

Abstract
The cell surface hyaluronan receptor CD44 promotes tumor growth and metastasis by mechanisms that remain poorly understood. We show here that CD44 associates with a proteolytic form of the matrix metalloproteinase-9 (MMP-9) on the surface of mouse mammary carcinoma and human melanoma cells. CD44-associated cell surface MMP-9 promotes cell-mediated collagen IV degradation in vitro and mediates tumor cell invasion of G8 myoblast monolayers. Several distinct CD44 isoforms coprecipitate with MMP-9 and CD44/MMP-9 coclustering is observed to be dependent on the ability of CD44 to form hyaluronan-induced aggregates. Disruption of CD44/MMP-9 cluster formation, by overexpression of soluble or truncated cell surface CD44, is shown to inhibit tumor invasiveness in vivo. Our observations indicate that CD44 serves to anchor MMP-9 on the cell surface and define a mechanism for CD44-mediated tumor invasion.
Keywords: CD44, tumor, invasion, MMP-9, cell surface
The cell surface hyaluronan (HA) receptor CD44 is expressed in most cell types as one or several of a series of isoforms generated by variable usage of at least 10 exons encoding a portion of the extracellular domain (Stamenkovic et al. 1991; Screaton et al. 1992, 1993; Lesley et al. 1993). The amino-terminal 142 residues of the extracellular domain form a B-loop that contains the hyaluronan-binding sequences (Aruffo et al. 1990; Peach et al. 1993) and is common to all CD44 isoforms. The polypeptide between the B-loop and the transmembrane domain, which is of variable length and composition depending on the number and combination of variant exon products it contains (Screaton et al. 1992), as well as cell type-specific glycosylation, has at least two functions. One function is to regulate the ability of the B-loop to bind hyaluronan, at least in part through O–glycosylation, which may modulate the conformation of the receptor (Bennett et al. 1995a). The other function is to provide binding sites for ligands unrelated to hyaluronan. Some of the variant exons, in particular exon v3, contain sequences that support covalent attachment of glycosaminoglycan side chains. Heparan sulfate associated with exon v3 can bind basic FGF (bFGF) and heparin-binding EGF (HBEGF, Bennett et al. 1995b; Sherman et al. 1998), chondroitin sulfate side chains of CD44 can mediate cell attachment to fibronectin (Jalkanen and Jalkanen 1992) and variant isoforms of CD44 have been proposed to bind osteopontin (Weber et al. 1996) and possibly additional cytokines (Tanaka et al. 1993).
Several lines of evidence indicate that CD44 expression may have a major implication in tumor growth and dissemination. Clinical studies have shown that a variety of tumor types expressing high levels of cell surface CD44 have a poorer prognosis than similar tumors in which CD44 expression is low or absent (Pals et al. 1989; Jalkanen et al. 1991). In experimental models, expression of CD44H, the common CD44 isoform that contains none of the variant exons, has been observed to augment the tumorigenic and metastatic proclivity of selected human lymphoma (Sy et al. 1991) and melanoma (Bartolazzi et al. 1994) cells in immunocompromised mice. Similarly, expression of a CD44 isoform containing variant exons v4–v7 has been shown to confer metastatic properties to a nonmetastasizing rat pancreatic carcinoma (Gunthert et al. 1991; Seiter et al. 1993).
In normal tissues, CD44 appears to play a prominent role in the regulation of hyaluronan metabolism. We have recently observed that targeted loss of CD44 expression in mouse skin results in disruption of local hyaluronan metabolism and impaired wound healing, hair regrowth, and keratinocyte proliferation in response to a variety of external stimuli (Kaya et al. 1997). In at least some tumors, CD44-associated increase in tumorigenicity and/or metastatic proclivity correlates with the ability of CD44 to mediate cell attachment to (Sy et al. 1991; Bartolazzi et al. 1994; Yu et al. 1997) and uptake and degradation of HA (Culty et al. 1994; Yu et al. 1997). Reduction of CD44-mediated HA internalization by expression of soluble CD44 isoforms in a mouse mammary carcinoma resulted in the inability of these cells to invade hyaluronan-rich myoblast monolayers and to survive in lung tissue of syngeneic mice following tail vein injection (Yu et al. 1997). However, the relationship between CD44-mediated HA binding and internalization and enhancement of tumor invasiveness remains to be elucidated. To gain insight into the mechanisms that underlie CD44-associated tumor invasion, we addressed the possibility that CD44 may promote tumor cell-mediated ECM degradation by regulating the activity of proteases capable of digesting ECM components.
Matrix metalloproteinases (MMPs) are the principal ECM-degrading enzymes whose function is required in tissue remodeling during development (Birkedal-Hansen, 1995; Alexander et al. 1996), bone resorption (Okada et al. 1995; Vu et al. 1998), wound healing (Sudbeck et al. 1997), and angiogenesis (Schnaper et al. 1993; Brooks et al. 1998). MMP activity is regulated at least in part by tissue inhibitors of matrix metalloproteinases (TIMPs, Ogata et al. 1995; Gomis-Ruth et al. 1997) and it is well established that MMPs are produced and used by leukocytes to infiltrate tissues (Wilhelm et al. 1989; Kitson et al. 1998), and by tumor cells (Liotta et al. 1991; Stetler-Stevenson et al. 1993; Mignatti and Rifkin 1994) to facilitate metastatic growth. However, several MMPs, including gelatinase B/MMP-9 (Vu and Werb 1998), which degrades collagen IV in basement membranes, are secreted and mechanisms that enable their association with the cell membrane are largely unknown. Using TA3 mouse mammary carcinoma cells, which constitutively express several CD44 isoforms, and human MC melanoma cells transfected with CD44 cDNA, we show that CD44 serves as an MMP-9 docking molecule to retain MMP-9 proteolytic activity on the cell surface. We provide evidence that the ability of TA3 and MC cells to degrade collagen IV and invade cell monolayers is related to CD44-associated MMP-9 activity on the cell surface, and that CD44/MMP-9 complex formation is associated with tumor invasiveness in vivo.
Results
Expression of soluble CD44 or overexpression of wild-type or tail-less cell surface CD44H decreases TA3 mammary carcinoma growth and invasiveness in vivo
Mammary carcinoma TA3 cells express multiple isoforms of CD44 on the cell surface, display CD44-mediated HA binding, and form lung tumors following tail vein injection into syngeneic mice (Yu et al. 1997). Expression of soluble CD44 in TA3 cells reduces their ability to bind, internalize, and degrade HA and inhibits HA-mediated CD44 capping (Yu et al. 1997). Following intravenous injection, these transfectants infiltrate lung tissue but undergo apoptosis shortly thereafter and fail to form lung tumors (Yu et al. 1997). These observations suggest that tumor formation by TA3 cells depends at least in part on cell surface CD44 function.
