

Abstract
Persistent viral infections can lead to disease such as myocarditis. Theiler's murine encephalomyelitis virus (TMEV) infects macrophages of SJL/J (H-2s) mice establishing persistent infections leading to demyelinating disease. In contrast macrophages from B10.S (H-2s) mice clear TMEV. Activation of the transcription factor IRF3 induces IFNβ, ISG56, and apoptosis for viral clearance, but also inflammatory cytokines, such as IL-23 and IL6, which contribute to disease. Here we identify polymorphisms in the IRF3 of SJL/J versus B10.S mice that are located in DNA binding, nuclear localization, and autoinhibitory domains. SJL-IRF3 expression in RAW264.7 macrophage cells with or without TMEV infection decreased IL-23p19 promoter activity compared with B10S-IRF3. In contrast SJL-IRF3 increased IL-6, ISG56 and IFNβ in response to TMEV. B10S-IRF3 expression augmented apoptotic caspase activation and decreased viral RNA in TMEV-infected macrophages while SJL-IRF3 increased viral replication with less Caspase activation. Therefore IRF3 polymorphisms contribute to viral persistence and altered cytokine expression.
Keywords: IRF-3, IFNβ, TMEV, ISG56, IL-6, Apoptosis, Macrophages, SJL/J, B10.S
Introduction
Humans become infected by viruses throughout life. After the acute stage of each infection, viruses are controlled by the innate and adaptive immune systems either by eliminating the viruses and virus infected cells entirely or by tolerating a persistent lifelong infection (Virgin, Wherry, and Ahmed, 2009). The bases for persistent viral infections are unclear, but virus interference with host cytokine production and apoptosis is thought to play a role. Macrophages are essential to anti-viral immunity because they destroy phagocytized viruses and produce cytokines that influence innate and adaptive immune responses. Cytokine production is induced when viral nucleic acids in the late endosome bind to pattern recognition receptors (PRRs), activate cell signaling pathways and transcription factors for cytokine responses. For instance, Toll-like receptor (TLR)-3 recognition of viral dsRNA (Diebold et al., 2004; Heil et al., 2003; Latz et al., 2004) leads to activation of the IRF3 transcription factor (Takeuchi, Hemmi, and Akira, 2004; Horng et al., 2002). However, macrophages are sometimes infected by viruses. Cytokine production can occur if macrophages are infected with viruses when cytoplasmic helicases recognize viral RNA. Retinoic Acid Inducible gene-I (RIG-I) recognizes uncapped 5′-triphosphorylated-RNA that is unique to viruses (Hornung et al., 2006; Pichlmair et al., 2006) and Melanoma Differentiation-associated gene (MDA)-5 recognizes RNA from picornaviruses that are triphosphorylated and capped with viral protein (VPg) (Kato et al., 2006;Yoneyama et al., 2005b). Activation of either RIG-I and MDA5 pathways via mitochondrial (Kawai et al., 2005; Seth et al., 2005) and peroxisomal (Dixit et al., 2010) MAVS (also known as IPS-1, CARDIF, VISA) also leads to activation of IRF3. As a result of IRF3 activation, IFNβ, IL-6, and interferon stimulated genes (ISG) such as ISG56 are expressed. We also found that IRF3 represses IL-12 p35 (Dahlberg, Auble, and Petro, 2006) and induces IL-23 p19 expression from macrophages challenged with Theiler's Murine Encephalomyelitis Virus (TMEV). Therefore, activation of IRF3 is a common feature of macrophage anti-viral responses following both phagocytosis of and infection by viruses in order to achieve the correct cytokine and anti-viral factors for innate and adaptive immunity.
In addition to its role in gene expression, IRF3 is also involved in virus induced apoptosis in cells infected by viruses (Heylbroeck et al., 2000). IRF3 activation induces proapoptotic caspase-3, 8, 9 activities. Induction of apoptosis is dependent on IRF3 interaction with Bax at aa366-427, the apoptosis activation domain (AAD) of IRF3 (Chattopadhyay et al., 2010). Following interaction of IRF3-AAD with Bax, the Bax/IRF3 dimer translocates to mitochondria causing release of cytochrome C, activation of the apoptosome and caspase 9, followed by activation of caspase 3. It is thought that induction of apoptosis in virus infected cells limits the viral infection while inhibition of apoptosis plays a role in viral persistence (Chattopadhyay et al., 2011b).
IRF3 is constitutively expressed in macrophages, is autoinhibited by two unphosphorylated inhibitory domains (ID), and is activated via hyperphosphorylation of the IDs. Activated TBK1/IKKε phosphorylates carboxy terminus serine/threonines of IRF3 positioned between aa385-405 at one of the inhibitory domains (ID1) (Fig. 1A). In addition, activated JNK-MAPK phosphorylates serine-173 of IRF3, which is near the other ID (ID2) and nuclear export a signal (NES) domain (Yoneyama et al., 1998). These phosphorylations rearrange the IDs and permit dimerization with IRFs at the IRF association domain (IAD), nuclear entry, interaction with the CBP/p300 transcription factor (Weaver, Kumar, and Reich, 1998), and binding to promoter elements via the N-terminal DNA binding domain (DBD). Promoters responsive to IRF3 after viral infection of cells include the IFNβ (Panne, 2008), IL-6 (Sweeney, Kimbler, and Firestein, 2010) and ISG56 (Grandvaux et al., 2002) genes. Therefore, activation of IRF3 is critical to the immediate/early response of macrophages to virus infection through expression of cytokines and ISGs, as well as induction of apoptosis. Because the human population has several polymorphisms at the IRF3 locus (rs79173361, rs2230667, rs968457, rs7251) that change single amino acids near the DBD and ID1 it is imperative to understand how polymorphisms at these IRF3 domains change the responses of macrophages to viruses.
Fig. 1.

Comparison of the predicted amino acid sequences of IRF3 from B10.S and IRF3 from SJL. A. Domains of the 419 aa IRF3 include the DNA binding domain (DBD), Nuclear Localization Signal (NLS), Autoinhibitory Domain (ID) 1 and 2, Nuclear Export Signal (NES), IRF association domain (IAD), Apoptosis Activation Domain (AAD), Signal Response Domain (SRD) and the sites phosphorylated by JNK and TBK/IKKε. The 33R(B10.S)>G(SJL/J), 147I>V, and the 169Q>R polymorphisms are noted. B. Alignment of SJL/J IRF3 amino acid sequence with B10.S IRF3 amino acid sequence. Following extraction from agarose and cloning into pTARGET expression vector, the inserted cDNA was sequenced and the predicted amino acid sequence was generated. The DNA binding domain (
 ), the autoinhibitory domain 1 (
), the autoinhibitory domain 1 (
 ), and the signal response/autoinhibitory domain 2 (
), and the signal response/autoinhibitory domain 2 (
 ) are noted.
) are noted.
