

Abstract
Activation of metabotropic- (mGluRs) or NMDA-type glutamate receptors (NMDARs) are each linked to inducing long-term depression (LTD) of synaptic transmission in CA1 hippocampal neurons. These two forms of LTD are triggered by diverse signaling pathways yet both are expressed by the internalization of AMPA-type glutamate receptors (AMPARs). An unanswered question remains as to whether the convergence of the mGluR and NMDAR signaling pathways on AMPAR endocytosis renders these two forms of plasticity functionally equivalent, with both pathways inducing endocytosis of the same population of synaptic AMPARs. We now report evidence that these pathways couple to the endocytosis of distinct populations of AMPARs defined by their mobility in the membrane surface NMDAR activation enhances removal of surface. AMPARs that rapidly cycle into and out of the membrane surface, while activation of mGluRs with DHPG results in the internalization of a non-mobile population of AMPARs. Glutamate Receptor Interacting Proteins 1 and 2 (GRIP1/2) play a key role in defining the non-cycling receptor population. GRIP1/2 knockdown with siRNA increases the proportion of rapidly cycling surface AMPARs and inhibits mGluR- but not NMDAR-mediated AMPAR internalization. Additionally, we find that mGluR activation dissociates surface AMPARs from GRIP1/2 while stimulation of NMDARs elicits the loss of membrane receptors not bound to GRIP1/2. We propose that these two receptor pathways can drive the endocytosis of distinct populations of AMPARs: NMDARs couple to the endocytosis of rapidly cycling surface AMPARs not directly associated with GRIP1/2 while mGluR activation induces the endocytosis of non-cycling GRIP-bound surface AMPARs.
Keywords: AMPAR, GRIP/ABP, endocytosis, LTD, cycling, plasticity
Introduction
Long-term depression (LTD) of neurotransmission at multiple synapses is mediated by the endocytosis of synaptic AMPARs (Carroll, Beattie et al. 2001; Snyder, Philpot et al. 2001; Xiao, Zhou et al. 2001; Malenka 2003; Shepherd and Huganir 2007). Regulated AMPAR internalization and the resulting synaptic depression can be triggered by diverse stimuli even within a single neuron type (Bear and Abraham 1996; Massey and Bashir 2007; Shepherd and Huganir 2007). In CA1 hippocampal pyramidal neurons, activation of Both N-Methyl-D-aspartic acid-type glutamate receptors (NMDARs) and metabotropic glutamate receptors (mGluRs) trigger AMPAR internalization( Beattie, Carroll et al. 2000; Daw, Chittajallu et al. 2000; Man, Lin et al. 2000; Kim, Chung et al. 2001; Snyder, Philpot et al. 2001; Nosyreva and Huber 2005). Surprisingly, an early study found that NMDAR and mGluR-mediated LTD in CA1 hippocampal neurons did not occlude each other(Oliet, Malenka et al. 1997). While this is normally indicative of independent expression mechanisms, the similar coupling of these pathways to AMPAR endocytosis raises a question as to how these two forms of depression may remain functionally independent. One explanation yet to be examined is if these LTD pathways actually mediate the internalization of different populations of AMPARs.
A number of factors distinguish the induction mechanisms of NMDAR and mGluR mediated LTD. While NMDARs couple to receptor endocytosis through activation of phosphatases (PP1, PP2B) (Mulkey, Herron et al. 1993; Mulkey, Endo et al. 1994), mGluR signaling couples through a cascade requiring tyrosine phosphatases MAPK, and PI3K-Akt-mTOR signaling(Gallagher, Daly et al. 2004; Hou and Klann 2004; Huang, You et al. 2004; Huang and Hsu 2006; Moult, Gladding et al. 2006). It has been recently shown that NMDAR-, but not mGluR-dependent LTD, and AMPAR endocytosis is blocked in the presence of an inhibitor of protein phosphatase 2A (PP2A) (Nicholls, Alarcon et al. 2008). Additionally, protein synthesis dependent mGluR-LTD, but not NMDAR-mediated LTD, was found to require Arc/Arg 3.1 translation leading to enhanced AMPAR endocytosis(Park, Park et al. 2008; Waung, Pfeiffer et al. 2008). Such discrepancies seem to be indicative of the diversity in the processes coupled to AMPAR endocytosis; however, they do not provide evidence of whether these pathways ultimately converge on the internalization of a common population of synaptic AMPARs in hippocampal neurons.
Previous studies have demonstrated that synaptic AMPARs can differ greatly in their mobility; some rapidly and constitutively cycle in and out of the synaptic membrane, while others remain somewhat stable in the synaptic membrane (Luscher, Xia et al. 1999; Luthi, Chittajallu et al. 1999). We find that mGluR- and NMDAR-activation differentially mediate the endocytosis of these two populations of AMPARs in hippocampal pyramidal neurons. NMDAR-activation drives the endocytosis of the subpopulation of GluA2 AMPARs which constitutively cycle rapidly into and out of the membrane surface while mGluR activation internalizes the less mobile surface AMPARs. GRIP proteins, which regulate GluA2 trafficking (Dong, O’Brien et al. 1997; Dong, Zhang et al. 1999; Osten, Khatri et al. 2000; Mao, Takamiya et al. 2010), appear to play a key role in defining these two populations of AMPARs. Knock down of GRIP by siRNA increases the population of cycling AMPARs and blocks mGluR, not NMDAR mediated receptor internalization. Finally activation of group I metabotropic glutamate receptors but not NMDARs, dissociates GluA2 from GRIP and removes these receptors from the membrane surface. Our results suggest that by inducing the internalization of different populations of AMPARs, NMDAR and mGluR activation couple to forms of synaptic depression that are likely to be functionally distinct.
Results
AMPAR internalization mediated by NMDAR and mGluR activation is additive
LTD mediated by mGluR and NMDAR activation have been reported to be non-occlusive suggesting different mechanisms of expression(Oliet, Malenka et al. 1997). Surprisingly, subsequent reports demonstrated that the internalization of AMPARs contributes to both forms of plasticity (Beattie, Carroll et al. 2000; Snyder, Philpot et al. 2001; Xiao, Zhou et al. 2001; Nosyreva and Huber 2005). Despite a common expression mechanism, non-occlusion could result if the two signaling pathways target different populations of AMPARs for internalization. We tested this possibility by measuring the agonist-induced internalization of GluA2 receptors, which are found in the majority of heteromeric AMPARs expressed in hippocampal pyramidal neurons (Wenthold, Petralia et al. 1996). Brief application of the agonists NMDA or DHPG induce internalization of AMPARs resulting in synaptic depressions in hippocampal slices (Lee, Kameyama et al. 1998; Huber, Roder et al. 2001) and cultures (Beattie, Carroll et al. 2000; Man, Lin et al. 2000; Snyder, Philpot et al. 2001). These agonists induced synaptic depressions share common properties with stimulus evoked forms of NMDAR- and mGluR-dependent LTD, respectively. Fifteen minutes following a 2 minute application of NMDA we observed a −37 +/− 1.1% (n=16, P<0.001) change from control in punctate surface AMPARs detected immunocytochemically in hippocampal culture. DHPG applied for 3 minutes caused a −35 +/− 4.9% change from control (n=16, P<0.001). When both were applied together, however, we found that there was a nearly additive decrease in punctate surface GluA2s of −65 +/− 5.4% (n=16) (Fig 1A, B). This result cannot be attributed to a lack of saturation in the activation of either pathway as two sequential rounds of agonist activation of either the NMDAR or mGluR pathways caused no further loss in surface AMPARs (1x NMDA: −44 +/− 7.9 %, 2x NMDA: −43 +/− 10.0%, 1x DHPG: −40 +/−8.0%, 2x DHPG: −37 +/− 7.1%).