As a first step toward elucidating the implication of CD44 in TA3 tumor formation, we compared the tumorigenicity and invasiveness of TA3 cells transfected with vector only (TA3c), or with one of several soluble or cell surface wild-type or mutant CD44 isoforms (Table 1). TA3sCD44v6-10 and TA3sCD44v6-10R43A transfectants have been described previously (Yu et al. 1997). The R43A mutation abrogates CD44-mediated HA binding, and expression of soluble CD44 bearing this mutation fails to block TA3 cell tumor development in the lungs of syngeneic mice following intravenous injection (Yu et al. 1997). To gain further insight into the aspects of CD44 function that are implicated in promoting tumor invasion and dissemination, we included TA3 cells transfected with cDNAs encoding a CD44H cytoplasmic deletion mutant (truncated, tr, or tail-less) and wild-type CD44H (Table 1). Equal numbers of viable cells from several independent isolates of each transfectant (selected for comparable levels of recombinant CD44 expression and comparable proliferation rate in culture, data not shown) were injected subcutaneously into syngeneic mice and the resulting tumors were monitored for growth and histologic appearance. All of the transfectants were able to support tumor development as assessed by histologic analysis 72 hr following injection. However, whereas tumors derived from TA3c and TA3sCD44R43A cells continued to grow rapidly, forming large masses within 14–21 days, TA3sCD44v6-10, TA3CD44tr, and TA3CD44H tumors displayed slow growth (Fig. 1A). Histologic analysis revealed that TA3c/TA3sCD44R43A tumors were poorly circumscribed and invaded adjacent adipose and muscle tissue, whereas TA3sCD44/TA3CD44tr/TA3CD44H tumors were characterized by a smooth, well circumscribed surface and absence of invasion (Fig. 1B, a–c; data not shown).
Table 1.
Characteristics of TA3 CD44 transfectants
|
cDNA
|
Transfectant
|
Tumor invasiveness
|
CD44 capping
|
HA binding (%)
|
HA uptake (%)
|
|---|---|---|---|---|---|
| Vector only | TA3c | + | + | 100 | 100 |
 |
TA3sCD44v6–10 | − | − | 33 ± 7 | 29 ± 5 |
 |
TA3sCD44v6-10R43A | + | + | 105 ± 10 | 95 ± 6 |
 |
TA3sCD44tr | − | − | 55 ± 7 | 38 ± 4 |
 |
TA3sCD44H | − | − | 140 ± 11 | 22 ± 6 |
CD44 cDNAs used to transfect TA3 cells and the nomenclature of the corresponding transfectants are indicated.
CD44 cDNA sequences shown are leader peptide ( ); HA-binding domain (□); non-HA binding domain common to all CD44 isoforms (
); HA-binding domain (□); non-HA binding domain common to all CD44 isoforms ( ); transmembrane domain (█); intracellular domain (
); transmembrane domain (█); intracellular domain (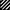 ); exons v6–10 (
); exons v6–10 ( ). The R43A mutation is indicated.
). The R43A mutation is indicated.
Tumor invasiveness was assessed by histology. HA-dependent CD44 capping was assessed by immunofluorescence microscopy. HA binding was assessed by FACS analysis of F1–HA binding to TA3 transfectants and quantified by the mean fluorescence intensity (MFI) as described (Yu et al. 1997). HA uptake was quantified using 3H-labeled HA as described previously (Yu et al. 1997). Binding and uptake values are expressed as a percentage of values obtained using TA3c cells. Values shown are the mean of three independent experiments ± s.d..
Figure 1.

Comparison of growth and invasiveness of tumors derived from TA3 CD44 transfectants. (A) Tumor weight 21 days following subcutaneous injection of 2 × 106 viable cells into syngeneic A/jax mice. At least six animals were injected with each transfectant. Mean weight ± s.d. of tumors derived from the different TA3 transfectants are shown. (B) Histology of a representative TA3c (a), TA3sCD44v6-10 (b), and TA3sCD44v6-10R43A (c) tumor. The TA3c and TA3sCD44v6-10R43A tumors (a,c) show irregular borders and invasion of adjacent adipose and muscle tissue. In contrast, the TA3sCD44v6-10 tumor (b) is characterized by smooth borders and absence of invasion. Bars, 400 μm.
In an effort to understand how overexpression of soluble or cell surface CD44H might inhibit TA3 tumor invasion, transfectants used in this study were compared for fluorescein-labeled HA (Fl–HA) binding by FACS analysis. Expression of soluble CD44v6-10 and truncated tail-less CD44H reduced the HA binding ability of TA3 cells while overexpression of wild-type CD44H increased Fl–HA binding (Yu et al. 1997; Table 1). In contrast, expression of CD44v6-10R43A did not significantly alter Fl–HA binding (Table 1). The observed loss of invasiveness by tumors overexpressing wild-type soluble or cell surface CD44 therefore appeared not to be directly related to the HA binding ability of the corresponding transfectants.
We had shown previously that TA3sCD44v6-10 cells display reduced HA uptake and degradation compared with TA3c cells (Yu et al. 1997). To determine whether TA3 cells overexpressing CD44H may have a defect in HA uptake and degradation, TA3c and TA3CD44H cells were incubated with Fl–HA (20 μg/ml) and Fl–HA binding and uptake were assessed by fluorescence microscopy at several time points. Twenty-four hours after Fl–HA addition, capping was observed in a significant fraction of TA3c cells (Fig. 2a). In contrast, Fl–HA was distributed evenly on the surface of TA3CD44H cells, indicating that overexpression of CD44H inhibits ligand-induced CD44 capping in TA3 cells (Fig. 2c). Endocytotic vesicles were formed in TA3c cells 48 hr after incubation with Fl–HA (Fig. 2b), whereas only few such vesicles could be observed in TA3CD44H cells (Fig. 2d). Reduction of HA uptake and degradation by CD44H overexpression in TA3 cells was confirmed by measuring 3H-labeled HA degradation (Table 1). HA-induced CD44 aggregates colocalized with F-actin, as revealed by anti-CD44 mAb and phalloidin double staining (data not shown), indicating that cytoskeletal components participate in CD44 aggregation through direct or indirect association with the CD44 cytoplasmic domain. This notion is further supported by the observation that cytochalasin D treatment of TA3c cells abolished HA-mediated CD44 capping (data not shown). Binding and internalization of Fl–HA was completely inhibited by the blocking anti-CD44 KM201 mAb, but not by the anti-ICAM-1 mAb HB233 (data not shown).
Figure 2.

Binding and internalization of Fl–HA by TA3c and TA3CD44H cells. Twenty-four hours (a,c) and 48 hr (b,d) after incubation with Fl–HA (20 μg/ml), capping and endocytotic vesicles were observed in TA3c cells, respectively (a,b, arrows); although TA3CD44H cells bind FL–HA, they fail to display capping or endocytotic vesicles at corresponding time points (c,d, respectively). Bar, 40 μm.