Theiler's murine encephalomyelitis virus (TMEV) persistently infects and causes Multiple Sclerosis-like neuroinflammatory disease in SJL/J mice (H-2s) but not B10.S mice (H-2s) (Aubagnac, Brahic, and Bureau, 2002; Monteyne et al., 1999; Vigneau et al., 2003). Thus comparison of TMEV induced responses in SJL mice with those of B10.S mice identifies non-MHC genes that are involved in persitent viral infections that lead to disease. One of the differences between these two strains is TMEV's chronic infection of macrophages that infiltrate the white matter of the CNS in SJL/J but not B10.S mice. We have found that the cellular and electrophoretic activity of IRF3 from SJL/J macrophages is distinctly different than that of IRF3 in B10.S macrophages (Dahlberg, Auble, and Petro, 2006). This is significant because viral replication in infected macrophages is higher, survival of infected macrophages is greater, macrophage expression of IL-12 is lower, and IFNβ expression is higher during innate anti-TMEV immune responses of SJL/J macrophages compared with B10.S macrophages (Petro, 2005a). We have previously seen that the IL-23 p19 promoter in RAW264.7 cells is activated following challenge with TMEV (Al-Salleeh and Petro, 2007). These data suggest that IRF3 polymorphisms could be the bases for the different phenotypes of SJL/J and B10.S macrophage responses to TMEV. The present study extends the analysis of these differences in IRF3 by comparing the amino acid sequences, the TMEV-induced transcriptional activities, and the TMEV-induced apoptotic activities of IRF3 from SJL/J and B10.S mice. IRF3 from SJL/J mice is different than that from B10.S mice in that there are three single nucleotide polymorphisms that predict three amino acid differences in the protein. RAW264.7 macrophage cells overexpressing IRF3 from SJL/J mice expressed more IL-6, ISG56, and IFNβ after TMEV challenge compared with RAW cells expressing B10.S IRF3. Therefore, TMEV RNA was higher in RAW cells expressing SJL/J IRF3 following challenge with TMEV compared to cells expressing B10.S IRF-3. In contrast, RAW cells with B10.S IRF3 exhibited significantly greater active Caspase-3/7 compared with cells expressing SJL/J IRF3, suggesting greater virus-induced apoptosis. Therefore, IRF3 is a significant factor in the differential response of SJL/J and B10.S mice to infection with TMEV.
Results
IRF3 from SJL/J mice has three amino acid polymorphisms compared with IRF3 from B10.S mice
IRF3 from SJL/J mice exhibits atypical activity in macrophages compared with IRF3 from B10.S mice (Dahlberg, Auble, and Petro, 2006). Because SJL/J and B10.S mice have distinct genealogical origins (Beck et al., 2000) polymorphisms are likely to exist between the two strains. To determine if differences in IRF3 exist between SJL/J and B10.S mice, IRF3 cDNA from macrophages of each strain was amplified by PCR using 5′ sense and 3′ anti-sense primers corresponding to the published mouse IRF3 coding sequence (NM_016849). The approximately 1200 base pair PCR products from each cDNA were inserted into the pTARGET expression vector to yield pSJLIRF3 and pB10S-IRF3. The insert sequence of pB10S-IRF3 was identical to the published mouse IRF3 coding sequence (NM_016849) (Fig. 1). However, the inserted pSJL-IRF3 sequence contains a G rather than an A at nt position 97 that predicts an arginine(R)>glycine (G) at aa 33, a G rather than an A at nt position 439 that predicts an isoleucine (I)>valine (V) change at aa 147, and a G rather than an A at nt position 503 that predicts a glutamine (Q)>arginine (R) change at aa 169 (Fig. 1). The R33G is located within the DBD of IRF3, the I47V is near the NLS, and the Q169R is in the ID1 autoinhibitory domain domain, near the CBP/p300 interacting domain (Weaver, Kumar, and Reich, 1998), nuclear export signal (NES), and the serine-173 that is phosphorylated by JNK-MAP-kinases following viral infection (Yoneyama et al., 1998). Therefore the SJL/J IRF3 has three SNP polymorphisms that could influence its activity.
IRF3 is constitutively expressed in an inactive form in macrophages (Servant, Grandvaux, and Hiscott, 2002). After viral challenge of macrophages IRF3 becomes active after post-translational hyperphosphorylation in a sequence of reactions at its carboxy terminal signal response and middle inhibitory domains. To initially evaluate SJL versus B10S IRF3, SJL-IRF3 pTARGET and B10S-IRF3 pTARGET expression vectors were transfected into RAW264.7 cells, a macrophage cell line, which were then left unchallenged or challenged with TMEV. After 9 h, cell lysates were western immunoblotted for IRF3 and tubulin production. Unchallenged RAW cells expressing SJL-IRF3 and B10S-IRF3 produced equivalent levels of IRF3 protein (Fig. 2A). This was confirmed with quantitative real-time PCR (data not shown). TMEV-challenged RAW cells expressing B10S-IRF3 exhibited a small increase in hyperphosphorylated IRF3 after 9 h. In contrast, TMEV-challenged RAW cells with SJL-IRF3 exhibited a much larger increase in hyperphosphorylated IRF3, as evidenced by its mobility shift. Therefore, IRF3 in RAW cells with SJL-IRF3 exhibits enhanced post-translational phosphorylation following TMEV challenge.
Fig. 2.
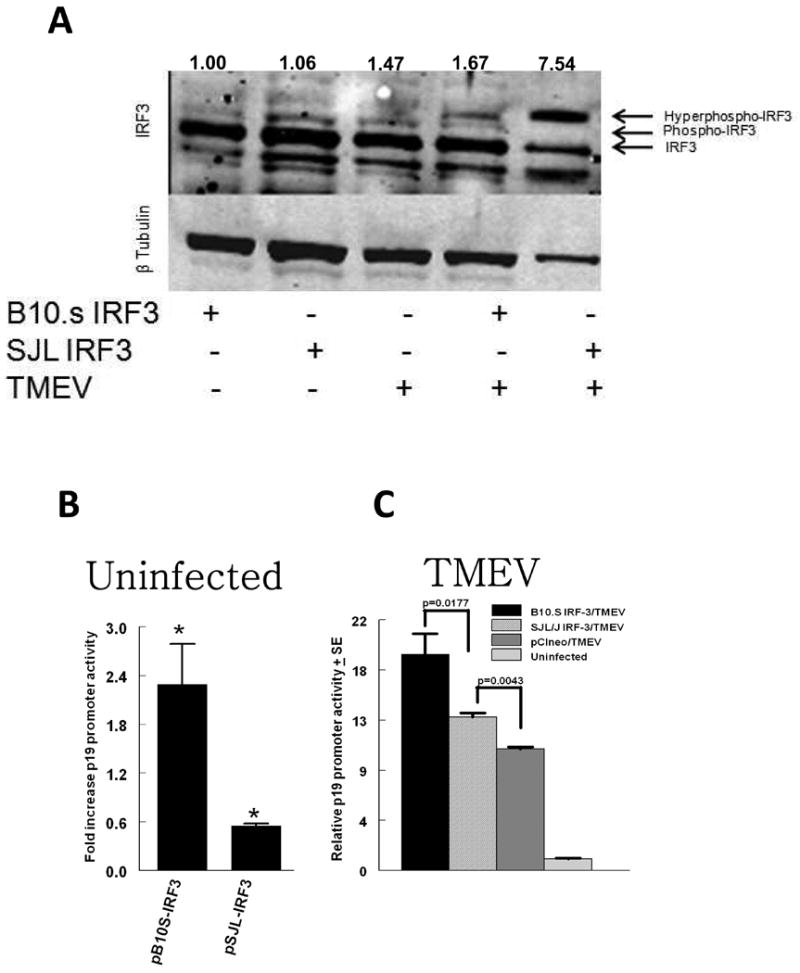
IRF3 protein and IL-23 p19 promoter activity in RAW cells expressing polymorphic IRF3. A. Western blot of IRF3 and tubulin in RAW cells following nucleofection with pCIneo (empty vector), SJLIRF3pTARGET or B10SIRF3pTARGET. Transfected RAW264.7 cells were left uninfected or infected with 3 × 105 TMEV PFU. Nine h after infection cells were lysed and 10 ug of protein immunoblotted. Numerical values are ratios of the sum of the densitometric values of three IRF3 bands divided by the densitometric value of the tubulin band. B, C. IL-23 p19 promoter activity in 3 × 105 RAW264.7 cells incubated overnight, were co-transfected with pCIneo (empty vector), SJLIRF3pTARGET or B10SIRF3pTARGET plus p19prompGL3 and pRL-SV40 vectors. Transfected RAW264.7 cells were left uninfected or infected with 3 × 105 TMEV PFU. Promoter activity in cells was measured as firefly luciferase-dependent luminescence expressed from p19prompGL3 normalized to Renilla luciferase-dependent luminescence expressed from pRL-SV40 in 24-h cell extracts divided by ratios obtained from uninfected cells. Data are means of 5 samples each of a representative experiment ± standard error; * indicates that the mean fold increase in promoter activity in uninfected cells with SJLIRF3pTARGET (pSJL-IRF3) or B10Sirf3pTARGET (pB10S-IRF3) is significantly different than promoter activity in uninfected cells with pCIneo control. Brackets indicate significant differences promoter activity in TMEV infected cells.
