychlewski L, Zhanwen L, Weizhong L, Godzik A. FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 2005;33:W284–W288. doi: 10.1093/nar/gki418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Li Z, Zhang Z, Baker ML, Prevelige PE, Chiu W. Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions. Nat. Struct. Biol. 2003;10(2):131–135. doi: 10.1038/nsb891. [DOI] [PubMed] [Google Scholar]
- Kang S, Hawkridge AM, Johnson KL, Muddiman DC, Prevelige PJ. Identification of subunit-subunit interactions in bacteriophage P22 procapsids by chemical cross-linking and mass-spectrometry. J. Proteome Res. 2006;5(2):370–377. doi: 10.1021/pr050356f. [DOI] [PubMed] [Google Scholar]
- Katsura I, Kobayashi H. Structure and Inherent Properties of the Bacteriophage Lambda Head Shell. J. Mol. Biol. 1990;(213):503–11. doi: 10.1016/S0022-2836(05)80211-6. [DOI] [PubMed] [Google Scholar]
- King J, Botstein D, Casjens S, Earnshaw W, Harrison S, Lenk E. Structure and assembly of the capsid of bacteriophage P22. Philos. Trans. R. Soc. London B. 1976;276:37–49. doi: 10.1098/rstb.1976.0096. [DOI] [PubMed] [Google Scholar]
- Lanman J, Tuma R, Prevelige PE., Jr. Identification and characterization of the domain structure of bacteriophage P22 coat protein. Biochemistry. 1999;38(44):14614–23. doi: 10.1021/bi9915420. [DOI] [PubMed] [Google Scholar]
- Lenk E, Casjens S, Weeks J, King J. Intracellular visualization of precursor capsids in phage P22 mutant infected cells. Virology. 1975;68(1):182–99. doi: 10.1016/0042-6822(75)90160-9. [DOI] [PubMed] [Google Scholar]
- Parent KN, Khayat R, Tu LH, Suhanovsky MM, Cortines JR, Teschke CM, Johnson JE, Baker TS. P22 coat protein structures reveal a novel mechanism for capsid maturation: Stability without auxiliary proteins or chemical cross-links. Structure accepted. 2010a doi: 10.1016/j.str.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent KN, Sinkovits RS, Suhanovsky MM, Teschke CM, Egelman EH, Baker TS. Cryo-reconstructions of P22 polyheads suggest that phage assembly is nucleated by trimeric interactions among coat proteins. Phys Biol. 2010b;7(4) doi: 10.1088/1478-3975/7/4/045004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent KN, Suhanovsky MM, Teschke CM. Polyhead formation in phage P22 pinpoints a region in coat protein required for conformational switching. Mol Microbiol. 2007;65(5):1300–10. doi: 10.1111/j.1365-2958.2007.05868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MH, Casjens S, Prevelige PE., Jr. Functional Domains of Bacteriophage P22 Scaffolding Protein. J. Mol. Biol. 1998;281:69–71. doi: 10.1006/jmbi.1998.1917. [DOI] [PubMed] [Google Scholar]
- Parker MH, Stafford WF, III, Prevelige PE., Jr. Bacteriophage P22 scaffolding protein forms oligomers in solution. J. Mol. Biol. 1997;268:655–665. doi: 10.1006/jmbi.1997.0995. [DOI] [PubMed] [Google Scholar]
- Poliakov A, Chang JR, Spilman MS, Damle PK, Christie GE, Mobley JA, Dokland T. Capsid Size Determination by Staphylococcus aureus Pathogenicity Island SaPI1 Involves Specific Incorporation of SaPI1 Proteins into Procapsids. J. .Mol. Biol. 2008;380:465–75. doi: 10.1016/j.jmb.2008.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad BVV, Prevelige PE, Jr., Marieta E, Chen RO, Thomas D, King J, Chiu W. Three-dimensional transformation of capsids associated with genome packaging in a bacterial virus. J. Mol. Biol. 1993;231:65–74. doi: 10.1006/jmbi.1993.1257. [DOI] [PubMed] [Google Scholar]