Expression of soluble or truncated cell surface CD44 decreases TA3 cell surface-associated MMP9 activity
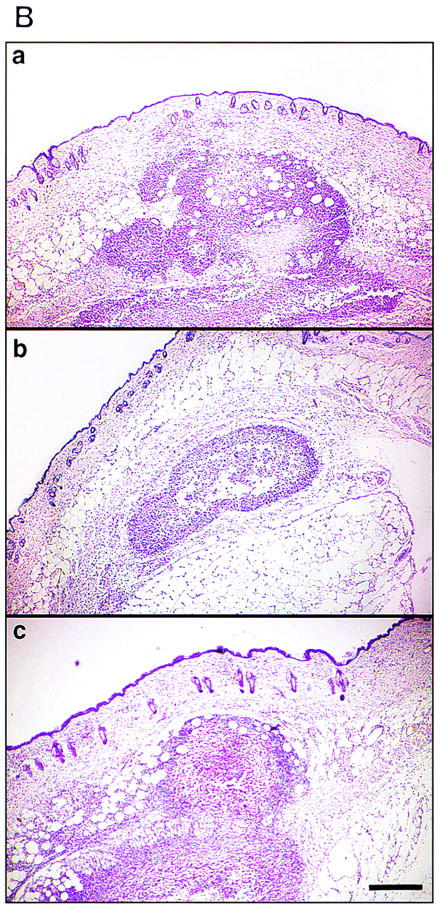
Tissue invasion by tumor cells is dependent in part on endogenously produced or stromal cell-derived soluble MMPs (Stetler-Stevenson et al. 1993). To determine whether TA3 cells express MMPs, we tested TA3 cell-derived RNA for the presence of MMP-2, MMP-3, MMP-7, and MMP-9 transcripts by RT–PCR, using appropriate oligonucleotide primers. All four proteinases were found to be expressed in TA3 cells (Fig. 3A). To assess MMP production, TA3 cell lysates and corresponding serum-free supernatants were tested for gelatinase (MMP-2 and MMP-9) and stromelysin (MMP-3) activity with gelatin and β-casein substrates, respectively. All of the TA3 transfectants were found to contain MMP2, MMP-3, and MMP-9 activity as determined by zymogram analysis of their supernatants (Fig. 3Ba,b; data not shown). Cell-associated metalloproteinase activity was addressed by comparing zymograms of water-soluble whole cell lysate fractions and detergent-soluble fractions of crude plasma membrane preparations. MMP-2 activity was detected primarily in the water soluble fraction of all TA3 transfectants (Fig. 3B,c). In contrast, TA3c cell detergent-soluble plasma membrane fractions were found to contain MMP-9 activity only (Fig. 3B,d). However, TA3sCD44v6-10-, TA3CD44tr-, and TA3CD44H-derived detergent-soluble membrane fractions displayed weak or undetectable MMP-9 activity (Fig. 3B,d; data not shown), whereas TA3sCD44R43A detergent-soluble membrane fractions showed MMP-9 activity comparable to that of TA3c counterparts (Fig. 3B,d). The membrane-associated, proteolytically active, MMP-9 displayed an apparent molecular mass of 83 kD, similar to that of the secreted form. No detectable cell-associated stomelysin activity was observed (data not shown).
Figure 3.

Expression of MMPs at the RNA and protein levels. (A) RT–PCR analysis of MMP transcripts in mRNA derived from TA3c cells. (Lane 1) Ladder of 100-bp DNA, ranging from 200–1500 bp; the top band corresponds to 2 kb; (lane 2) negative control PCR; (lane 3) MMP-2; (lane 4) MMP-3; (lane 5) MMP-7; (lane 6) MMP-9. (B) Zymograms of TA3c and TA3sCD44v6-10, TA3CD44tr, and TA3CD44H transfectant-derived supernatants, lysates, and crude membrane preparations. Gelatin (a,c,d) and β-casein (b) gels were used to test, respectively gelatinase A/MMP-2 (open arrow) and B/MMP-9 (solid arrow) and stromelysin (MMP-3, small arrows) activity in serum-free supernatants (a,b), water-soluble whole cell lysates (c) and 1% Triton-soluble crude membrane extracts of TA3 transfectants (d). Two independent isolates of each transfectant were tested. TA3 cells were transfected with the following: (Lanes 1,2) CD44tr; (lanes 3,4) CD44H; (lanes 5,6) sCD44v6-10; (lanes 7,8) sICAM-1; (lanes 9,10) sCD44v6-10R43A; (lanes 11,12), vector only. Molecular markers corresponding to 83 and 49 kD are indicated by arrowheads at left in a, c, and d; a molecular marker corresponding to 49 kD is indicated by an arrowhead at left in b. Bands corresponding to specific MMPs are shown.
CD44 and MMP-9 colocalize on the TA3 cell surface
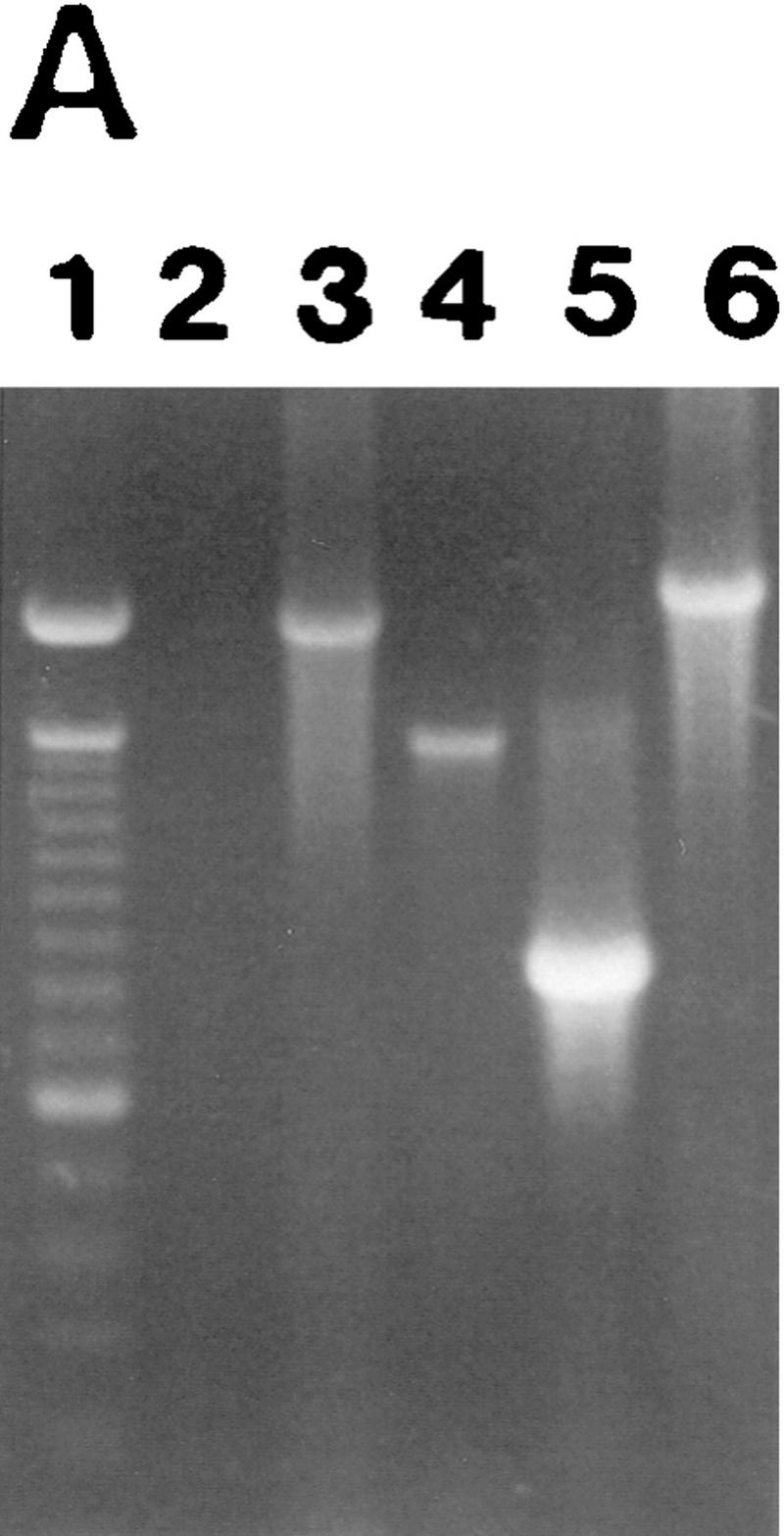
To determine whether MMP-9 on the TA3 cell surface is associated with CD44, we incubated TA3 cells with anti-CD44 mAb IM7.8 and polyclonal goat anti-MMP-9 antibody, followed by noncross-reactive fluorescein-conjugated rabbit anti-rat and rhodamine (TRITC)-conjugated donkey anti-goat secondary antibodies. In a fraction of TA3 cells, CD44 spontaneously forms caps on the cell surface as a result of binding endogenously produced HA, and CD44 capping in these cells is abolished by hyaluronidase treatment. Heterogeneity with respect to spontaneous capping is probably related, at least in part, to differences in HA production and/or turnover among individual cells. Interestingly, CD44 and MMP-9 were observed to colocalize in the caps (Fig. 4A,B). However, cell surface CD44 expression in TA3 cells was not limited to the capping structures, whereas MMP-9 was primarily detected in the caps, with only barely perceptible staining throughout the rest of the cell membrane. Disruption of the CD44 clusters by hyaluronidase treatment resulted in loss of the corresponding MMP-9 aggregates (Fig. 4C,D) but CD44/MMP-9 cocapping was not inhibited by excess exogenous HA (data not shown), suggesting that MMP-9 does not directly bind HA. In TA3sCD44v6-10, TA3CD44tr, and TA3CD44H cells, little if any CD44 clustering was observed on the cell surface. Consistent with the above observations, MMP-9 capping on the surface of these transfectants was scarce or absent (Fig. 4E–H; data not shown). In contrast, TA3sCD44R43A cells displayed CD44/MMP-9 clusters similar to those observed in TA3c cells (Fig. 4I,J).