SJL/J IRF3 does not induce or enhance IL-23 p19 promoter activity as well as B10.S IRF3
Using a reporter vector that contains the mouse IL-23 p19 promoter (Petro, 2005b), we have shown that IL-23 p19 expression in RAW cells after TMEV challenge peaks at 24 h and IRF3 is involved in IL-23 p19 promoter activity due to a potential IRF3 binding element (Al-Salleeh and Petro, 2008). To compare transcriptional activity of SJL-IRF3 with B10S-IRF3, we transfected each expression vector along with the IL-23 p19 promoter-reporter vector (p19prompGL3) into RAW cells. Transfection efficiencies were approximately 50% based upon expression of GFP from parallel transfections with pmaxGFP, which constitutively expresses GFP. Transfection of pB10S-IRF3 increased p19 promoter activity significantly more than empty vector (Fig. 2B). In contrast pSJL-IRF3 significantly decreased p19 promoter activity compared with cells with empty vector. Transfection of either pSJL-IRF3 or pB10S-IRF3 significantly increased p19 promoter activity following challenge of RAW cells with TMEV (Fig. 2C). However, enhancement of p19 promoter activity due to challenge with TMEV was significantly greater in RAW cells with pB10S-IRF3 compared with pSJL-IRF3. Therefore transcriptional activity of SJL-IRF3 for the IL-23 p19 promoter is significantly different than B10S-IRF3.
SJL/J IRF3 augments IFNβ, ISG56, and IL-6 expression but also TMEV RNA replication during TMEV infection of RAW cells
In addition to IL-23 p19, we have seen that challenge of macrophages with TMEV induces expression of IFNβ (Petro, 2005b) and IFNβ expression was greater in macrophages from SJL/J mice infected with TMEV compared with B10.S macrophages (Dahlberg, Auble, and Petro, 2006). To determine if the dichotomy in IRF3 between SJL/J and B10.S mice could be responsible for these conflicting results, we transfected pSJL-IRF3, pB10S-IRF3, or control vector into RAW cells, challenged transfected cells with TMEV, and then determined IFNβ expression by qRT-PCR after 9h. Expression of IFNβ was significantly greater in TMEV-challenged RAW264.7 cells with pSJL-IRF3 compared with those with pB10S-IRF3 or control vector (Fig. 3A). Therefore, SJL-IRF3 promotes increased IFNβ expression in macrophages.
Fig. 3.
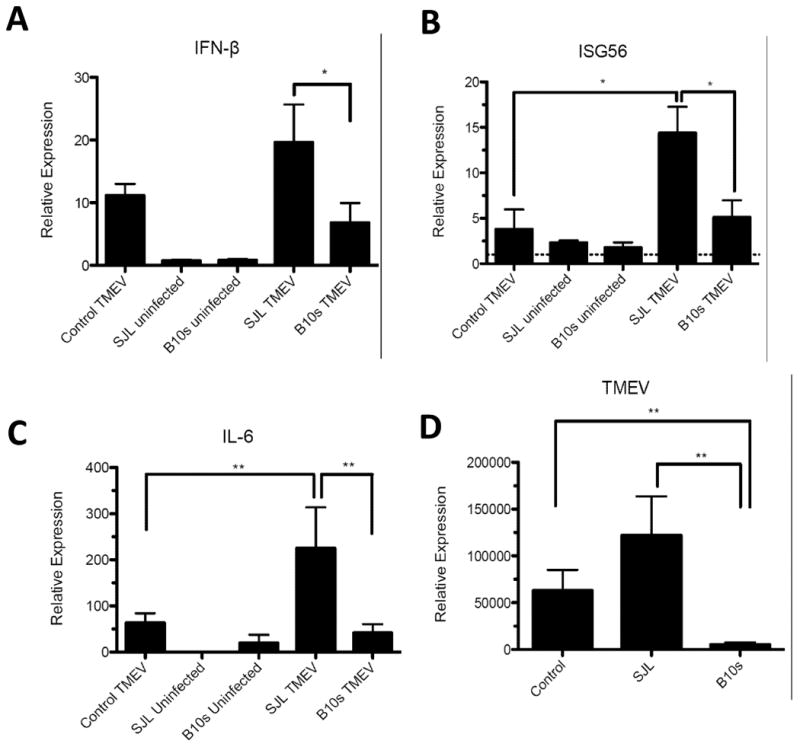
Effect of SJL-IRF3 and B10.S IRF3 on relative expression of IFNβ, IL-6, ISG56, and TMEV RNA after TMEV infection. Real-time PCR of IFNβ (A), ISG56 (B), IL-6 (C), and TMEV RNA (D) in uninfected or TMEV-infected RAW264.7 cells transfected with pCIneo (control), SJLIRF3pTARGET (SJL), B10SIRF3pTARGET (B10.S) as measured after 9 h. Levels of IFNβ, IL-6, ISG56, and TMEV RNA relative to GAPDH RNA were evaluated by real-time PCR. Individual means were evaluated with the Wilcoxon-Mann-Whitney rank-sum U test. Comparisons of means in which p values < 0.05 are bracketed. *P<.05, **P<.01. Data are an average + standard error of 3 replicates per group and two different experiments.
In addition to IRF3, several other transcription factors cooperate with IRF3 at the IFNβ enhancesome (Panne, Maniatis, and Harrison, 2007). In contrast, ISG56 expression in response to viral infection of cells takes place with IRF3 activation independent of other factors (Grandvaux et al., 2002). We compared ISG56 expression in RAW cells transfected with pSJL-IRF3, pB10S-IRF3, or control vector 9 h after challenge with TMEV. Similar to IFNβ, expression of ISG56 was significantly greater in TMEV-challenged RAW cells with SJL-IRF3 compared with those with pB10S-IRF3 or control vector (Fig. 3B).