- Prevelige PE, Jr., King J. Assembly of bacteriophage P22: a model for ds-DNA virus assembly. Prog. Med. Virol. 1993;40:206–221. [PubMed] [Google Scholar]
- Schägger H, von Jagow G. Tricine-Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis for the Separation of Proteins in the Range from 1 to 100 kDa. Anal. Biochem. 1987;166(2):368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Serwer P, Pichler ME. Electrophoresis of bacteriophage T7 and T7 capsids in agarose gels. J. Virol. 1978;28(3):917–928. doi: 10.1128/jvi.28.3.917-928.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhanovsky MM, Parent KN, Dunn DE, Baker TS, Teschke CM. Determinants of bacteriophage P22 polyhead formation: the role of coat protein flexibility in conformational switching. Mol Microbiol. 2010;77(6):1568–1582. doi: 10.1111/j.1365-2958.2010.07311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Parker MH, Weigele P, Casjens S, Prevelige PEJ, Krishna NR. Structure of the Coat Protein-binding Domain of the Scaffolding Protein from a Double-stranded DNA Virus. J. Mol. Biol. 2000;297:1195–1202. doi: 10.1006/jmbi.2000.3620. [DOI] [PubMed] [Google Scholar]
- Teschke CM, Fong DG. Interactions between coat and scaffolding proteins of phage P22 are altered in vitro by amino acid substitutions in coat protein that cause a cold-sensitive phenotype. Biochemistry. 1996;35(47):14831–40. doi: 10.1021/bi960860l. [DOI] [PubMed] [Google Scholar]
- Teschke CM, King J, Prevelige PE., Jr. Inhibition of viral capsid assembly by 1,1'-bi(4-anilinonaphthalene-5-sulfonic acid) Biochemistry. 1993;32(40):10658–65. doi: 10.1021/bi00091a016. [DOI] [PubMed] [Google Scholar]
- Teschke CM, Parent KN. Let the phage do the work: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology. 2010 doi: 10.1016/j.virol.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuman-Commike PA, Greene B, Jakana J, McGough A, Prevelige PE, Chiu W. Identification of additional coat-scaffolding interactions in a bacteriophage P22 mutant defective in maturation. J. Virol. 2000;74(8):3871–73. doi: 10.1128/jvi.74.8.3871-3873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuman-Commike PA, Greene B, Malinski JA, King J, Chiu W. Role of the scaffolding protein in P22 procapsid size determination suggested by T = 4 and T = 7 procapsid structures. Biophys. J. 1998;74:559–568. doi: 10.1016/S0006-3495(98)77814-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma R, Parker MH, Weigele P, Sampson L, Sun Y, Krishna NR, Casjens S, Thomas GJJ, Prevelige PEJ. A helical coat protein recognition domain of the bacteriophage P22 scaffolding protein. J. Mol. Biol. 1998;281(1):81–94. doi: 10.1006/jmbi.1998.1916. [DOI] [PubMed] [Google Scholar]
- Weigele PR, Sampson L, Winn-Stapley DA, Casjens SR. Molecular genetics of bacteriophage P22 scaffolding protein's functional domains. J. Mol. Biol. 2005;348:831–44. doi: 10.1016/j.jmb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Duda RL, Hendrix RW, Johnson JE. Crystallization and preliminary X-ray analysis of the dsDNA bacteriophage HK97 mature empty capsid. Virology. 1998;243(1):113–8. doi: 10.1006/viro.1998.9040. [DOI] [PubMed] [Google Scholar]
- Winston R, Botstein D, Miller JH. Characterization of amber and ochre suppressors in Salmonella typhimurium. Journal of Bacteriology. 1979;137:433–439. doi: 10.1128/jb.137.1.433-439.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zopf D, Ohlson S. Weak-affinity chromatography. Nature. 1990;346:87–88. [Google Scholar]