Figure 4.
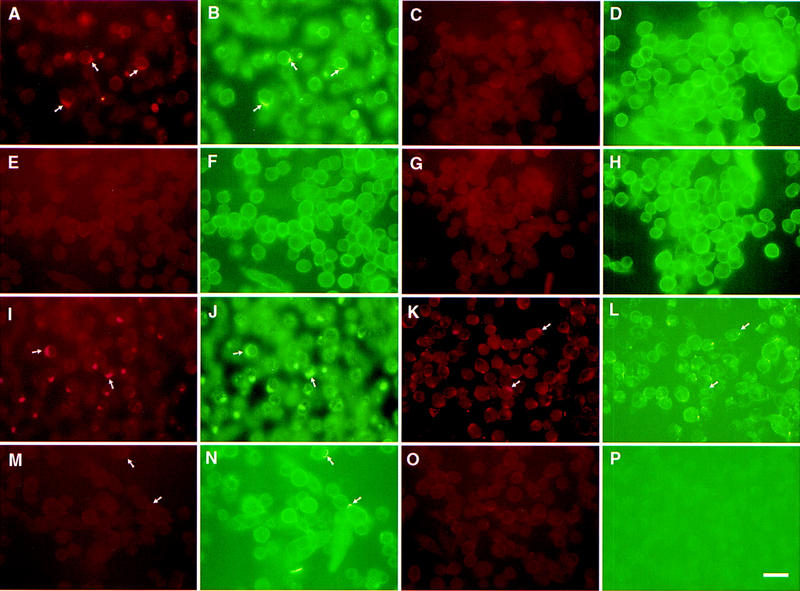
Colocalization of CD44 and MMP-9 in TA3 cells. TA3 transfectants were incubated with rat anti-mouse CD44 mAb IM7.8 and polyclonal goat anti-mouse MMP-9 antibody followed by FITC-labeled anti-rat antibody (green fluorescence) and TRITC-labeled donkey antigoat antibody (red fluorescence). (A,B) TA3c cells; (C,D) hyaluronidase-treated TA3c cells; (E,F) TA3sCD44v6-10 transfectants; (G,H) TA3CD44tr transfectants; (I,J) TA3sCD44v6-10R43A transfectants; (K,L) TA3c cells stained with anti-CD44 mAb KM201, which fails to recognize CD44/MMP-9 containing clusters; (M,N) TA3c cells stained with anti-TIMP-1 mAb (M), which fails to recognize CD44/MMP-9 clusters (N). (O,P) TA3c cells incubated with secondary antibodies only. (Arrows) Corresponding and noncorresponding capping. Bar, 43 μm.
Most CD44/MMP-9 clusters on fixed TA3 cells failed to stain with anti-CD44 mAb KM201 (Fig. 4K,L), which recognizes an epitope within or close to the HA-binding site (Zheng et al. 1995). Stabilization of the HA–CD44–MMP-9 complex by fixation probably reduced the ability of KM201 to displace the complex-associated HA, because KM201 can displace CD44-bound HA from live cells (Zheng et al. 1995). Moreover, CD44-MMP-9 cocapping could be induced by cross-linking CD44 with KM201 mAb and a secondary antibody on the surface of live TA3 cells (Fig. 5). These observations suggest that different regions of CD44 may be responsible for HA binding and MMP-9 association.
Figure 5.
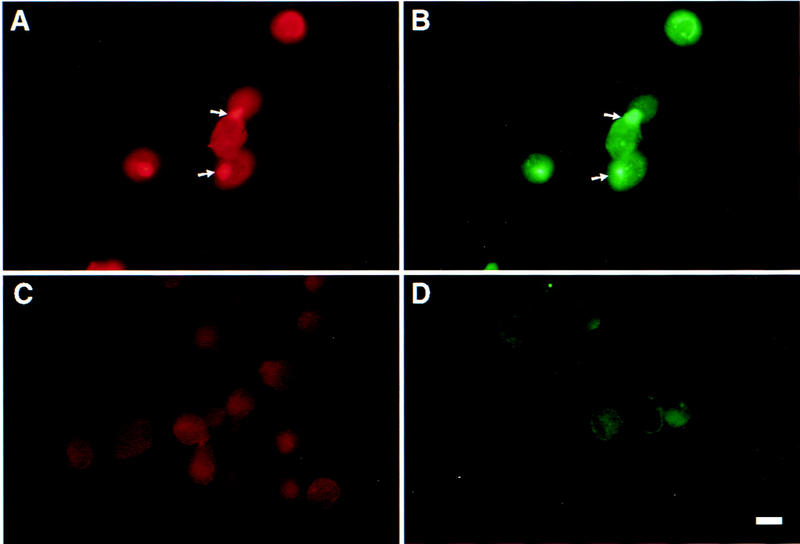
Anti-CD44 antibody-induced CD44-MMP-9 cocapping. TA3 cells were detached with EDTA, replated, and incubated with KM201 mAb, and FITC-labeled rabbit-anti-rat secondary antibody. The cells were then fixed and incubated with anti-MMP-9 antibody and TRITC-labeled donkey anti-goat antibodies. Staining shown is with anti-MMP-9 antibody (A); anti-CD44 mAb (B); secondary TRITC labeled antibody (C); secondary FITC-labeled antibody (D). Bar, 22 μm.
MMP activity is regulated by specific inhibitors known as TIMPs. Because MMP-9 is known to have high affinity for and to be inhibited by TIMP-1 (Ogata et al. 1995), we tested TA3 cells and the corresponding CD44 transfectants for cell surface association between TIMP-1 and the CD44–MMP-9 complex. Although TIMP-1 was found to be expressed in TA3 cells, as assessed by RT–PCR (data not shown), anti-TIMP-1 mAb failed to stain CD44–MMP-9 clusters on the TA3 cell surface (Fig. 4M,N).
Incubation of TA3 cells with IM7.8 mAb followed by TRIC-conjugated donkey anti-goat secondary antibody failed to stain the capping sites, as did incubation with anti-MMP-9 mAb followed by FITC-conjugated goatanti-rat antibody, confirming the absence of secondary antibody cross-reactivity with the inappropriate primary antibody (data not shown). Staining with secondary antibodies alone (Fig. 4O,P) and staining with anti-MMP-2 polyclonal antibody, used as a control (data not shown), was negative.