Recently, it was shown that SJL/J mice infected with TMEV are susceptible to neuroinflammatory autoimmune disease due in part to high IL-6 expression by TMEV infected SJL/J APCs, which was not seen in TMEV-resistant C57Bl/6 mice (Hou, Kang, and Kim, 2009). Recent data suggests that IRF3 plays an important role in expression of IL-6 in inflammatory disease (Sweeney, Kimbler, and Firestein, 2010). To compare the influence of SJL-IRF3 and B10S-IRF3 on this cytokine, we quantified IL-6 expression in RAW cells transfected with pSJL-IRF3, pB10S-IRF3, or empty vector 9 h after challenge with TMEV. Expression of IL-6 was significantly greater in TMEV-challenged RAW cells transfected with pSJL-IRF3 compared with those with pB10S-IRF3 or empty vector (Fig. 3C). These data indicate that SJL/J-IRF3 could play a role in the enhanced susceptibility of SJL/J mice to TMEV-induced neuroinflammatory autoimmune disease.
TMEV replication is greater in SJL/J macrophages than in B10.S macrophages (Petro, 2005b). Similarly replication of TMEV RNA is significantly greater in TMEV-infected RAW cells with pSJL-IRF3 compared with cells with empty vector or pB10S-IRF3 (Fig. 3D). In addition, RAW cells with pB10S-IRF3 exhibited significantly less TMEV RNA than RAW cells with empty vector. Therefore, B10S-IRF3 leads to decreased while SJL-IRF3 leads to increased TMEV RNA production in macrophages.
RAW cells with SJL/J IRF3 have lower ISG56 and IL-6 in response to polyIC
In order to activate IRF3 for IFNβ and ISG56 expression macrophages that encounter RNA-viruses like TMEV use TLR3 to sense viral RNA during phagocytosis of virus (Al-Salleeh and Petro, 2007) but use cytoplasmic RIG-1 and MDA5 to sense viral RNA during infection with virus (Pichlmair et al., 2006). We next wanted to compare the effects of SJL-IRF3 with B10S-IRF3 on IL-6, IFNβ, and ISG56 expression during TLR3 pathway activation. RAW cells were transfected with pSJL-IRF3, pB10S-IRF3, or control vector. Twenty four h after transfection cells were stimulated with the TLR3 agonist, polyIC, which will not stimulate RIG-I/MDA5 pathways unless transfected into the cytoplasm (Diebold et al., 2003; Yoneyama et al., 2005a) or will not stimulate IFNβ expression in TLR3 knockdown RAW cells (Al-Salleeh and Petro, 2007). In contrast to the response to TMEV challenge, neither SJL-IRF3 nor B10S-IRF3 significantly influenced expression of IFNβ in response to polyIC at 9 h (Fig. 4A). However, pSJL-IRF3 significantly decreased IL-6 (Fig. 4B) and ISG56 (Fig. 4C) expression in response to polyIC compared with pB10S-IRF3 or control vector. These data suggest that the increased expression of IL-6, IFNβ, and ISG56 during the TMEV infection of macrophages with SJL-IRF3 is not dependent upon TMEV stimulation of the TLR3 pathway.
Fig. 4.
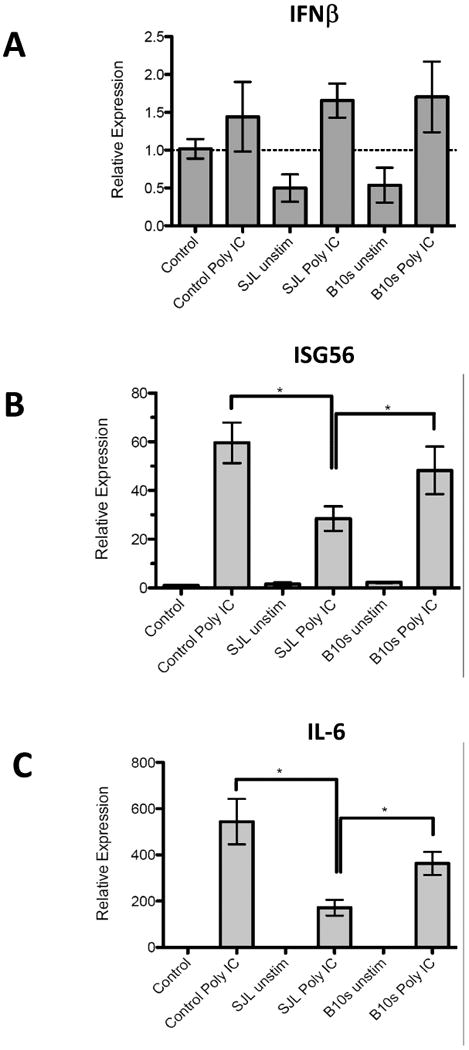
Effect of SJL-IRF3 and B10.S IRF3 on relative expression of IFNβ, ISG56, and IL-6 after polyIC stimulation. Real-time PCR of IFNβ (A), ISG56 (B), and IL-6 (C) in RAW264.7 cells transfected with pCIneo (control), SJLIRF3pTARGET (SJL), B10SIRF3pTARGET (B10.S) as measured after 9 h. Levels of IFNβ, IL-6, and ISG56 RNA relative to GAPDH RNA were evaluated by real-time PCR. Individual means were evaluated with the Student t test. Comparisons of means in which p values < 0.05 are bracketed. Data are an average +/- standard error of 3 replicates per group from a representative experiment.
TMEV-induced Caspase 3 activation and Bax levels are lower in RAW cells with SJL/J IRF3
In addition to its role in gene expression, virus-induced IRF3 activation induces proapoptotic caspase-3, 8, 9 activities (Heylbroeck et al., 2000) indicating a role for this protein in virus-induced apoptosis. We have seen that SJL/J macrophages infected with TMEV are more viable after infection compared with B10.S macrophages (Dahlberg, Auble, and Petro, 2006). To determine if the SJL-IRF3 polymorphisms hinder virus-induced caspase-3/7 activation in TMEV-infected macrophages, RAW264.7 cells were transfected with pSJL-IRF3, pB10S-IRF3, or control vector before challenge with TMEV. Caspase 3/7 activity in cells was quantified by measuring amino-luciferin release from a substrate containing Z-DEVD-Luciferin using a luciferase assay. Following TMEV infection Caspase 3/7 activity was greater in RAW cells expressing B10S-IRF3 at 16 (Fig. 5a) and 35 h (Fig. 5b) PI compared with uninfected RAW cells with B10S-IRF3 or infected RAW cells with SJL-IRF3. Therefore, SJL-IRF3 diminishes TMEV-induced Caspase activation compared with B10S-IRF3 due its polymorphisms. Bax is a proapoptotic 21 kDa cellular protein which can homo- or heterodimerize with BH3-domain containing proteins, including IRF3, to induce mitochondria-associated apoptosis (Chattopadhyay et al., 2010; Chattopadhyay et al., 2011a). To confirm, the impact of SJL-IRF3 on virus induced apoptosis, Bax protein was evaluated by immunoblot followed by densitometry. Immunoreactive Bax protein was detected at approximately 75 kDa, indicating its multimerization with other BH3-containing proteins. Following TMEV infection relative Bax levels increased more in RAW cells overexpressing B10S-IRF3 than in RAW cells overexpressing SJL-IRF3 or non-transfected RAW cells (Fig. 5c, 5d). Therefore TMEV-induced Bax-associated apoptosis is likely diminished in macrophages with polymorphic SJL-IRF3.