Figure 1. AMPAR internalization mediated by NMDAR and mGluR activation is additive.

A. Images depict surface GluA2 receptors on dendrites of cultured hippocampal neurons treated with ACSF (control), NMDA (50uM, 2 min), DHPG (20 μM, 3 min), or both agonists together. Surface GluA2s were labeled in live neurons prior to treatment. Fifteen minutes after agonist addition cells were fixed and immunolabeled for remaining surface-expressed receptors. (Scale bar = 5μm) B. Quantitation of surface GluA2 AMPAR immunofluorescence from cells in experiments described in A. Activation of both pathways resulted in a nearly additive loss of surface AMPARs. (n=16, *** P<0.001). C. Images depict the expression of surface GluA2s and synapsin on dendrites of cultured hippocampal neurons treated with ACSF (control), NMDA (50uM, 2 min), DHPG (20 μM, 3 min), or both agonists together. DHPG and NMDA application together caused a much greater reduction in synapsin puncta colocalized with GluA2 than did either agonist alone (Scale bar = 5μm) D. Quantitation of surface GluA2 AMPAR immunofluorescence from cells in experiments described in C. Activation of both pathways resulted in a nearly additive loss of surface AMPARs (n=5, * P<0.05). E. Images depict internalized GluA2s in dendrites of cultured hippocampal neurons treated with ACSF (Control) NMDA (50uM, 2 min), DHPG (20 μM, 3 min), or both agonists together. Surface GluA2s were labeled in live neurons prior to agonist addition. Following treatments antibodies bound to GluA2s remaining at the membrane surface were stripped with an acidic solution. Cells were then fixed and immunolabeled for antibody bound receptors internalized from the surface with agonist stimulation. (Scale bar = 5μm) F. Quantitation of internalized surface GluA2 AMPAR immunofluorescence from cells in experiments described in E. Activation of both pathways resulted in a nearly additive increase of internalized surface AMPARs (n= 5, ** P<0.01). G. Western blots were probed for GluA2 to detect internalized surface (top) and total (bottom) GluA2 after agonist stimulation in hippocampal acute slices. Surface proteins were labeled with cleavable biotin prior to agonist treatment. After biotin was cleaved from remaining surface proteins, lysates were subjected to avidin precipitation to isolate internalized protein. H. Quantitation of blots in G. The increase in internalized GluA2 labeling intensity is plotted as a percent change from control. Activation of both NMDAR and mGluR pathways resulted in a slightly greater than additive increase in internalized GluA2 (n=4, * P<0.05).
To ascertain if the decrease in surface GuA2 puncta represents a loss in synaptic receptors, we immunolabeled surface GluA2s and the presynaptic marker synapsin in control and agonist treated neurons. Following application of NMDA we observed a −30 +/− 4.7% change from control in the intensity of surface AMPAR puncta colocalized with synapsin in hippocampal cultures. DHPG caused a −29 +/− 2.0% change. When both agonists were applied together, we found that there was a nearly additive decrease in synaptic surface GluA2s −53 +/− 10.4% (n=5). We also examined whether there was a change in the proportion of synapses colocalized with GluA2 puncta and found a similar additive decrease when both signaling pathways were activated (NMDA −18 +/− 6.7%, DHPG −19 +/− 2.7%, NMDA + DHPG −33 +/− 4.1, n=5; Fig 1C, D).
It is possible that the reduced intensity in punctate surface GluA2 staining may reflect a diffusion of receptors away from the synapse rather than an internalization of AMPARs (Borgdorff and Choquet 2002; Tardin, Cognet et al. 2003). To determine if the loss of receptors could be due to lateral diffusion we analyzed the surface GluA2 labeling from experiments described in Figure 1A and B with a lower detection threshold to highlight all surface GluA2 labeling on the dendrite. Results were similar to what we observed with the analysis of punctate labeling indicating there was not simply a redistribution, but rather an overall loss in surface AMPARs due to receptor internalization (NMDA −21 +/− 5.6%, DHPG −32 +/− 8.6% NMDA + DHPG −41 +/− 5.8% (n=5).
To more directly assay the internalization of GluA2 we labeled surface GluA2 immunocytochemically prior to agonist stimulation. After 15 minutes, remaining surface bound antibodies were removed with an acid stripping protocol to enable subsequent detection of only Ab–bound AMPARs internalized from the membrane surface (Carroll, Beattie et al. 1999). NMDA induced a 223 +/− 66.9% (n=5) increase in internalized AMPARs and DHPG caused a 243 +/− 69.4% increase (n=5). When both agonists were applied together, we observed a 472 +/− 81.4% increase in internalized surface GluA2s of (n=5) (Fig 1E, F). A comparable experiment was performed in acute hippocampal slices. Slices (300μm) prepared from 14–21 day old rats were incubated in cleavable biotin to label all surface proteins. Slices were then treated at 37°C with NMDA, DHPG or both. Fifteen minutes after agonist application, remaining surface biotin was cleaved (4°C) and lysates were prepared. Biotin-labeled proteins (proteins internalized from the membrane surface only) were isolated by precipitation with streptavidin-coated beads and assayed on immunoblot for GluA2 expression. While these stimuli did not cause any change in total protein expression (NMDA 90 +/− 7%, DHPG 101 +/− 19%, NMDA + DHPG 104 +/− 18% of control, n=4), activation of NMDARs and mGluRs enhanced internalization of AMPARs over controls to an extent that was nearly additive to that elicited by either NMDAR or mGluR activation alone (NMDA 236 +/− 49%, DHPG 295 +/− 1.14%, NMDA/DHPG 937 +/− 2.6% of control, n=4). Together these results suggest that while both NMDAR and mGluR activation mediate the endocytosis of AMPARs, they apparently couple to the internalization of a largely non-overlapping pool or receptors.
Disruption of AMPAR cycling blocks NMDAR-, but not mGluR-mediated receptor internalization
We next sought to identify factors which could differentiate the apparently separable pools of AMPARs internalized following NMDAR and mGluR activation. It has been previously reported that a pool of AMPARs which rapidly cycles in and out of the synaptic membrane is likely internalized during NMDAR-dependent LTD as depletion of the cycling pool with agents that block the NSF-dependent AMPAR exocytosis occludes this form of depression (Luscher, Xia et al. 1999; Luthi, Chittajallu et al. 1999). However, rapidly cycling receptors appear to make up only a portion of all surface receptors, with the rest residing more stably in the synaptic membrane(Luscher, Xia et al. 1999). We therefore investigated whether cycling or immobilized receptors are targeted for endocytosis by mGluR activation. Hippocampal neurons were incubated with a TAT-conjugated NSF peptide (NSF-TAT) that disrupts the NSF-SNAP interaction. The cycling of AMPARs was monitored by labeling live neurons with an antibody recognizing an extracellular epitope of GluA2 and, following removal of the antibody; cells were fixed at various time points. Remaining surface levels were imaged following immunolabeling of fixed, non-permeabilized cells with a fluorescently-tagged secondary Ab. In control peptide treated neurons, surface receptor labeling decreased over time in untreated neurons as surface AMPARs were constitutively internalized during cycling and replaced with unlabeled receptors (Fig 2A, 55 +/− 2.7% of 0 minute control at 30 min, n=5; P<0.01). Disruption of NSF function resulted in a modest decrease in the surface expression of AMPARs compared to control peptide treated neurons measured at the 0 time point (−25 +/− 5.9 %, n=7, P<0.01). However these cells exhibited little further loss of surface AMPAR labeling over 30 minutes (Fig 2A, 88.9 +/− 6.6% of 0 minute control at 30 min., n=5; P<0.01) indicating that the constitutive internalization of AMPARs associated with rapid receptor recycling had been disrupted.