CD44 and MMP-9 physically interact on the TA3 cell surface
To address the possible interaction between CD44 and MMP-9, TA3 cells were transfected with constructs encoding CD44 immunoglobulin fusion proteins (CD44Rg, for receptor globulin, Aruffo et al. 1990). Because TA3 cells express multiple CD44 isoforms with potentially different biologic properties, several individual CD44 isoforms were tested for their ability to bind MMP-9. Sequences encoding the extracellular domain of CD44H, CD44v7, CD44v8-10, and CD44v7-10 were ligated to human IgG1 Fc sequences, to generate four CD44Rg isoforms. Each receptor globulin was protein A purified from serum-free supernatants of the corresponding transfectants (Fig. 6A) and tested for its ability to immunoprecipitate MMP-9 from TA3 cell lysates as assessed by zymogram analysis. Gelatin zymogram analysis of CD44Rg precipitates indicated the presence of proteolytically active 83-kD MMP-9 but absence of MMP-2 (Fig. 6B). All of the CD44Rgs immunoprecipitated MMP-9, suggesting that common CD44 structures may be required for the interaction.
Figure 6.

Interaction between CD44 and MMP-9. (A) Western blot analysis of CD44Rgs derived from supernatants of TA3 cells transfected with corresponding CD44–Igs. Receptor globulins were immunoprecipitated with protein A beads and blotted with anti-CD44 mAb IM7.8. Supernatants were from TA3c cells (lane 1), and from TA3 cells transfected with immunoglobulin fusions of CD44H (lane 2), CD44v7 (lane 3), CD44v8-10 (lane 4), and CD44 v7-10 (lane 5). (B) Gelatin zymogram of TA3 cell lysates immunoprecipitated with protein A-purified human IgG (lane 1), CD44HRg (lane 2), CD44v7Rg (lane 3), CD44v8-10Rg (lane 4), CD44v7-10Rg (lane 5). (C,D) Gelatin zymogram (C), and Western blot analysis (D) of immunoprecipitates from TA3 cell lysates by use of anti-ICAM-1 mAb HB233 (lanes 1,2), anti-CD44 mAb KM81 (lanes 3,4), and anti-CD44 mAb KM201 (lanes 5,6). Immunoprecipitates in D were blotted with anti-MMP-9 antibody. (E) Supernatants of TA3 cells transiently transfected with constructs encoding soluble CD40-v5 (lane 1) and MMP-9-v5 (lanes 2,3) fusion proteins blotted with anti-v5 mAb. (F) Western blot analysis with anti-CD44 mAb IM7.8 of anti-v5 mAb immunoprecipitates from lysates of TA3 cells transiently transfected with v5-tagged soluble CD40 (lane 1) and MMP-9 (lanes 2,3, corresponding to two independent transfectants) constructs. (G) Gelatin zymogram of TA3 cell lysates immunoprecipitated with CD40Rg (lanes 1,2), CD44HRg (lanes 3,4), CD44HR43ARg (lanes 5,6). In each case, Rg fusion proteins from two independent transfectants were used. (Arrowheads) Molecular mass markers of 203, 116, and 83 kD (A), 116, 83, and 49 kD (B–F) and 116 and 83 kD (G). Bands corresponding to CD44Rgs, MMP-9, sCD40v5, and CD44 are indicated.
To determine whether endogeous CD44 and MMP-9 interact in TA3 cells, lysates from TA3 cells were immunoprecipitated with anti-CD44 mAb KM201 or KM81 and agarose bead-conjugated goat anti-rat antibody, and the immunoprecipitates tested for the presence of MMP-9 by zymogram and Western blot analysis. Proteolytic MMP-9 was found to be present in the anti-CD44 mAb immunoprecipitates of TA3 cell lysates, as demonstrated by both zymogram (Fig. 6C) and Western blot (Fig. 6D) analysis, but not in anti-ICAM-1 mAb immunoprecipitates used as controls (Fig. 6C,D).
Demonstration that CD44 coimmunoprecipitates with MMP-9 was obtained with a simian virus V protein peptide-tagged (v5, Southern et al. 1991) MMP-9 construct transiently expressed in TA3 cells. Expression and production of MMP-9-v5tag and a control soluble CD40-v5tag construct were verified by Western blot analysis of supernatants of the transfected TA3 cells (Fig. 6E). Transfectant lysates were immunoprecipitated with anti-v5 tag mAb and the resulting immunoprecipitates were tested for the presence of CD44 by Western blot analysis by use of mAb IM7.8. Anti-v5 tag mAb immunoprecipitates from lysates of TA3 cells transiently expressing the MMP-9-v5 fusion protein contained multiple CD44 isoforms (Fig. 6F), consistent with the notion that several CD44 isoforms can associate with MMP-9 on the cell surface. The specificity of the interaction was supported by the failure of CD44 to coimmunoprecipitate with v5-tagged soluble CD40 (Fig. 6F).
The ability of MMP-9 to bind HA was tested in a ligand-binding assay, in which gelatin bead-purified MMP-9 immobilized on membrane filters was blotted with biotinylated HA (bHA). Whereas membrane filter-immobilized CD44Rg bound bHA, MMP-9 failed to do so (data not shown). Furthermore, CD44Rg containing the R43A mutation, which abolishes HA binding, produced in transiently transfected TA3 cells, was able to immunoprecipitate MMP-9 from TA3 cell lysates, as assessed by zymogram analysis (Fig. 6G). These observations suggest that HA does not serve as a molecular bridge for the interaction between CD44 and MMP-9.
CD44 expression can augment collagen IV degradation by tumor cells
To demonstrate that the ability of CD44 to localize MMP-9 to the cell surface is not a cell type-specific phenomenon, we tested human MC melanoma cells transfected with different CD44 isoforms for cell surface expression of MMP-9. We had shown previously that MC cells, which do not constitutively express CD44 and are poorly tumorigenic in immunocompromised mice, become highly tumorigenic when transfected with CD44 isoforms that bind HA (Bartolazzi et al. 1994). FACS analysis of cell surface MMP-9 expression by use of anti-MMP-9 mAb in MC transfectants expressing vector only, CD44H and CD44v3 revealed that both CD44 transfectants tested positive for anti-MMP-9 reactivity, whereas vector only transfected cells did not (data not shown).
To address the functional significance of CD44–MMP-9 association on the cell surface, we tested the ability of CD44-transfected MC melanoma cells to degrade radiolabeled collagen IV. After 16 hr of incubation on radiolabeled collagen IV-coated dishes, released radiolabel was measured in the supernatant of each transfectant. MC44H and MC44v3 transfectants were observed to release a three-to-five fold greater amount of radiolabel into the supernatant than MC cells transfected with vector only (Fig. 7A; data not shown). Collagen IV release could be be blocked by 1-10 phenanthroline, which inhibits most MMPs (Brooks et al. 1998), the broad spectrum MMP inhibitor peptide I (Odake et al. 1994) as well as a functional blocking anti-human MMP-9 antibody (Fig. 7A; Schnaper et al. 1993). Aprotinin, MMP-3 inhibitor peptide (Fotouhi et al. 1994) and anti-MMP-2 antibody (Fig. 7A) used as controls had no inhibitory effect.
Figure 7.