Fig. 5.
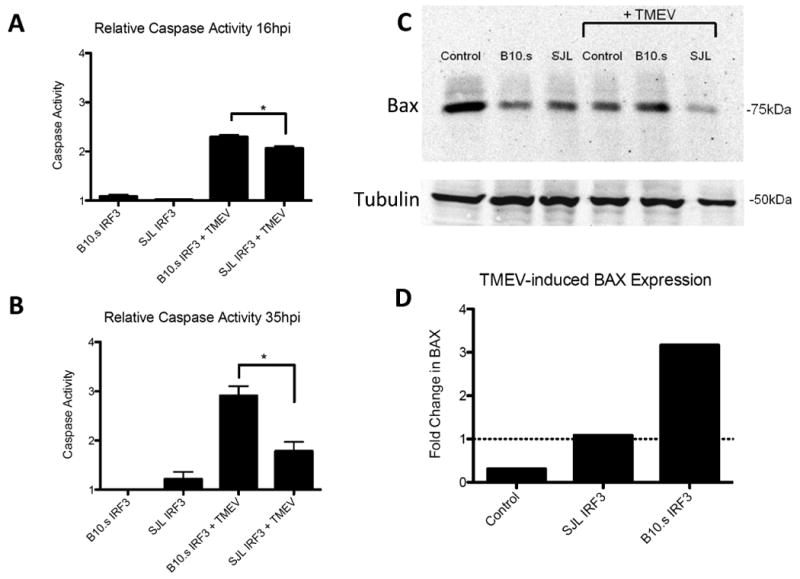
Effect of SJL-IRF3 and B10.S IRF3 on virus induced caspase activation and Bax protein. Caspase activation after 16 h (A) and 35 h (B) in uninfected or TMEV-infected RAW264.7 cells transfected with pCIneo (control), SJLIRF3pTARGET (SJL), B10SIRF3pTARGET (B10.S). Levels of caspase activation were evaluated by the release of luciferin from Z-DEVD-luciferin substrate and then measuring luminescence by adding UltraGlo™luciferase from Promega. Individual means of 5 samples each of a representative experiment were evaluated with the Student t test. Comparisons of means in which p values < 0.05 are bracketed. Data are an average ± standard error of 3 replicates per group from a representative experiment. (C) Western blots of Bax and beta-tubulin protein in uninfected or TMEV-infected RAW264.7 cells transfected with pCIneo (Control), SJLIRF3pTARGET (SJL), or B10SIRF3pTARGET (B10.S). (D) Densitometric ratio of Bax in 9 h TMEV-infected RAW cells with pCIneo (Control), SJLIRF3pTARGET (SJL-IRF3), or B10SIRF3pTARGET (B10.S-IRF3) with Bax uninfected RAW cells with pCIneo, SJLIRF3pTARGET, or B10SIRF3pTARGET, respectively.
Discussion
Chronic viral infection is a common human condition that oftentimes leads to disease in the infected host beyond that which is seen with the initial infection. Like humans, mice experience chronic viral infections that sometimes lead to disease. Strains of TMEV infect most feral mice at some point in their natural life, but it was noticed about 75 years ago that the SJL/J laboratory mouse strain that becomes infected with TMEV develops a chronic infection followed by a demyelinating disease (Theiler, 1937). Several studies set out to determine the basis for susceptibility to TMEV-induced demyelinating disease (Bihl, Brahic, and Bureau, 1999; Vigneau et al., 2003) by comparing susceptible SJL/J (H-2s) with B10.S (H-2s) mice, the latter being resistant to persistent TMEV infection. This approach permitted the discovery of non-MHC chromosome regions responsible for differential susceptibility to TMEV. These studies have revealed that the telomeric end of Chromosome 10 in mice in a region that is near the IFNγ and IL-22 genes predisposes SJL/J mice to greater susceptibility to chronic TMEV infection in macrophage lineage cells. However, no protein product of a gene was definitively identified from these studies for viral persistence in SJL/J macrophages. Our previous studies comparing SJL/J and B10.S macrophage responses to TMEV showed that the cellular localizations of IRF3 in macrophages and the electorphoretic mobilities of IRF3 differ between these two strains of mice (Dahlberg, Auble, and Petro, 2006). The present study confirms that SJL/J and B10.S IRF3 are different between these two mouse strains and these differences confer greater susceptibility of SJL/J macrophages to persistent infection with TMEV. We show here that the SJL-IRF3 gene has three single nucleotide polymorphisms that are predicted to lead to three amino acid differences compared with B10-IRF3. One of the polymorphisms R33G is located in the DNA binding domain of IRF3 near the amino acids W38 and L42, R43, and Q44 which are known to contact DNA elements of the IFNβ enhancesome (Panne, Maniatis, and Harrison, 2007). It remains to be seen if the R33G alters the ability of IRF3 to physically bind to promoter elements for either enhancement or repression of gene expression. However, in the present study, overexpression of SJL-IRF3, unlike B10S-IRF3, failed to induce IL-23 p19 promoter activity and exhibited significantly impaired ability to enhance IL-23 p19 promoter activity during TMEV infection of RAW cells. The IL-23 p19 promoter, which contains a putative IRF3 binding element, was activated by overexpression of B10S-IRF3, but not by SJL-IRF3, even without TMEV infection. So far the R33G polymorphism has not been found in other strains of mice. However, a human polymorphism in IRF3 in which glutamine at aa 48 is changed to glutamic acid is found in 2% of west African population samples tested (rs76863988) would be expected to influence the DBD.
These data suggest that SJL/J macrophages by virtue of their polymorphic IRF3 should exhibit depressed IL-23 p19 expression following TMEV challenge compared with B10.S macrophages. However, CD4+T cells from TMEV-susceptible SJL/J mice develop more Th17 compared to CD4+ T cells from TMEV-resistant C56Bl/6 mice following challenge with TMEV. Because of the involvement of IL-23 in Th17 development, this would imply that SJL/J macrophages might express higher levels of IL-23. We have indeed shown previously that SJL/J macrophages do express IL-23 p19 mRNA and produce IL-23 protein during TMEV infection (Al-Salleeh and Petro, 2008). While IL-23 has been linked to Th17, its role is to sustain the Th17 phenotype following Th17 differentiation in response to TGFβ and IL-6 while. Furthermore, Hou et al. (Hou, Kang, and Kim, 2009) have shown that enhanced Th17 by SJL/J mice during TMEV infection is due to increased IL-6 and not IL-23 expression. Furthermore we have shown that knockdown of IRF3 with RNAi approaches did not alter TMEV-induced IL-23 p19 (Al-Salleeh and Petro, 2008) and there was little impact on IL-23 p19 expression in IRF3 knockout mice (Johnson et al., 2011). Altogether these results suggest that in the presence of IRF3 IL-23 p19 expression increases but in its absence its expression is unaffected. This may be due to the fact that IL-23 p19 expression in response to TMEV, unlike IRF3, is totally dependent on other transcription factors such as ATF2, SMAD3, and NF-κB at the IL-23 p19 promoter (Al-Salleeh and Petro, 2008).