Figure 2. Disruption of AMPAR cycling blocks NMDAR but not mGluR-mediated receptor internalization.

A. Disruption of NSF blocks AMPAR cycling. Surface AMPARs in live neurons were immunolabeled, after which neurons were washed and incubated for different time intervals to allow constitutive receptor internalization to occur. Remaining surface AMPARs were labeled with 2° Ab following fixation. Loss of surface receptors over time provides a measure of constitutive AMPAR endocytosis. Graph depicts constitutive endocytosis of AMPARs over 30 minutes. The loss of surface AMPAR immunolabeling in control cells reveal the basal endocytosis associated with rapid receptor cycling. Neurons in which NSF-SNAP function has been disrupted with a TAT-tagged peptide (NSF-TAT) exhibit significantly reduced receptor endocytosis (n=5, **P< 0.01). B. Disruption of NSF prevents NMDAR- mediated AMPAR endocytosis. Representative images of surface GluA2 levels in dendrites of control peptide and NSF-TAT peptide-treated neurons with and without treatment with NMDA or DHPG to induce receptor internalization (Scale bar= 5 μm). C. Quantitation of experiments in B (n=7, **P<0.01) shows that the NSF-TAT peptide blocks the NMDA but not DHPG induced loss of surface GluA2s. D, E mEPSCs were recorded in cultured neurons during a 10 minute baseline period and 15 minutes after bath application of NMDA (D) or DHPG (E). Scale bar 500ms/20 pA. Cumulative distribution plots of mEPSC inter event intervals (top) and amplitude (bottom) demonstrate a reduction in the size and frequency of events with NMDA and DHPG treatment (NMDA n=6, Interval P< 0.05, Amplitude P< 0.05 KS test; DHPG, n=5, Interval P<0.001; Amplitude P<0.05 KS test). Inset shows series resistance at 5 minutes before (−5) and 15 minutes (15) after agonist application. F,G. The amplitude of mEPSCs was measured in neurons loaded with a peptide that disrupts NSF function (NSF peptide) through the recording pipette. mEPSC amplitude and inter event intervals were measured before and 15 minutes after a 1 minute application of NMDA (F) or DHPG (G). NMDA application caused no shift in the cumulative probability plots for mEPSC amplitudes and intervals in the presence of the NSF peptide (n=5, ns). DHPG application caused similar shifts the cumulative probability of mEPSC interval and amplitudes to controls when recorded in the presence of NSF peptide (n=5, Interval P<0.05; Amplitude P<0.01 KS test). The shift in amplitude was similar to controls but did not achieve significance. H. Graph showing the effects of various treatments on mean mEPSCs frequency (P< 0.05) demonstrates that disruption of NSF function blocks NMDA but not the DHPG induced depression in mEPSCs. I. Graph depicts changes in mEPSC amplitudes in response to agonist treatments (P< 0.05).
To examine the regulated internalization of AMPARs following NMDAR and mGluR activation in cells in which cycling had been blocked, we measured the loss of surface receptors following NMDA or DHPG treatments in cells exposed to the NSF-TAT peptide. While mGluR activation reduced surface GluA2 levels similar to controls, NMDA did not induce AMPAR endocytosis in the presence of the NSF-TAT peptide (NMDA/Control −39 +/− 7.6%, NMDA/NSF-TAT 10 +/− 7.1% of untreated control, n=7, P<.01, DHPG/Control −36 +/− 6.6%, DHPG/NSF-TAT peptide −36 +/− 6.9% of untreated control, n=7; P<0.01; Fig 2B, C). These findings indicate that rapidly recycling AMPARs are necessary for NMDAR−, but not mGluR-mediated AMPAR endocytosis and provides additional evidence that these two receptor signaling pathways can target the internalization of two different populations of AMPARs, the non-cycling versus the rapidly cycling population.
Electrophysiological experiments were performed to confirm a differential role of cycling AMPARs in NMDAR and mGluR-mediated synaptic depression. Miniature excitatory post synaptic currents (mEPSCs) were recorded in whole cell patch clamp in the presence of TTX. Baseline currents were recorded during a 10 minute period followed by application of agonist. Fifteen minutes after washout mEPSCs were again recorded. In NMDA-treated neurons (2 minutes, 50 μM) we also observed a rightward shift in the cumulative distribution of AMPAR mEPSC inter event intervals (Fig 2D (left) n=6, P< 0.05 KS test), and a leftward shift in amplitudes (Fig. 2D (right), n=6, P<0.05 KS test) along with a decrease in the mean frequency (−24 +/− 7.4 %, n=6, P< 0.05) and amplitude (−13 +/− 1.8%, n=6, P< 0.05) of mEPSCs (Fig 2H,I). Application of DHPG (3 minutes, 20 μM) resulted in similar shifts in the distribution of mEPSC intervals and amplitudes (Fig 2F, G, n=5 P<0.05 KS test) with a decrease in the overall size and frequency of events (Amplitude −34 +/− 7.2%, n=5, P<0.05, Frequency −23 +/− 4.3%, n=5, P<0.05). When we included an unmodified NSF disrupting peptide (NSF peptide) similar to that used in our immunocytochemical studies, the depression of mEPSCs by NMDA was blocked (Amplitude −2 +/− 1.2%, Frequency +29 +/− 15.9%, Fig 5F,H,I), however, DHPG application still caused a reduction in mEPSCs (Amplitude −20 +/− 7.2%, Frequency −39 +/− 11.0%, n=5, P<0.05, Fig 5G, H,I).
Figure 5. Knockdown of GRIP1/2 blocks mGluR-mediated AMPAR internalization and synaptic depression.

A. Surface GluA2 levels imaged after NMDA or DHPG treatment in neurons following knockdown of GRIP1/2 by siRNA. Neurons were transfected with scrambled or GRIP1/2 targeted siRNA and treated with NMDA or DHPG 72 hours later. While NMDAR-mediated reductions in surface GluA2 are unaffected by GRIP1/2 siRNA, the DHPG-induced decreases are blocked (Scale bar=10 μm). B. Graph shows quantitation of experiments represented in A. (n=8, ***P<0.001). C. The inter event interval (top) and amplitude (bottom) of mEPSCs was measured in GRIP siRNA transfected neurons before and 15 minutes after a 2 minute application of NMDA. As in control neurons, cumulative distribution plots show that NMDA caused an increase in intervals and decrease in amplitudes of mEPSCs in GRIP siRNA transfected neurons (n=5, Interval P<0.05; Amplitude P<0.01). Inset shows series resistance at 5 minutes before (−5) and 15 minutes (15) after agonist application. Scale bar for traces 500ms/20 pA. D. mEPSCs were recorded in cultured neurons during a 10 minute baseline period and 15 minutes after bath application of DHPG in GRIP siRNA transfected neurons. DHPG application caused no change in mEPSC inter event intervals (top) or amplitudes (bottom) in siRNA transfected neurons as indicated by the cumulative distribution plots. (n=5). E. Quantitation of median mEPSC frequency and amplitude experiments in C and D (*P<0.05).