CD44-dependent cell-mediated degradation of collagen IV. Twenty-four-well plates were coated with 3H-labeled collagen type IV (5000 cpm/well), and TA3 and MC transfectants were seeded onto the plates at 2 × 105 cell/well. Following a 16 hr incubation at 37°C, cell culture supernatants were recovered, centrifuged, and the radiolabel quantified from one-fifth of each supernatant in a β-counter. Cells were either untreated or preincubated with antibody, MMP-inhibitor peptides, or phenantroline as described in Materials and Methods. (A) Collagen IV degradation by MCwt and MCCD44 transfectants pretreated or not with MMP-3 inhibitor peptide, MMP-inhibitor peptide I, anti-MMP-2, and anti-MMP-9 mAb; (B) TA3c and TA3sCD44v6-10 cells pretreated or not with 1-10 phenanthroline, aprotinin, anti-ICAM mAb HB233, blocking anti-CD44 mAb KM201, and weakly blocking anti-CD44 mAb IM7.8. (C) Collagen degradation by two independent TA3c and TA3sCD44v6-10 isolates transfected with scrambled (vector) or antisense MMP-9 cDNA. The results are expressed as the mean ± s.d. of triplicate values.
Comparison of the ability of TA3c and TA3sCD44v6-10 cells to degrade radiolabeled collagen IV bound to plastic dishes revealed similar results to those provided by MC transfectants. The amount of radiolabel released into the supernatants of TA3c cells was two-to-three fold higher than that released into the supernatants of TA3sCD44v6-10, TA3CD44tr, or TA3CD44H cells (Fig. 7B; data not shown). The observed release of radiolabel could be blocked by MMP inhibitor peptide I and by 1-10 phenanthroline but niether by MMP-3 inhibitor peptide nor by aprotinin (Fig. 7B; data not shown). In addition, preincubation of TA3c cells with the blocking anti-CD44 mAb KM201 had an inhibitory effect on collagen IV degradation, whereas preincubation with mAb IM7.8, which is non- or weakly blocking depending on the cell type (Zheng et al. 1995), had only a minimal inhibitory effect (Fig. 7B). The notion that MMP-9 is required for the observed collagen IV degradation is supported by the observation that TA3 cells expressing an MMP-9 construct in antisense orientation showed markedly reduced collagen IV degradation (Fig. 7C).
MC cell invasion of G8 myoblast monolayers is associated with CD44 expression and MMP-9 activity
To assess the role of CD44–MMP–9 association in regulating tumor cell invasiveness, we compared the ability of wild-type and CD44-transfected MC cells to invade G8 myoblast monolayers. G8 monolayer invasion provides a simple and highly reproducible assay to test the ability of tumor cells to invade a monolayer that is enriched in a variety of ECM components including HA. We had shown previously that TA3c cells invade G8 monolayers, whereas TA3sCD44 cells do not (Yu et al. 1997). MC cells transfected with vector only failed to invade the G8 monolayers (Fig. 8a), whereas expression of CD44H or CD44v3,7-10, promoted invasion (Fig. 8b; data not shown). The observed invasion could be blocked by MMP inhibitor peptide I, 1-10 phenanthroline and anti-MMP-9 antibody (Fig. 8c,e; data not shown) but not by MMP-3 inhibitor peptide or an isotype matched antibody (Fig. 8d,f).
Figure 8.
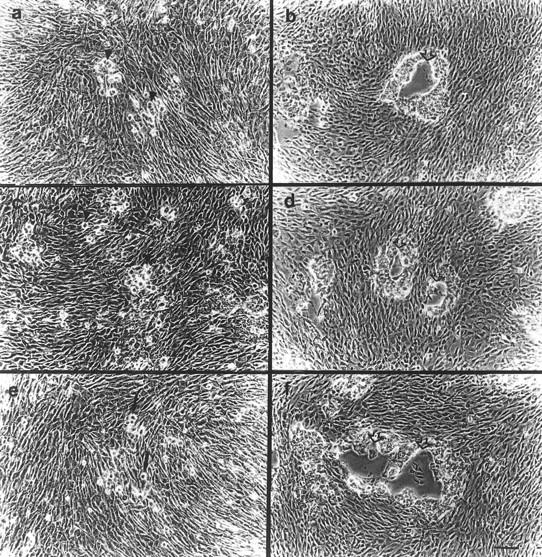
G8 myoblast monolayer invasion by MC cell transfectants. G8 myoblast monolayers were prepared and fixed as described in Materials and Methods. MC cell transfectants were seeded onto the monolayers in six-well plates at 5 × 103 cells/well with or without preincubation with antibody, phenanthroline, and MMP-inhibitor peptides. After 7–10 days of incubation in the presence or absence of these reagents, cell invasiveness was documented under an inverted microscope. G8 monolayer invasion was as follows: MC cells transfected with vector only (a); (b) MC44H cells; (c–f) MC44H cells pretreated with MMP inhibitor peptide I (c), MMP-3 inhibitor peptide (d), anti-MMP-9 mAb (e), and isotype matched mouse IgG (f). Bar, 128 μm.
Discussion
Adhesion and regulated proteolysis are critical functions in tumor development, angiogenesis, invasion, and metastasis. Although several adhesion receptors and matrix metalloproteinases have been shown to play a role in tumor invasion and dissemination, relatively little is known about mutual regulation and coordinated function of tumor cell-associated adhesive and proteolytic processes. Various isoforms of the hyaluronan receptor CD44 have been implicated in the regulation of both primary and metastatic tumor growth (Gunthert et al. 1991; Sy et al. 1991; Seiter et al. 1993; Yu et al. 1997), but their mode of action has been poorly understood despite a good correlation between the ability of several isoforms to bind HA and promote tumor invasion (Bartolazzi et al. 1994; Yu et al. 1997). Similarly, expression of various metalloproteinases correlates with enhanced tumor invasion, but with the exception of membrane-type MMPs (MT-MMPs), which are integral membrane proteins (Basbaum and Werb 1996), MMPs implicated in tumor invasion lack a membrane anchoring domain and are secreted (Kleiner and Stetler-Stevenson 1993). Recent work has shown that the αvβ3 integrin can localize proteolytically active MMP-2 to the cell membrane (Brooks et al. 1996), raising the possibility that adhesion receptors may provide a means to retain soluble MMPs on the cell membrane, where their proteolytic activity is most likely to promote cell invasion.
In the present paper, we provide evidence that CD44 associates with MMP-9 in mouse and human tumor cells and helps localize MMP-9 activity to the cell surface. Evidence supporting this notion is based on the observations that (1) overexpression of soluble or truncated or wild-type cell surface CD44, which disrupts ligand-induced aggregation of endogenous CD44, reduces cell surface MMP-9 activity in TA3 cells; (2) cell surface CD44 and MMP-9 colocalize in hyaluronan-induced aggregates; (3) CD44 and MMP-9 coimmunoprecipitate whether anti-CD44 or anti-MMP-9-associated tag mAb are used. The functional relevance of the CD44–MMP-9 association on the cell surface is demonstrated by the observations that (1) the ability of TA3 cells to degrade collagen IV and to invade G8 monolayers is abrogated by soluble CD44 overexpression and that similar inhibition is provided by MMP inhibitors, including 1-10 phenanthroline and MMP inhibitor peptide I, and by antisense MMP-9 cDNA expression; (2) expression of different CD44 isoforms in MC cells, which are constitutively CD44-negative, promotes MC cell-mediated degradation of collagen IV and invasion of G8 myoblast monolayers, both of which are inhibited by 1-10 phenanthroline, MMP inhibitor peptide I, and, more specifically, by MMP-9 blocking antibody.