In contrast to IL-23 p19, expressions of IFNβ, ISG56, and IL-6, all of which are known to be dependent upon IRF3, were enhanced during TMEV infection of macrophages when SJL-IRF3 was overexpressed. Therefore, the transcriptional effect of the SJL/J polymorphic IRF3 is not the same for each IRF3 responsive gene. This may be due to the fact that IRF3 collaborates with other transcription factors such as IRF7, ATF2, NF-κB, and c-Jun at the IFNβ enhancesome, with ATF2, SMAD3, and NF-κB at the IL-23 p19 promoter (Al-Salleeh and Petro, 2008), but can induce promoter activity at the ISG56 gene independent of other transcription factors (Grandvaux et al., 2002).
Paradoxically we found here TMEV replication is increased in cells with higher IFNβ and ISG56 expression. These results are similar to our previous finding comparing TMEV-infected SJL/J macrophage with B10.S macrophages (Petro, 2005a). In that report TMEV replication and IFNβ expression was higher in SJL/J macrophages than in B10.S macrophages. IRF3-induced ISG56 was originally thought to have anti-viral activity. Its enhanced expression in RAW264.7 cells with SJL-IRF3 would be expected to decrease TMEV RNA replication. However, enhanced ISG56 expression in these cells was accompanied by increased TMEV RNA replication. More recently ISG56 was shown to also regulate anti-viral responses, inhibit cellular protein synthesis, and even enhance viral replication (Li et al., 2009; Stawowczyk et al., 2011). Therefore, augmentation of ISG56 by IRF3 may dampen or negate some of the other anti-viral effects initiated by IRF3. Moreover, Hou et al. (Hou, Kang, and Kim, 2009) showed that TMEV-induced pathogenesis in SJL/J mice is related to heightened IL-6 expression despite an elevated IFNβ response. We show here that the polymorphic IRF3 of SJL/J mice may be related to the TMEV-induced pathogenesis in SJL/J mice due to its ability to increase the expression of IL-6 and perhaps ISG56.
Another IRF3 polymorphism described here, 169Q>R, is located in a region that is part of the first ID, which with the second ID results in IRF3 latency (Takahasi et al., 2003). Hyperphosphorylation of serines and threonines in the second ID by TBK/IKKε and phosphorylation of serine-173 in the first ID by JNK-MAPK (Zhang et al., 2009) results in rearrangement of the IDs, IRF3 dimerization, and nuclear localization. It is possible that the proximity of 169Q>R to serine-173 affects the first ID resulting in rearranged IRF3 with a different electrophoretic mobility and constitutive nuclear localization previously reported (Dahlberg, Auble, and Petro, 2006). Earlier nuclear localization could also explain the heightened expression of IFNβ, ISG56, and IL-6 observed here during macrophage responses to TMEV with SJL-IRF3. It should be noted that this polymorphism in SJL/J mice has been confirmed (rs37133231), is also present in other strains such as NZW and BPL/1J but is absent in B10, C57Bl/6, and Balb/c mice. It remains to be seen if these other strains of mice with the same IRF3 polymorphism exhibit similar responses to viral infection.
The results presented herein suggest that the IRF3 polymorphisms are responsible for several phenotypes during TMEV infection in SJL/J macrophages that do not occur in B10.S macrophages. Our previous results showed that macrophages from SJL/J mice expressed more IFNβ before and following infection with TMEV than macrophages from B10.S mice. The present investigation shows that RAW cells overexpressing either SJL-IRF3 or B10S-IRF3 exhibit the same dichotomy in IFNβ phenotype following TMEV infection. Interestingly, in our previous studies (Petro, 2005a; Dahlberg, Auble, and Petro, 2006) enhanced IFNβ expression was not seen when SJL/J macrophages were stimulated with polyIC, which will stimulate IFNβ through TLR3 but will not stimulated IFNβ through RIG-I/MDA5 pathways unless transfected into the cytoplasm (Diebold et al., 2003; Yoneyama et al., 2005a). We have shown that RAW cells with knocked down TLR3 expression will not express IFNβ in response to polyIC (Al-Salleeh and Petro, 2007). Here, polyIC induced expression of IFNβ, ISG56, and IL-6 but RAW cells expressing SJL-IRF3 did not exhibit higher expression of these factors in response to polyIC compared with RAW cells with B10S-IRF3. The IFNβ response of macrophages challenged with TMEV is in part dependent upon TLR3 (So, Kang, and Kim, 2006; Al-Salleeh and Petro, 2007). TLR3 pathways are activated by viral RNA or polyIC in the late endosome in which downstream TRIF activation leads to activation of TBK1/IKKε and then IRF3. The results here suggest that even though polyIC stimulates IFNβ expression through the TLR3 pathway the enhancement of IFNβ expression due to polymorphic SJL-IRF3 are not dependent on the TLR3 pathway.
While IRF3 plays an important role in cytokine and ISG expression following virus infection of macrophages it also has an important role in induction of virus induced apoptosis (Sharif-Askari et al., 2007). Activated IRF3 associates with Bax and localizes to mitochondria, thus inducing cytochrome C release into the cytoplasm ultimately leading to caspase activation (Chattopadhyay et al., 2010). Hou et al. (Hou, Kang, and Kim, 2009) showed that SJL/J macrophages infected with TMEV exhibit decreased virus-induced Annexin V, which is expressed by cells during apoptosis (Hou, Kang, and Kim, 2009). We have seen that survival of SJL/J macrophages following TMEV infection is greater than that of TMEV-infected B10.S macrophages (Dahlberg, Auble, and Petro, 2006). These data raise the possibility that TMEV-induced apoptosis is diminished in SJL/J macrophages. We show herein for the first time that expression of SJL-IRF3 in RAW cells during TMEV infection led to significantly decreased caspase activation, another characteristic of apoptosis, compared with caspase activation in cells with B10S-IRF3. Similarly expression of the proapoptotic Bax protein was lower in cells expressing SJL-IRF3 compared with B10S-IRF3. Interestingly we found that Bax protein in RAW cells forms a 75 kDa hetero- or homo-multimer complex other proteins most likely containing the BH3 association domain. It remains to be seen if this is the native endogenous configuration of Bax multimers in RAW cells or is an artifact of western blot. Previous studies have shown that detergents in western blot lysing buffers stabilize or enhance formation of Bax multimers even under denaturing condition(Hsu and Youle, 1997; Hsu and Youle, 1998). Nevertheless, these data indicate that not only do the polymorphic IRF3 of SJL/J mice play a role in cytokine phenotype following TMEV infection but it also is deficient in inducing apoptosis in TMEV-infected macrophages. Since virus-induced apoptosis reduces viral replication and persistence (Peters, Chattopadhyay, and Sen, 2008; Chattopadhyay et al., 2011b), these data suggest that polymorphisms in SJL-IRF3 may decrease virus-induced apoptosis leading to increased virus replication and persistence.
It is noteworthy that chronic TMEV infection of SJL/J mice leads to demyelinating disease similar to human Multiple Sclerosis (MS). Several meta-analyses have identified one MS susceptibility locus at human chromosome region 19q13 (Bonetti et al., 2009; Haines et al., 2002). Interestingly, the human IRF3 gene is located at chromosome region 19q13. However, it will take additional investigations to determine if one or more of the human IRF3 polymorphisms (rs79173361, rs2230667, rs968457, rs7251) are basis for the MS susceptibility locus at 19q13. In summary, the results of this report clearly show that the IRF3 encoded in SJL/J mice contains three amino acid polymorphisms that alter TMEV-induced cytokine expression and virus-induced apoptosis in macrophages. It remains to be seen which of the polymorphisms are required for the effects upon cytokines and apoptosis.