NMDAR, but not mGluR-mediated AMPAR internalization reduces the population of cycling surface AMPARs
Our aforementioned results suggest that NMDAR (but not mGluR activation) couples to the internalization of cycling AMPARs. Therefore, NMDA treatment would be predicted to reduce the numbers of cycling surface AMPARs, while DHPG would not. To test this prediction we induced the endocytosis of AMPARs with the agonists NMDA or DHPG and then measured the extent of constitutive endocytosis in the remaining surface receptors by immunolabeling as previously described. Fifteen minutes after both agonist, treatments we observed a decrease in surface AMPARs (Fig 3B inset, NMDA −39+/4.8% control, DHPG −36 +/− 4.7%, n=6; P< 0.05). However, when we measured the constitutive endocytosis of surface AMPARs (beginning 15 minutes after agonist application) we observed that while DHPG treated-cells exhibited AMPAR cycling comparable to untreated control cells, surface AMPARs in the NMDA-treated cultures exhibited a reduced endocytosis of GluA2, indicative of a decrease in AMPAR cycling (Fig 3A, B, Control @ 40 min −61 +/− 2.5% 0 min value, DHPG @ 40 min −50 +/− 4.9%, NMDA @ 40 min −25 +/− 11.2%; P< 0.01). These data are consistent with cycling AMPARs being removed from the membrane surface following activation of NMDARs while mGluR activation predominantly removes the more stable surface receptors, leaving the mobile population of receptors in tact.
Figure 3. NMDAR, but not mGluR-mediated AMPAR internalization reduces pool of cycling surface AMPARs.
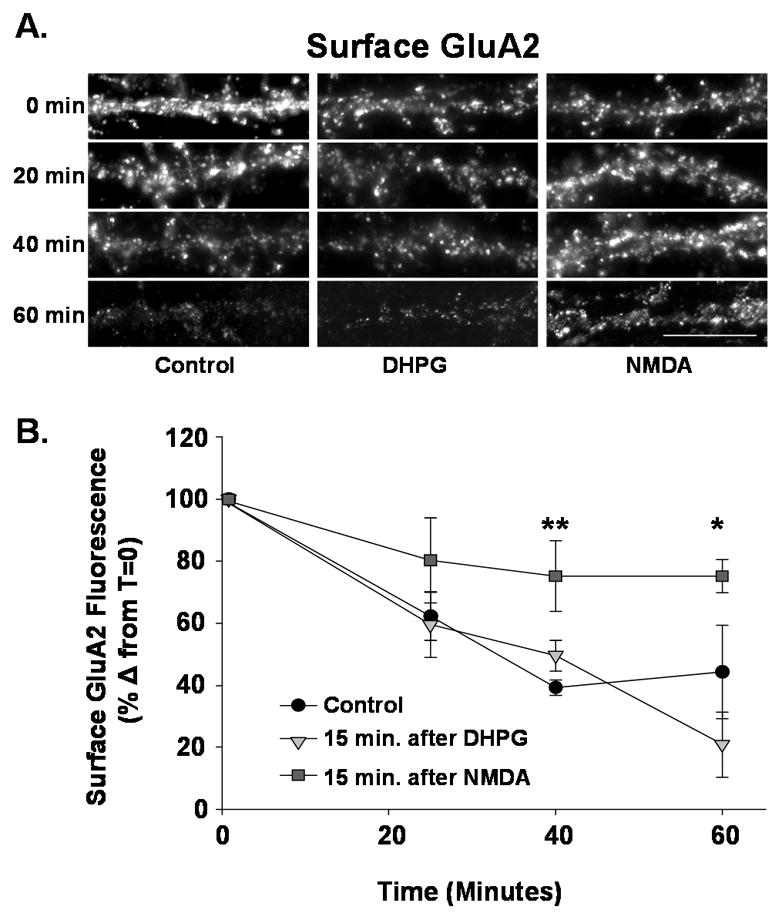
Cultured hippocampal neurons were treated with NMDA or DHPG to induce endocytosis of surface AMPARs. Fifteen minutes following agonist treatment constitutive AMPAR endocytosis was measured as described in Figure 2. A. Images are shown of surface GluA2 labeling in dendrites of control; NMDA- or DHPG-treated neurons fixed after 0, 20, 40 or 60 minutes. NMDA and DHPG treated neurons exhibit reduced GluA2 labeling at 0 minutes due to regulated endocytosis of receptors. (Scale bar= 5 μm) The constitutive loss of AMPARs over time is reduced in NMDA treated neurons. B. Quantitation of experiment in A. While DHPG-treated and control neurons exhibit a similar level of constitutive GluA2 endocytosis, NMDA treated neurons undergo little constitutive endocytosis suggesting a reduced population of rapidly cycling receptors (n=7, * P< 0.05, ** P<0.01).
Knockdown of GRIP1 increases the cycling population of AMPARs
Our data indicate that disrupting the rapid recycling of AMPARs blocks NMDAR- but not mGluR-mediated internalization of AMPARs. These results suggest that disruption of the population of non-recycling AMPARs might interfere with AMPAR endocytosis after mGluR activation. Immobilization of AMPARs at synapses is likely to be mediated by a number of factors including their interaction with associated proteins. Glutamate Receptor Interacting Protein/AMPAR Receptor Binding Protein (GRIP/ABP), found at a subset of excitatory synapses in hippocampal neurons (Wyszynski, Kim et al. 1998) has been implicated in the stabilization of surface AMPARs (Srivastava, Osten et al. 1998; Osten, Khatri et al. 2000). We first examined the impact of knocking down GRIP expression with siRNA on AMPAR recycling. After 72 hours we found that average GRIP levels were reduced by about 60% both as measured immunocytochemically and by Western analysis of protein levels (Fig 4A, B) The GRIP siRNA also caused a 26 +/− 7.2% decrease in surface GluA2 levels. We compared the extent of cycling of remaining surface AMPAR, as indicated by constitutive AMPAR endocytosis in control and GRIP siRNA treated neurons. The proportion of cycling AMPARs in control siRNA treated neurons was similar to that observed in naïve neurons (Fig 4C,D; Control siRNA min 38 +/− 12.2% of 0 min time point remaining @ 40min value; n=5, P<0.01). However, GRIP siRNA treated neurons exhibit a higher proportion of surface AMPARs subject to constitutive endocytosis (Fig 4C,D; GRIPsi 19 +/− 8.1% of 0 min time point remaining @ 40 min; n=5, P<0.01s). This suggests that GRIP1/2 mediates the stabilization of a population of surface receptors and reduced GRIP protein expression enables these receptors to become part of a cycling pool and readily internalize.
Figure 4. Knockdown of GRIP1/2 increases the proportion of surface AMPARs that rapidly cycle.
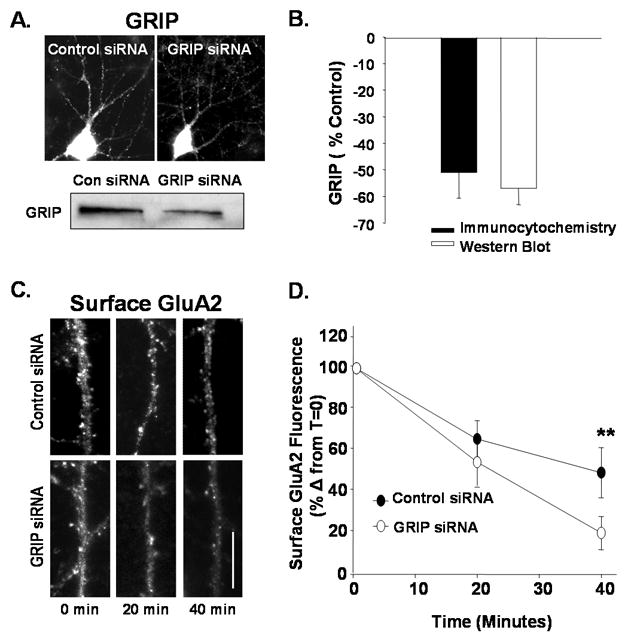
A. GRIP levels in cultured hippocampal neurons were monitored by immunocytochemistry (top) and Western blotting (bottom) 72 hours after transfection with control or siRNA targeting GRIP ½ (GRIP siRNA) (Scale bar= 10 μm). B. Graph represents changes in the levels of GRIP in experiments represented in A. C. Constitutive AMPAR endocytosis was measured in control and GRIP siRNA treated cells. Images depict remaining surface GluA2 labeling measured at 0, 20, and 40 minutes (Scale bare = 5 μm). D. Quantitation of GluA2 endocytosis from experiments described in C (n=4, ** P< 0.01). GRIP siRNA-treated cells exhibited a significantly greater proportion of constitutively cycling GluA2s.