TA3 cell-mediated degradation of collagen IV could also be blocked by preincubation of the cells with anti-CD44 mAb KM201, which competes with HA for the same or a closely related binding site (Zheng et al. 1995). Although KM201 could not bind HA-induced CD44 clusters on fixed TA3 cells, preincubation of live TA3 cells with KM201 inhibited HA binding, HA-mediated CD44 clustering, and cell surface MMP-9 association, consistent with the notion that KM201 can displace CD44-bound HA from live cells (Zheng et al. 1995). As might be expected, cross-linking of KM201 on the surface of TA3 cells with secondary antibody induced CD44–MMP-9 cocapping, thus mimicking the effect of HA.
The mechanism by which MMP-9 is recruited to sites of CD44 aggregation remains to be elucidated. It is conceivable, however, that HA- or antibody-mediated aggregation may alter CD44 conformation in a manner that promotes MMP-9 binding. It is also possible that some CD44 isoforms may aggregate by mechanisms that do not require ligand binding. For example, CD44 isoforms containing exons v6 and v7, may aggregate on the basis of their isoform-specific glycosylation (Sleeman 1996a). Expression of such isoforms in appropriate cells may result in their ability to recruit MMP-9 without requiring HA-mediated clustering, which may explain the ability of certain CD44 isoforms to promote invasion and metastasis of some tumor types without binding HA (Sleeman et al. 1996b). Whatever the mechanism responsible for CD44 clustering, cell surface-associated MMP-9 proteolytic activity appears to be more closely associated with CD44 aggregation than with HA binding per se. This notion is supported by the observation that wild-type cell surface CD44H overexpression in TA3 cells, despite augmenting their adhesion to HA, inhibits HA-mediated CD44 aggregation, cell surface MMP-9 association, and the ability of the cells to form invasive or metastatic tumors in syngeneic mice. Ligand-induced CD44 aggregation may provide a natural means to concentrate the proteolytic activity of MMP-9 at foci of cell-matrix contact and possibly render MMP-9 inaccessible to inactivation by TIMPs. By degrading ECM proteins and thereby disrupting ECM architecture, MMP-9 may help liberate ECM protein-bound HA and facilitate tumor cell HA uptake and degradation. The association between CD44 and MMP-9 may therefore provide a link between cell adhesion to ECM, ECM degradation and HA metabolism.
Elucidation of the mechanism underlying the disruption of ligand-induced aggregation of endogenous CD44 by overexpression of soluble or cell surface CD44 will require further investigation. It seems plausible, however, that soluble and truncated CD44 molecules may play the role of decoy receptors that can compete for HA binding and possibly even form partial aggregates with endogenous CD44 on the cell surface while disrupting physiologic CD44 clustering that requires appropriate communication with the cytoskeleton. Similarly, overexpression of wild-type CD44H may result in a disproportionate number of receptors with respect to cytoskeletal-interacting protein availability. Reduction in the fraction of HA-binding receptors that associate with the cytoskeletal components required for ligand-mediated capping may lead to suboptimal CD44 aggregation. Analogous observations have been made using receptors for inducers of apoptotic cell death. Naturally occuring decoy receptors for the trigger of apoptosis TRAIL/Apo2L have been described (Pan et al. 1997), and the requirement for an optimal number of cell surface TNF or Fas receptors for the generation of signals that initiate apoptosis has been suggested (Clement and Stamenkovic 1994).
The increase in tumor growth associated with CD44 expression may be due to MMP-9-mediated release of ECM-sequestered growth and/or angiogenic factors. MMP-9 has been implicated in promoting angiogenesis (Vu et al. 1998b) and our own preliminary data suggest that CD44-associated cell surface MMP-9 activity may stimulate angiogenesis (Q. Yu and I. Stamenkovic, unpubl.). However, the precise mechanism by which CD44/MMP-9 may enhance tumor growth, and the potential role, if any, of HA internalization by tumor cells remain to be resolved. The results of the present study show that CD44 can promote tumor invasion by helping localize proteolytically active MMP-9 to the tumor cell surface. These observations help define a novel function of CD44, propose a mechanism for retention of MMP-9 on the cell surface and provide new insight into the role of CD44 in regulating tumor invasiveness and dissemination.
Materials and methods
Cells and antibodies
G8 mouse fetal myoblasts were obtained from American Type Culture Collection (ATCC, Rockville, MD), and cultured in DMEM with 10% FBS (both from Irvine Scientific, Santa Ana, CA) and 10% horse serum (GIBCO-BRL, Gaithburg, MD). MC human melanoma cells (Thomas et al. 1992) were cultured in 10% FBS DMEM medium. TA3 murine mammary carcinoma cells were maintained in DMEM supplemented with 10% FBS. All TA3 transfectants were cultured in DMEM supplemented with 10% FBS and 0.5 mg/ml G418 (GIBCO-BRL). The hybridomas KM201, KM81, IM7.8, and HB-233 (ATCC), which produce rat–anti-mouse CD44 and ICAM-1 mAb, respectively, were cultured in DMEM supplemented with 10% FBS (Irvine Scientific). Partially purified mAbs were obtained by 50% (NH4)2SO4 precipitation of the culture medium of KM201, KM81, IM7.8, and HB-233 hybridomas, and were used for immunohistochemistry and immunocytochemistry. Affinity-purified KM201, KM81, IM7.8, and HB-233 mAb were obtained by purification of serum-free DMEM supernatants collected from cultures of the corresponding hybridomas with goat anti-rat IgG agarose beads (Sigma, St. Louis, MO).