Matierials and Methods
Experimental animals, cell line, virus
Female B10.S and SJL/J mice were obtained from Jackson Laboratories (Bar Harbor, Maine). RAW264.7 cells were originally obtained from the American Type Culture Collection (Rockville, MD). The DA strain of TMEV was obtained from Dr. Kristen Drescher, Department of Medical Microbiology and Immunology, Creighton University, Omaha, Nebraska. TMEV was grown in BHK-21 cells. The titer of stock cultures of TMEV was 1 × 106 PFU/ml.
IRF3 cDNA cloning
RNA was isolated from SJL/J and B10.S thioglycollate-elicited peritoneal macrophages, IRF3 cDNA was amplified by RT-PCR, cloned into the pTARGET expression vector, and sequenced. To analyze IL-23 p19 promoter activity, RAW264.7 cells were seeded at 3 × 105 per well of a 6-well plate in DMEM cell culture medium containing 10% fetal bovine serum and 50μg/ml gentamycin and transfected using lipofectamine with p19prompGL3 promoter reporter vector (Petro, 2005b) plus pRL renilla luciferase normalization reporter vector plus pCIneo (empty vector), SJLIRF3pTARGET or B10SIRF3pTARGET. After 24 h 106 PFU of TMEV (DA strain) were added. After an additional 24 h, luciferase was measure using the dual reporter Luciferase kit of Promega.
Measurement of lFNβ, IL-6, ISG56, and TMEV RNA
RAW264.7 cells were nucleofected with pCIneo(empty vector), SJLIRF3pTARGET or B10Sirf3pTARGET using an Amaxa nucleofector kit and then seeded at 3×105 cells per well of a 6 well plate. To evaluate transfection efficiencies GFP positive cells were evaluate microscopically following parallel nucleofections with pmaxGFP vector (Amaxa), which constitutively expresses GFP. After 48 h, cells were left untreated (control) or challenged with TMEV or stimulated with 50 μg/mL polyinosine-polycytidylic acid (polyIC) (InvivoGen) an agonist of TLR-3 but not RIG-I when polyIC is not transfected into the cytoplasm (Al-Salleeh and Petro, 2007; Diebold et al., 2003; Yoneyama et al., 2005a). RNA was collected at 9 h for qRT-PCR using the RNAeasy kit of Qiagen (Valencia, CA). The primer pairs of genes of interest were: IFNβ sense 5′ ATGAACAACA GGTGGATCCTCC 3′ and anti-sense 5′ AGGAGCTCCT GACATTTCCGAA 3′; IL-6 sense 5′ ATGAAGTTCCTCTCTGCAAGAGACT 3′ and antisense 5′ CACTAGGTTTGC CGAGTAGATCTC 3′, ISG56 sense 5′ CAGAAGCAC ACATTGAAGAAGC 3′ and antisense 5′ TGTAAGTAGCCAGAGGAAGGTG 3′; TMEV sense 5′ CTTCCCATTC TACTGCAATG 3′ and antisense 5′ GTGTTCCTGG TTTACAGTAG3′; and GAPDH sense 5′-TTGTCAGCAA TGCATCCTGCAC-3′ and antisense 5′-ACAGCTTTCCAGAGGGGC CATC-3′. Quantitative PCR reactions were run on an ABI Prism 7000 thermal cycler at 50 °C for 2 min, 95 °C for 10 min, 45 cycles of 95 °C for 15 s/60 °C for 30 s. Relative levels of mRNA for each factor were normalized to GAPDH determined by using the Ct value and the formula: 2 ΔΔCt.
Measurement of Apoptosis
RAW264.7 cells were nucleofected with pCIneo(empty vector), SJLIRF3pTARGET or B10Sirf3pTARGET using an Amaxa Nucleofector Kit V and then seeded at 6×104 cells per well of a 96 well plate. After 48 h, cells were left untreated (control) or challenged with TMEV. Activation of Caspase was measured after 16 and 35 h using the Promega Apo-Tox-Glow assay which measures intracellular caspase 3/7 activity by quantifying release of luciferin from a luciferin- (Z-DEVD)-tetrapeptide product and measuring luminescence activity after adding UltraGlo™ Luciferase.
PAGE and Western blot analysis of IRF-3 and Bax
Ten μg of protein from cell lysates of RAW cells expressing SJL-IRF3 or B10S-IRF3 with or without challenge with TMEV were added to a sample buffer containing 63 mM Tris-HCl, 10% glycerol, 2% SDS, 2.5% 2-ME, and 0.0025% bromophenol blue. Protein concentrations in lysates were determined using the BioRad (Hercules, CA) DC protein assay kit. Twenty μl of each sample containing 10 μg of protein was run on a 10% SDS, Tris-glycine-polyacrylamide gel and transferred to a nitrocellulose membrane as previously described (Dahlberg, Auble, and Petro, 2006). The membrane was treated with blocking buffer for 1 h at room temperature, followed by incubation in 1:1000 dilution of rabbit IgG anti-IRF-3 (Invitrogen), mouse anti-IgG anti-Bax (Invitrogen), or 1:500 mouse anti-tubulin E7 (Developmental Studies Hybridoma Bank, University of Iowa, Department of Biological Sciences, Iowa City, IA) and then 1:1000 dilution of IRDye® 800CW Goat Anti-Rabbit IgG (Rockland Immunochemicals, Inc., Gilbertsville, PA) or Alexa Fluor680-labeled anti-Mouse IgG (Rockland Immunochemicals). The membrane was washed three times and then scanned with a LICOR Odyssey® Infrared Imaging System and densitometry of individual bands was done with the LICOR imaging software.
Statistical Analysis
All data were analyzed by the Student t test or the Wilcoxon-Mann-Whitney rank-sum U test to determine the significance of differences between the sample means or medians, respectively. P values of less than 0.05 were considered to be significant.