GRIP is required for mGluR-mediated AMPAR internalization and synaptic depression
If GRIP1/2 expression can impact the proportion of non-recycling AMPARs, then it could also modulate the ability of neurons to express mGluR-dependent AMPAR endocytosis which we predict to be linked to this receptor population. Neurons exposed to control or GRIP siRNA for 72 hours were treated with NMDA or DHPG. After fifteen minutes, the remaining surface GluA2 AMPARs were immunolabeled. Decreasing GRIP1/2 levels did not affect the NMDA-induced decrease in surface AMPARs, but blocked the mGluR-mediated AMPAR reduction (control siRNA/NMDA: −47 +/− 6.3%, GRIP siRNA/NMDA: −46 +/− 7.8% of control, n=8, ns; control siRNA/DHPG: 56 +/− 6.7%, GRIP siRNA/DHPG: 3 +/− 12%, n=8, P<0.001; Fig 5A, B). Normal endogenous levels of GRIP1/2, therefore, appear to be essential for AMPAR endocytosis following mGluR activation.
To confirm the functional impact of reduced GRIP expression on NMDAR- and mGluR-mediated endocytosis, we performed electrophysiological recordings on our cultured neurons. In neurons transfected with GRIP1/2 siRNA, the NMDA-induced decrease in mEPSC frequency was not affected (Frequency: baseline 7 +/− 1.1 Hz, NMDA 5 +/− .9 Hz, −28 +/− 2.2 % change, n=4 P<0.05; Amplitude: baseline 13 +/− 2.4 pA, NMDA 12 +/− 2.1 pA, −10 +/− 3.0 % change, n=4; Fig 5C,E). However, we found no depression in synaptic currents following DHPG application (Frequency: baseline 7 +/− 0.9 Hz, NMDA 7 +/− 1.1 Hz, n=5 ns; Amplitude: baseline 15 +/− 2.7 pA, NMDA 14 +/− 2.5 pA, n=5; Fig 5D, E) in the GRIP siRNA treated neurons. Together these findings suggest that GRIP1/2 may maintain a non-cycling population of AMPARs that are necessary for mGluR-mediated internalization AMPARs.
mGluR, not NMDAR activation decreases GRIP-surface GluA2 association and colocalization
Our results are consistent with the possibility that GRIP normally stabilizes a population of surface GluA2s that is internalized following activation of mGluRs but not NMDARs. We tested this using immunocytochemistry to monitor NMDAR- or mGluR-dependent changes in colocalization of GRIP with surface expressed GluA2s. In untreated cultures, 61 +/− 2.6% of GRIP clusters were colocalized with puncta of surface expressed GluA2s (n=9). Activation of both mGluRs and NMDARs resulted in an overall reduction in the intensity of surface AMPAR labeling, as well as in the complete loss of a portion of detectable dendritic clusters (DHPG −22 +/− 2.5%, NMDA −21 +/− 6% change in clusters per length of dendrite compared to control, n=6, P<0.05). However, mGluR activation caused a decrease primarily in GRIP-colocalized AMPAR clusters (colocalized −32 +/−7.7% of control, n=6, P<0.05; not colocalized −6 +/− 2.9% of control, n=6, ns; Fig 6A, B). Conversely, NMDAR activation preferentially caused a loss in AMPARs not colocalized with GRIP (colocalized −12 +/− 7.1% of control, n=6, ns, not colocalized −25 +/− 4.7% of control, n=6 P<0.05; Fig 6A, B).
Figure 6. mGluR, not NMDAR activation decreases GRIP-GluA2 association and colocalization.
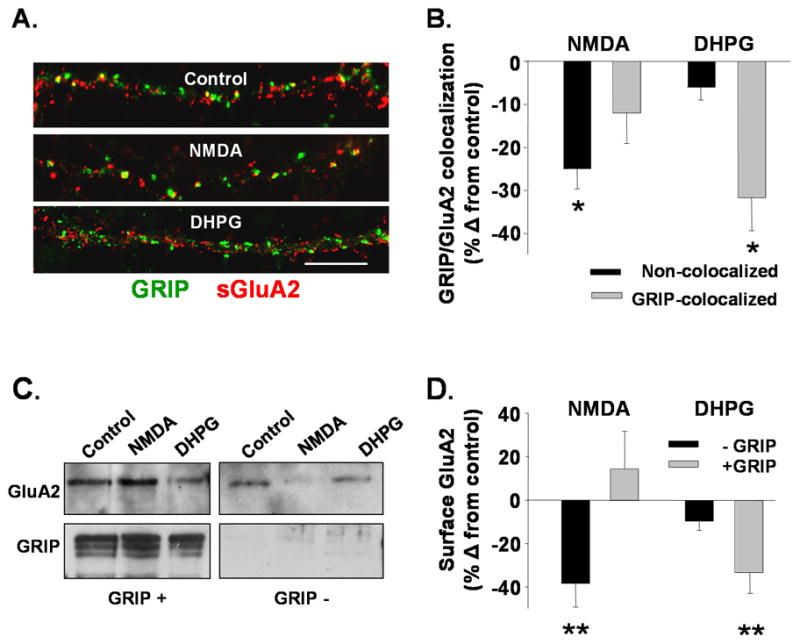
A. Control, NMDA or DHPG treated neurons were immunolabeled to detect surface GluA2 (red) and GRIP (green) (Scale bar =5 μm). B. Graph presents the percent change from control in the number of surface GluA2s found colocalized with (GRIP-colocalized) or not (non-colocalized) in NMDA or DHPG treated neurons (n=6, *P<0.05 using ANOVA). NMDA reduces the numbers of surface GluA2s not colocalized with GRIP while DHPG reduces the GluA2s found colocalized with GRIP. C. Western blot shows surface-expressed GluA2s, either bound to GRIP (left, GRIP+) or not (right, GRIP−) Western Blots were first probed for GluA2, then reprobed for GRIP to confirm the purity of the GRIP+ and GRIP− fractions. D. Graph shows quantitation of immunoprecipitated surface GluA2 receptors in the GRIP-bound, and non-GRIP bound (n=5, **P<0.01) fractions. Results provide evidence that mGluR activation preferentially reduces the levels of surface GluA2 bound to GRIP, whereas NMDAR stimulation decreases surface GluA2s not associated with GRIP.