Acknowledgments
This work was supported by funding from the University of Nebraska Medical Center College of Dentistry and University of Nebraska Lincoln, School of Biological Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Salleeh F, Petro TM. TLR3 and TLR7 are involved in expression of IL-23 subunits while TLR3 but not TLR7 is involved in expression of IFN-beta by Theiler's virus-infected RAW264.7 cells. Microbes Infect. 2007;9(11):1384–92. doi: 10.1016/j.micinf.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Al-Salleeh F, Petro TM. Promoter Analysis Reveals Critical Roles for SMAD-3 and ATF-2 in Expression of IL-23 p19 in Macrophages. J Immunol. 2008;181(7):4523–4533. doi: 10.4049/jimmunol.181.7.4523. [DOI] [PubMed] [Google Scholar]
- Aubagnac S, Brahic M, Bureau JF. Bone marrow chimeras reveal non-H-2 hematopoietic control of susceptibility to Theiler's virus persistent infection. J Virol. 2002;76(11):5807–12. doi: 10.1128/JVI.76.11.5807-5812.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, Fisher EM. Genealogies of mouse inbred strains. Nat Genet. 2000;24(1):23–5. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- Bihl F, Brahic M, Bureau JF. Two loci, Tmevp2 and Tmevp3, located on the telomeric region of chromosome 10, control the persistence of Theiler's virus in the central nervous system of mice. Genetics. 1999;152(1):385–92. doi: 10.1093/genetics/152.1.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti A, Koivisto K, Pirttilä T, Elovaara I, Reunanen M, Laaksonen M, Ruutiainen J, Peltonen L, Rantamäki T, Tienari PJ. A follow-up study of chromosome 19q13 in multiple sclerosis susceptibility. J Neuroimmunol. 2009;208(1-2):119–124. doi: 10.1016/j.jneuroim.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BRG, Sen GC. Viral apoptosis is induced by IRF-3-mediated activation of Bax. Embo J. 2010;29:1762–73. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. The IRF-3/Bax-Mediated Apoptotic Pathway, Activated by Viral Cytoplasmic RNA and DNA, Inhibits Virus Replication. J Virol. 2011a;85(8):3708–3716. doi: 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. The IRF-3/Bax mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol. 2011b;85(8):3708–16. doi: 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg A, Auble MR, Petro TM. Reduced expression of IL-12 p35 by SJL/J macrophages responding to Theiler's virus infection is associated with constitutive activation of IRF-3. Virology. 2006;353(2):422–432. doi: 10.1016/j.virol.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, R e Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424(6946):324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- Dixit E, Boulant S, Zhang Y, Lee ASY, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes Are Signaling Platforms for Antiviral Innate Immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. 2002;76(11):5532–9. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines JL, Bradford Y, Garcia ME, Reed AD, Neumeister E, Pericak-Vance MA, Rimmler JB, Menold MM, Martin ER, Oksenberg JR, Barcellos LF, Lincoln R, Hauser SL. Multiple susceptibility loci for multiple sclerosis. Hum Mol Genet. 2002;11(19):2251–6. doi: 10.1093/hmg/11.19.2251. [DOI] [PubMed] [Google Scholar]
- Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol. 2003;33(11):2987–97. doi: 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
- Heylbroeck C, Balachandran S, Servant MJ, DeLuca C, Barber GN, Lin R, Hiscott J. The IRF-3 Transcription Factor Mediates Sendai Virus-Induced Apoptosis. J Virol. 2000;74(8):3781–3792. doi: 10.1128/jvi.74.8.3781-3792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420(6913):329–33. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–7. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Hou W, Kang HS, Kim BS. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J Exp Med. 2009;206(2):313–28. doi: 10.1084/jem.20082030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272(21):13829–34. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273(17):10777–83. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- Johnson J, Molle C, Aksoy E, Goldman M, Goriely S, Willems F. A conventional protein kinase C inhibitor targeting IRF-3-dependent genes differentially regulates IL-12 family members. Mol Immunol. 2011;48(12-13):1484–93. doi: 10.1016/j.molimm.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5(2):190–8. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- Li Y, Li C, Xue P, Zhong B, Mao AP, Ran Y, Chen H, Wang YY, Yang F, Shu HB. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc Natl Acad Sci, USA. 2009;106(19):7945–7950. doi: 10.1073/pnas.0900818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteyne P, Bihl F, Levillayer F, Brahic M, Bureau JF. The Th1/Th2 balance does not account for the difference of susceptibility of mouse strains to Theiler's virus persistent infection. J Immunol. 1999;162(12):7330–4. [PubMed] [Google Scholar]
- Panne D. The enhanceosome. Curr Opin Struct Biol. 2008;18(2):236–42. doi: 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129(6):1111–23. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters K, Chattopadhyay S, Sen GC. IRF-3 Activation by Sendai Virus Infection Is Required for Cellular Apoptosis and Avoidance of Persistence. J Virol. 2008;82(7):3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petro TM. Disparate expression of IL-12 by SJL/J and B10.S macrophages during Theiler's virus infection is associated with activity of TLR7 and mitogen-activated protein kinases. Microbes Infect. 2005a;7(2):224–32. doi: 10.1016/j.micinf.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Petro TM. ERK-MAP-kinases differentially regulate expression of IL-23 p19 compared with p40 and IFN-beta in Theiler's virus-infected RAW264.7 cells. Immunol Lett. 2005b;97(1):47–53. doi: 10.1016/j.imlet.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Servant MJ, Grandvaux N, Hiscott J. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol. 2002;64(5-6):985–92. doi: 10.1016/s0006-2952(02)01165-6. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-[kappa]B and IRF3. Cell. 2005;122(5):669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Sharif-Askari E, Nakhaei P, Oliere S, Tumilasci V, Hernandez E, Wilkinson P, Lin R, Bell J, Hiscott J. Bax-dependent mitochondrial membrane permeabilization enhances IRF3-mediated innate immune response during VSV infection. Virology. 2007;365(1):20–33. doi: 10.1016/j.virol.2007.03.011. [DOI] [PubMed] [Google Scholar]
- So EY, Kang MH, Kim BS. Induction of chemokine and cytokine genes in astrocytes following infection with Theiler's murine encephalomyelitis virus is mediated by the Toll-like receptor 3. Glia. 2006;53(8):858–67. doi: 10.1002/glia.20346. [DOI] [PubMed] [Google Scholar]
- Stawowczyk M, Van Scoy S, Kumar KP, Reich NC. The Interferon Stimulated Gene 54 Promotes Apoptosis. J Biol Chem. 2011;286(9):7257–7266. doi: 10.1074/jbc.M110.207068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney SE, Kimbler TB, Firestein GS. Synoviocyte innate immune responses: II. Pivotal role of IFN regulatory factor 3. J Immunol. 2010;184(12):7162–8. doi: 10.4049/jimmunol.0903944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol. 2003;10(11):922–7. doi: 10.1038/nsb1001. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hemmi H, Akira S. Interferon response induced by Toll-like receptor signaling. J Endotoxin Res. 2004;10(4):252–6. doi: 10.1179/096805104225005896. [DOI] [PubMed] [Google Scholar]
- Theiler M. Spontaneous Encephalomyelitis of Mice, a New Virus Disease. J Exp Med. 1937;65(5):705–19. doi: 10.1084/jem.65.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneau S, Rohrlich PS, Brahic M, Bureau JF. Tmevpg1, a candidate gene for the control of Theiler's virus persistence, could be implicated in the regulation of gamma interferon. J Virol. 2003;77(10):5632–8. doi: 10.1128/JVI.77.10.5632-5638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Wherry EJ, Ahmed R. Redefining Chronic Viral Infection. Cell. 2009;138(1):30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- Weaver BK, Kumar KP, Reich NC. Interferon Regulatory Factor 3 and CREB-Binding Protein/p300 Are Subunits of Double-Stranded RNA-Activated Transcription Factor DRAF1. Mol Cell Biol. 1998;18(3):1359–1368. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Akira S, Yonehara S, Kato A, Fujita T. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J Immunol. 2005a;175(5):2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005b;175(5):2851–8. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17(4):1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Li M, Chen L, Yang K, Shan Y, Zhu L, Sun S, Li L, Wang C. The TAK1-JNK cascade is required for IRF3 function in the innate immune response. Cell Research. 2009;19:412–428. doi: 10.1038/cr.2009.8. [DOI] [PubMed] [Google Scholar]