To test whether reductions in the colocalization of AMPARs and GRIP following mGluR-mediated AMPAR endocytosis represent reduced binding of GluA2 and GRIP, coimmunoprecipitation experiments were performed. As GRIP proteins have been found to be localized both at synapses and in intracellular compartments (DeSouza, Fu et al. 2002) we performed an experiment designed to specifically measure changes in the levels of surface expressed GluA2 receptors bound to GRIP. Lysates were prepared from hippocampal slices treated with NMDA or DHPG followed after 15 minutes by biotinylation of surface proteins. Using immunoprecipitation with an anti-GRIP antibody, GRIP-bound proteins were separated by Protein G agarose binding from non-GRIP bound proteins. GRIP-bound (eluted from bead pellet) and non-GRIP bound (supernatant) proteins were subjected to streptavidin pull-down in order to isolate surface proteins. Surface proteins from both fractions were run on SDS-PAGE gels and analyzed for GluA2 on Western blot. If either the NMDAR or mGluR signaling pathways induce the endocytosis of surface GluA2s not associated with GRIP a decrease in GluA2 levels is predicted in the non-GRIP bound fraction. If GluA2s bound to GRIP at the surface are internalized a decrease of GluA2 in the GRIP bound fraction is expected. Analysis showed that in the DHPG- but not the NMDA-treated lysates there was a decrease in surface GluA2 in the GRIP-bound fraction (NMDA 15 +/− 17%, DHPG −34 +/− 9.5% of control, n=4, P<0.01Fig 6C left upper panel, 6D). In contrast there was a decrease in the surface GluA2 levels in the non-GRIP bound fraction following NMDAR activation (NMDA −38 +/− 10.8%, DHPG −10 +/− 4.1% of control, n=4, P<0.01; Fig 6C, D). The blots, when reprobed with anti-GRIP antibody, confirmed the presence of GRIP1/2 in the GRIP-bound but not the non-GRIP bound fraction. These results provide further evidence that NMDAR stimulation primarily targets the internalization of non-GRIP bound AMPARs, while mGluR activation drives the endocytosis of GRIP-bound AMPARs.
Discussion
Our results demonstrate that NMDAR and mGluR activation induce the internalization of two distinct populations of AMPARs. NMDAR activation results in the loss of a population of rapidly cycling surface AMPARs not directly associated with GRIP1/2. In contrast, mGluR activation decreases GRIP1/2-GluA2 association and internalizes surface GluA2s previously bound by GRIP1/2. By demonstrating that NMDA and mGluR-activation couple to the endocytosis of different subpopulations of GluA2 AMPARs, these findings provide an explanation for previously proposed differences in expression mechanisms between NMDAR- and mGluR- induced LTD (Oliet, Malenka et al. 1997).
The hypothesis that these two forms of LTD induce endocytosis of different populations of surface GluA2-receptors is supported by the observed additivity of GluA2 endocytosis when both NMDAR and mGluR are activated simultaneously. While it is possible that the simultaneous activation of these two receptors could result in an interaction of the downstream signaling pathways that could impact AMPAR endocytosis, analysis of the link between rapidly cycling AMPARs and NMDAR- or mGluR-mediated AMPAR endocytosis further supports the internalization of different populations of receptors by NMDAR and mGluR activation. Blockade of the rapidly cycling population of AMPARs prevented the NMDAR-mediated changes in surface AMPARs, consistent with previous electrophysiological data. Additionally, constitutive GluA2 endocytosis was selectively reduced after NMDAR-induced AMPAR endocytosis consistent with NMDAR activation alone depleting the pool of rapidly cycling GluA2s. As these measurements are of endocytosis in isolation (i.e. not exocytosis as well), they are indirect measurements of constitutive AMPAR cycling. However, in most of our experiments we have measured endocytosis when surface AMPAR expression is expected to be stabilized, and therefore endocytosis is in balance with exocytosis, making it a reasonable indicator of receptor turnover (Carroll, Beattie et al. 2001).
The mechanistic link between NMDAR activation and cycling AMPARs is still not clear. One possibility is that constitutive endocytosis during rapid cycling delivers these receptors into endosomal compartments where they can then be sequestered through protein interactions, possibly PICK1 (Sossa, Court et al. 2006; Citri, Bhattacharyya et al. 2010) or GRIP1/2 (DeSouza, Fu et al. 2002). The inability of mGluR activation to alter constitutive AMPAR cycling or for mGluR-mediated AMPAR internalization to be affected by disrupting AMPAR cycling indicated that this receptor pathway is more closely linked to receptors which are maintained out of this highly mobile population.
Following activation of mGluRs, GRIP-stabilized AMPARs appear to be the primary target for endocytosis. Consistent with this model, we find that using siRNA to reduce GRIP1/2 expression blocks the AMPAR internalization and the synaptic depression mediated by mGluRs. Reductions in GRIP1/2 expression, however, do not interfere with the NMDAR-triggered reductions in surface GluA2s. Other studies suggest a role for GRIP1 in modulating AMPARs following their internalization by NMDAR activation by either maintaining internalized AMPARs (DeSouza, Fu et al. 2002) or in facilitating the reinsertion of the receptors (Mao, Takamiya et al. 2010). A recent study further indicates that the ATPase thorase can modulate AMPAR recycling though modulation of GRIP1/AMPAR interactions (Zhang, Wang et al. 2011). Our finding that NMDAR-mediated AMPAR internalization occurs despite reduced GRIP expression are not inconsistent with these results which identify a role for GRIP, not in the initial regulated endocytosis of receptors, but rather in the stabilization of internalized receptors in intracellular pools. Our data does support a role for GRIP binding to AMPARs prior to LTD induction to maintain a population of surface AMPARs that can be released for endocytosis following mGluR activation. This model is consistent with that which has been proposed to occur in the cerebellum, whereby activation of the mGluR pathway mediates a dissociation of AMPARs from GRIP resulting in their enhanced endocytosis (Matsuda, Launey et al. 2000; Takamiya, Mao et al. 2008). Taken together, surface AMPARs bound to GRIP may be essential to mGluR –mediated receptor endocytosis, while AMPARs bound to GRIP after their endocytosis appear to be important for maintaining and regulating intracellular AMPAR pools following NMDAR-mediated endocytosis.
The existence of two different populations of AMPARs internalized following NMDAR or mGluR activation has important functional implications. Local synaptic expression of GRIP 1/2 could alter the extent of synaptic depression mediated by AMPAR endocytosis triggered by NMDAR or mGluR activation. It has, in fact, been shown that GRIP 1/2 is not apparently expressed at all synapses, suggesting some synapses would not be competent to express mGluR-LTD at all. Further studies will certainly reveal additional complexities of AMPAR internalization in regulating receptor-driven changes in surface AMPAR expression mediated by the these and other proteins.
Methods
Primary hippocampal cultures
Hippocampi were isolated from P0 Sprague-Dawley rat pups and the dentate gyri were removed. The tissue was treated in papain (20 Units/ml, Worthington Biochemical Corp, Lakewood, NJ) for 45 minutes. Papain was removed and replaced with trypsin inhibitor (2.5 mg/ml, Sigma-Aldrich, St. Louis, MO). Cells were dissociated by mechanical trituration with a pasteur pipette, and plated at a density of ~ 75,000 cells per 12mm well on poly-L-lysine coated coverslips (0.2mg/ml for >1 hour, Sigma-Aldrich). Cultures were incubated in MEM (GIBCO, Invitrogen Corp, Grand Island, NY) with 5% fetal bovine serum (GIBCO) for 24–36 hours and subsequently maintained in Neurobasal media (GIBCO) supplemented with 1X B27 (GIBCO) and Glutamax (GIBCO). Half of the media was exchanged on a weekly basis. Experiments were performed on cultures from 14 to 21 DIV.
Antibodies and reagents
Antibodies (Abs) used in the study included: GluA2 (for IP, ICC mouse monoclonal, Chemicon International, Inc, Temecula, CA; for WB rabbit polyclonal, Chemicon or rabbit polyclonal, Proteintech Group), GRIP1/2 (mouse monoclonal, BD Transduction Laboratories, San Jose, CA), synapsin 1 (rabbit polyclonal: Chemicon). Cy3, FITC and HRP-conjugated Abs raised in donkey were obtained from Jackson ImmunoResearch Laboratories, Inc (West Grove, PA). All agonists and antagonists were purchased from Tocris Cookson (Ellisville, MO). NSF-SNAP inhibition peptides (NSF-TAT) were synthesized by AnaSpec Corp, (San Jose, CA). All drugs were diluted in ddH20 except CNQX (DMSO).
Cell Treatments
Hippocampal cells were treated with NMDA (20μM), with CNQX (10μM), MPEP (1μM), and LY367385 (1μM) for 2 min at 37°C as specified per experiment. MGluRs were activated using 20μM DHPG with APV (50μM) for 5min at 37°C. We also used a cell permeable TAT-conjugated peptide (Marsden, Beattie et al. 2007)designed to inhibit NSF-mediated delivery of AMPARs by blocking NSF-SNAP association. The NSF peptide (NSF-TAT) mimics the SNAP binding site for NSF (Lledo, Zhang et al. 1998).
Immunocytochemical methods
Assays for constitutive and agonist mediated changes in surface AMPARs
To examine surface GluA2 after agonist application, live hippocampal neurons were incubated in growth media with an anti-GluA2 Ab (1:100) recognizing an N-terminal epitope for 30 min (37°C). After the 30 minute incubation, antibodies were washed off and agonist was applied as described above. Fifteen minutes after agonist application neurons were fixed for 10 min in 4% paraformaldehyde (PFA) and blocked with Tris-buffered saline (TBS) containing 4% bovine serum albumin (BSA) without permeabilization. A mouse secondary Ab conjugated with Cy3 was used to label surface GluA2-containing receptors. The effects of the NSF-TAT peptide on GluA2 surface expression after agonist treatments was measured by incubating neurons with the control or NSF-TAT peptide 15 minutes prior to and during surface AB labeling.
To measure constitutive AMPAR endocytosis, surface GluA2 receptors were labeled as above. After antibody washout, coverslips were incubated at 37°C for 0, 15, or 30 minutes) before fixation, block and labeling of remaining surface GluA2s with secondary antibody. To examine the effects of the NSF-TAT peptide on constitutive GluA2 endocytosis, control-TAT or NSF-TAT peptides (100 μM) were added into media 30 minutes prior to antibody incubation (as described above). For analysis of the effects of the agonist induced LTD on AMPAR cycling, live labeling of GluA2 surface receptors was completed before agonist addition. Fifteen minutes after agonist application slips were fixed after another 0, 20, 40 or 60 minutes.
To examine synaptic colocalization of surface GluA2 after LTD stimulus, surface GluA2 receptors were labeled as above followed by permeabilization with Tris-buffered saline (TBS) containing 4% bovine serum albumin (BSA) and 0.1% Triton-X 100. Cells were then incubated with anti- synapsin1 (1:1000) followed by a rabbit secondary Ab conjugated with FITC to label synapses.
Assays for internalized surface AMPARs
Internalized AMPARs were detected by live labeling of GluA2 surface receptors as above. Fifteen minutes after the agonist application, Abs bound to non-internalized GluA2 were stripped with acetic acid solution according to protocol Beattie et al 2000. Tris-buffered saline (TBS) containing 4% bovine serum albumin (BSA) and 0.1% Triton-X 100 was used to permeabilize neurons. A mouse secondary Ab conjugated with Cy3 was used to label internalized surface GluA2-containing receptors.
Image acquisition and analysis
Neurons were imaged using a Hamamatsu Orca ER camera mounted on an inverted Nikon fluorescent microscope with a 60X Plan Apo lens. Exposures were adjusted to ensure signals throughout the neuron fell within the linear range of the camera. Images were analyzed using Metamorph software (Universal Imaging, Inc, Downingtown, PA). Images were background-subtracted and thresholded to include signals more than 2-fold greater than the diffuse labeling in dendritic shafts to highlight punctate labeling. Integrated signal intensity values of fluorescence were determined for dendrites, normalized to area, and were graphed as a percent change from labeling in untreated controls. In one series of control experiments thresholds were lowered to highlight all surface GluA2 labeling as described in the Results.
To measure the intensity of synaptic GluA2 puncta, regions of interest were created around synapses puncta and transferred to background-subtracted, thresholded GluA2 images. Integrated labeling intensity within the synaptic regions was normalized to the total synaptic area analyzed.
The colocalization of GluA2 puncta with synapsin was analyzed in overlayed images by an investigator blinded to the treatment conditions. Positive colocalization was scored for discrete puncta of similar size with ~50% or more overlap.
siRNA knockdown of GRIP
Hippocampal cultures, 11–13 DIV, were transfected with negative control siRNA or siRNAs against GRIP1 and 2 using lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to a procedure specified by the manufacturer. Two sets of pre-designed siRNA sequences targeted to each GRIP protein family coding sequence (GRIP1 exons 5 and 21; and GRIP2 exons 7 and 25) were purchased from Ambion, Inc (Austin, TX). Following incubation for 2 hrs, the cells were washed in conditioned media and kept at 37°C. Neurons were treated with drugs as specified and immunocytochemically processed for labeling of GRIP or surface GluA2 after 3 days.
Biochemical Methods
Hippocampal slices and drugtreatments
Hippocampal slices (400 μm) were prepared from 17–21 day old Sprague Dawley rats. Slices were kept in artificial cerebrospinal fluid [ACSF: (in mM) 119 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2] saturated with 95% O2/5% CO2. After 1 hour equilibration to 37°C, slices were incubated in ACSF alone (control), NMDA (20μM with 10μM CNQX, 1μM MPEP, 1μM LY367385 in ACSF) for 3min or DHPG (20μM with 10μM CNQX, 50μM APVin ACSF) for 15min.
Surface protein biotinylation
Following 1 hour recovery at 37°C, acute hippocampal slices (300μm) were incubated in ACSF with 2mg/ml EZ-link Sulfo NHS-SS biotin (Pierce) for 30 min. Unbound biotin was washed out and slices were incubated in ACSF alone (control), NMDA (20μM for 3min, DHPG (20μM for 5min) or both at the same time. Drugs were washed out and after 20 minutes slices were transferred to 50 mM DTT on ACSF for 2 hours at 4°C to cleave remaining surface biotin. At 4°C, slices were washed in PBS. Protein extracts were prepared by homogenization in HEPES based lysis buffer (20mM HEPES, 150mM NaCl, 5mM EDTA, 0.1% SDS, 1% Tx-100). Lysates were spun at 6000 rpm, and the supernatant was retained and assayed for protein concentration. 700 μg of protein extract were incubated with 100ul Streptavidin Utra-Link Resin (Pierce) for 2 hrs at 4°C. The protein-bound beads were washed in lysis buffer and pelleted by centrifugation for 2 min at 2000 rpm. The beads were resuspended in SDS sample buffer and the immune-complexes were eluted by boiling. Total and immunoprecipitated proteins were separated by electrophoresis on 10% SDS-PAGE gels and were transferred onto nitrocellulose membranes and probed with anti- GluA2 Ab (1:2,000, Proteintech Group).
Coimmunoprecipitation with surface AMPARs
Hippocampal slices were treated with ACSF, NMDA or DHPG to induce AMPAR endocytosis followed by biotinylation of remaining surface proteins for 1 hr. at 4°C using non-cleavable EZ-Link Sulfo-NHS-LC Biotin (Pierce/Thermo Fisher Scientific, Inc.; Rockford, IL). Protein extracts were prepared by homogenization in Tris-HCl based lysis buffer (140mM NaCl, 50mM Tris-HCl, 0.1%Triton X-100, pH 7.6), and subject to centrifugation at 6000 rpm. Using immunoprecipitation with an anti-GRIP antibody, GRIP bound proteins were isolated with Protein G agarose beads. The supernatant was used as the non-GRIP bound fraction. The GRIP-bound proteins were eluted from beads in low pH (0.2M Glycine/HCl buffer pH 2.5) followed by neutralization using 1M Tris/HCl buffer (pH 9). Samples of these fractions were run on SDS-PAGE gels and probed on Western blot with anti GRIP Abs to test the integrity of these fractions. Remaining proteins in both the GRIP-bound and non-GRIP bound fractions of lysates were further incubated with streptavidin beads in order to isolate surface proteins thereby isolating GRIP-bound surface proteins and non-GRIP-bound surface proteins. Both fractions were run on SDS-PAGE gel, transferred to nitrocellulose and probed with an anti-GluA2 antibody.
Western Blot Analysis
In membranes probed with the appropriate HRP-conjugated secondary antibodies, the ECL Western Blotting Detection System (GE Biosciences Corp.; Piscataway, NJ) was used to visualize the bound antigens. The chemiluminescent signal was captured using Kodak BioMax light film. The films were imaged using Epson Perfection 1240U scanner and the intensity of the bands was quantified with Metamorph software. Protein signals in the bound fractions were normalized to total protein signals or to immunoprecipitated GluA2 as specified in the text and presented as a percent change from control.
Electrophysiology
Whole cell patch recordings were made from 2–3 week old cultured hippocampal neurons. Miniature EPSCs were recorded with a Multiclamp Amplifier (Molecular Devices, Sunnyvale, CA) using low-resistance electrodes (3–5 MΩ). The pipette solution contained: (in mM) 125 K gluconate, 10 NaCl, 10 HEPES, 10 EGTA, 5 Sucrose, 4 MgATP, 0.3 NaGTP pH 7.2. The extracellular solution contained 120 NaCl, 26 NaHCO3, 10 Glucose, 2.5 KCl, 1 NaH2PO4, 20 Glucose, 2.5 CaCl2, 1.3 MgSO4 and adjusted to pH 7.4. Extracellular solution was infused with 95% O2/5% CO2 and contained 100 μM lidocaine. The stability of series and input resistances were confirmed throughout the experiment using Igor Pro Software (Wavemetrics, Lake Oswego, OR). mEPSCs were collected at −70 mV during a 10 minute baseline followed by a 3 min application of DHPG or 2 minutes of NMDA while the cell was held in current clamp. Ten minutes following agonist washout, a further 10 minutes of mEPSCs was acquired. mEPSCs were analyzed using the Mini Analysis Program (Synaptosoft, Decatur, GA). For cumulative distribution plots inter event intervals and amplitudes from 400 mEPSCs from each experiment were normalized to the median value of the baseline events in each experiment, pooled and plotted. In one series of experiments an unmodified NSF-disrupting peptide as described above was included in the recording pipette (10 mM). Baseline mEPSCs were recorded beginning 10 minutes after break-in.
Statistical Analysis
For immunology experiments data and statics are performed on n values which refer to each experiment performed using and independent culture in which 7 to 14 cells were imaged. Data was analyzed using two tailed Student’s t-tests. One-way ANOVA analysis was performed where indicated. For electrophysiological studies n refers to the number of recorded cells. Cumulative distribution plots were compared by Kolmogorov-Smirnov Tests. All error bars represent the standard error of the mean (SEM).
Acknowledgments
This work was supported by NIH/NINDS NS 049661. KGS was supported by NIH Training Grant5T32NS007439. GD was supported by an Albert Einstein College of Medicine Neuroscience Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, et al. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3(12):1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, et al. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Borgdorff AJ, Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417(6889):649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, et al. Role of AMPA receptor endocytosis in synaptic plasticity. Nature reviews Neuroscience. 2001;2(5):315–324. doi: 10.1038/35072500. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, et al. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96(24):14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Bhattacharyya S, et al. Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long-term depression. J Neurosci. 2010;30(49):16437–16452. doi: 10.1523/JNEUROSCI.4478-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, et al. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28(3):873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- DeSouza S, Fu J, et al. Differential palmitoylation directs the AMPA receptor-binding protein ABP to spines or to intracellular clusters. J Neurosci. 2002;22(9):3493–3503. doi: 10.1523/JNEUROSCI.22-09-03493.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, O’Brien RJ, et al. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386(6622):279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhang P, et al. Characterization of the glutamate receptor-interacting proteins GRIP1 and GRIP2. J Neurosci. 1999;19(16):6930–6941. doi: 10.1523/JNEUROSCI.19-16-06930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, et al. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24(20):4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24(28):6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Sustained activation of metabotropic glutamate receptor 5 and protein tyrosine phosphatases mediate the expression of (S)-3,5-dihydroxyphenylglycine-induced long-term depression in the hippocampal CA1 region. J Neurochem. 2006;96(1):179–194. doi: 10.1111/j.1471-4159.2005.03527.x. [DOI] [PubMed] [Google Scholar]
- Huang CC, You JL, et al. Rap1-induced p38 mitogen-activated protein kinase activation facilitates AMPA receptor trafficking via the GDI.Rab5 complex. Potential role in (S)-3,5-dihydroxyphenylglycene-induced long term depression. J Biol Chem. 2004;279(13):12286–12292. doi: 10.1074/jbc.M312868200. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, et al. Chemical induction of mGluR5- and protein synthesis--dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86(1):321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, et al. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci U S A. 2001;98(20):11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, et al. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, et al. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279(5349):399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- Luscher C, Xia H, et al. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24(3):649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Luthi A, Chittajallu R, et al. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF-GluR2 interaction. Neuron. 1999;24(2):389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity and AMPA receptor trafficking. Ann N Y Acad Sci. 2003;1003:1–11. doi: 10.1196/annals.1300.001. [DOI] [PubMed] [Google Scholar]
- Man HY, Lin JW, et al. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25(3):649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Man HY, Lin JW, et al. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25(3):649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Mao L, Takamiya K, et al. GRIP1 and 2 regulate activity-dependent AMPA receptor recycling via exocyst complex interactions. Proc Natl Acad Sci U S A. 2010;107(44):19038–19043. doi: 10.1073/pnas.1013494107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden KC, Beattie JB, et al. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. J Neurosci. 2007;27(52):14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Bashir ZI. Long-term depression: multiple forms and implications for brain function. Trends in Neurosciences. 2007;30(4):176–184. doi: 10.1016/j.tins.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Launey T, et al. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000;19(12):2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, et al. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci. 2006;26(9):2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, et al. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, et al. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Nicholls RE, Alarcon JM, et al. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58(1):104–117. doi: 10.1016/j.neuron.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25(11):2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, et al. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18(6):969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Osten P, Khatri L, et al. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27(2):313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Park S, Park JM, et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59(1):70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annual review of cell and developmental biology. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, et al. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4(11):1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Sossa KG, Court BL, et al. NMDA receptors mediate calcium-dependent, bidirectional changes in dendritic PICK1 clustering. Mol Cell Neurosci. 2006;31(3):574–585. doi: 10.1016/j.mcn.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Osten P, et al. Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron. 1998;21(3):581–591. doi: 10.1016/s0896-6273(00)80568-1. [DOI] [PubMed] [Google Scholar]
- Takamiya K, Mao L, et al. The glutamate receptor-interacting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J Neurosci. 2008;28(22):5752–5755. doi: 10.1523/JNEUROSCI.0654-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardin C, Cognet L, et al. Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J. 2003;22(18):4656–4665. doi: 10.1093/emboj/cdg463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, et al. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59(1):84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, et al. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16(6):1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Kim E, et al. Biochemical and immunocytochemical characterization of GRIP, a putative AMPA receptor anchoring protein, in rat brain. Neuropharmacology. 1998;37(10–11):1335–1344. doi: 10.1016/s0028-3908(98)00120-8. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Zhou Q, et al. Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology. 2001;41(6):664–671. doi: 10.1016/s0028-3908(01)00134-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wang Y, et al. The AAA+ ATPase Thorase regulates AMPA receptor-dependent synaptic plasticity and behavior. Cell. 2011;145(2):284–299. doi: 10.1016/j.cell.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]