RT–PCR and expression constructs
Total RNA was isolated from mouse placenta and TA3 cells by use of TRIzol reagent (GIBCO-BRL) according to the manufacturer’s instructions. cDNA was synthesized from 5 μg of total RNA with Superscript II RNase H− reverse transcriptase (GIBCO-BRL). PCR was performed with Taq DNA polymerase (Perkin Elmer, Norwalk, CT) for 30 cycles at 94°C for 30 sec, 55°C for 30 sec, and 72°C for 90 sec, followed by a final 7 min at 72°C. To generate double-stranded cDNA of mouse mmp-9 with cDNA generated from mouse placenta, PCR was performed by use of the forward primer, 5′-CACGACGATATCATGAGTCCCTGGCAGCCCCTGCTCCTG-3′ and the reverse primer, 5′-CACGATGATGGCGGCCGCAGGGCACTGCAGGAGGTCGTAGGT-3′. Soluble human CD40 was generated by PCR from a CD40 expression plasmid by use of the forward primer, 5′-CACGATGATATCATGGTTCGTCTGCCTCTGCAGTGC-3′ and the reverse primer, 5′-CACGGGATCCAGCCGATCCTGGGGACCACAGACAAC-3′. MMP-9 and soluble CD40 were cloned into pcDNA3.1/v5/His-Topo eukaryotic expression vectors engineered to generate carboxy terminal v5 epitope tag fusions (Invitrogen Corp., Carlsbad, CA). To generate soluble CD44-Rgs, four different soluble CD44 fragments were amplified by PCR using cDNA from TA3 cells, a forward primer corresponding to sequences in the first exon of CD44: 5′-CACACAAAGCTTATGGACAAGGTTTTGGTGGCACACAGCT-3′, and a reverse primer corresponding to sequences in exon 17 located immediately 5′ to sequences encoding the transmembrane domain: 5′-CACACAAGATCTTTCTGGAATCTGAGGTCTCCTCATAGG-3′. These fragments were cloned into pCR 3-Uni eukaryotic expression vectors (Invitrogen), and sequence analysis indicated that they correspond to sequences encoding the extracellular domain of CD44H, CD44v7, CD44v8-10, and CD44v7-10. A human IgG1 Fc BamHI–XbaI fragment was then inserted into BglII–XbaI-cut vector containing the CD44 fragment and ligated in frame to the 3′ end of each CD44 sequence. To generate CD40Rg, the human IgG1 Fc BamHI–XbaI-cut fragment was inserted into a BamHI–XbaI-cut vector containing sequences encoding the extracellular domain of CD40. Generation of soluble ICAM-1, CD44R43A, and CD44 isoforms containing variant exons v6–v10 were described elsewhere (Yu et al. 1997). CD44R43A–Ig was made by inserting the immunoglobulin Fc fragment into the expression vector containing the extracellular domain of CD44R43A. The CD44 cytoplasmic deletion mutant (CD44tr) and CD44H were generated by PCR with cDNA from TA3 cells and the forward primer, 5′-CGCCATGGACAAGTTTTGGT-3′ and the reverse primer 5′-CTACACCCCAATCTTCATGTCCACACT-3′. The v3 containing CD44 isoforms were obtained by screening successive CD44-positive clones generated by PCR amplification of cDNA derived from TA3 cells by use of the forward primer, 5′-CGCCATGGACAAGTTTTGGT-3′ and reverse primers, 5′-CAGGAGAGATGCCAAGATGATGAG-3′ and 5′-CTACACCCCAATCTTCATGTCCACACT-3′, respectively. These PCR-generated fragments were all cloned into pCR 3-Uni eukaryotic expression vectors (Invitrogen). The authenticity and orientation of the above-cloned inserts were confirmed by DNA sequencing by the dideoxy chain termination method with Sequenase (U.S. Biochemical Corp., Cleveland, OH), according to the manufacturer’s recommendations. The antisense MMP-9 constructs consisted of 513 nucleotides encoding the amino-terminal portion of the enzyme. The fragment was amplified from TA3 cell RNA by RT–PCR with the forward primer 5′-ATGAGTCCCTGGCAGCCCCTGCTCCTG-3′ and the reverse primer 5′ACCAAACTGGATGACAATGTCCGC-3′ and inserted into the pCR3.1 Uni expression vector in reverse orientation. To detect MMP and TIMP transcripts in TA3 cell RNA, RT–PCR was performed with TA3 cell-derived cDNA by use of the following pairs of primers: MMP2F, 5′ATGGAGGCACGAGTGGCCTGGGGAGCG-3′, MMP-2R, 5′-GCAGCCCAGCCAGTCTGATTTGAT-3′; MMP-3F, 5′-ATGAAGGGTCTTCCGGTCCTGCTGTGGCTG-3′, MMP3R, 5′-ACAATTAAACCAGCTATTGCTCTT-3′; MMP-7F, 5′-ATGCAGCTCACCCTGTTCTGC-3′, MMP-7R, 5′-AGCGTGTTCCTCTTTCCATATAA-3′; MMP-9F, 5′-ATGAGTCCCTGGCAGCCCCTGCTCCTG-3′, MMP-9R, 5′-AGGGCACTGCAGGAGGTCGTAGGT-3′. TIMP-1F, 5′-ATGGCCCCCTTTGCATCTCTGGCATCT-3′, TIMP-1R, 5′-GGCCCCAAGGGATCTCCAAGTGCA-3′.The RT–PCR products were inserted into the pCR3.1 Uni vector (Invitrogen) and their identity was confirmed by dideoxy sequencing.
Transient and stable transfection
TA3 cells were transfected by use of Lipofectamine (GIBCO-BRL) with plasmids containing soluble CD40–Ig and CD44–Ig fusions, CD44H, CD44tr, sense, and antisense MMP-9, and sCD40 and expression vectors alone. MC melanoma cells were transfected with CD44v3, CD44v3,v7-10 isoforms, and expression vectors only. TA3sCD44 and MC44H transfectants were described previously (Bartolazzi et al. 1994; Yu et al. 1997). In transient transfection assays, TA3 cells were used 72 hr following transfection, whereas stable transfectants were selected for G418 resistance (1.5 mg/ml G418), and resistant colonies were picked after 2–3 weeks of growth in selection medium. Culture supernatants and lysates of transfectants were tested by Elisa and/or Western blot analysis for expression of the approrpriate gene products.
FACS analysis of cell surface CD44 and MMP-9
TA3 cells were detached from plates with EDTA, washed, and incubated with mAb IM7.8 (∼10 μg/ml) and polyclonal goat antimouse MMP-9 antibody M-17 (5 μg/ml, Santa Cruz Biotechnology, Santa Cruz, CA), on ice for 1 hr, washed extensively with PBS, and incubated with fluorescein (FITC)-conjugated rabbit anti-rat, and TRITC-conjugated donkey anti-goat secondary antibody (Sigma) for 30 min on ice. MC cells were incubated with 5 μg/ml mouse anti-human MMP-9 (Neomarkers, Fremont CA) and a secondary FITC-conjugated goat-anti-mouse antibody. Cells were washed extensively and analyzed on a FACScan (Becton Dickson, Mountain View, CA).
Sample preparation, zymogram, and Western blot analysis, and bHA-binding assay
Serum-free supernatants of cultured TA3 transfectants were collected, and attached cells were extracted with 5 mm HEPES, 2 mm MgCl2, and 75 mm NaCl (pH 7.4) containing 5 mm EDTA, 2 mm PMSF, and 2 μg/ml leupeptin, 0.5 units/ml aprotinin, by homogenizing the cells in a Dounce homogenizer. After centrifugation at 6300g to remove nuclei and large cell debris, the supernatants were centrifuged again at 14,000g to pellet the membranes. The supernatant was then retained as the water-soluble fraction of the cell lysates, whereas the crude membrane preparations were lysed in RIPA buffer, 50 mm Tris-HCl (pH 7.4), containing 150 mm NaCl, 5 mm EDTA, 1% Triton, 0.1% SDS, 2 mm PMSF, 2 μg/ml leupeptin, and 0.5 units/ml aprotinin. Following removal of RIPA buffer-insoluble materials, the remaining supernatant was considered to represent the detergent-soluble fraction of the extract. Gelatin and casein zymograms were performed as described previously (Herron et al. 1986). Briefly, 50 μl of serum-free supernatant from the transfected TA3 cells and 50 μg of proteins from water-soluble or detergent-soluble TA3 cell extracts were separated by 10% SDS-PAGE containing 1 mg/ml gelatin (Fisher, Columbia, MD) or β-casein (Sigma). Following electrophoresis, gels were washed with 2.5% Triton X-100 to remove SDS, and incubated with 50 mm Tris-HCl (pH 8.0), containing 5 mm CaCl2, and 0.02% sodium azide at 37°C for 24 hr. Gelatin and stromelysin activity was visualized by staining the gels with 0.5% Coomassie blue. For Western blots, gels subjected to electrophoresis were blotted onto Hybond-ECL membranes (Amersham Corp., Arlington Heights, IL). mAb IM7.8, polyclonal goat antimouse MMP-9 antibody, and anti-v5 epitope tag mAb (Invitrogen) were used to detect CD44/CD44Rgs, MMP-9, and v5 epitope-tagged proteins, respectively. bHA binding assays were performed as described previously (Yu and Toole 1995).
Immunoprecipitation
Protein A beads were preincubated with serum-free medium derived from CD44–Ig, CD44R43A–Ig, and CD40–Ig-transfected cells, then incubated overnight at 4°C with RIPA lysates of TA3 cells, transiently or stably transfected with different CD44Rg isoforms, CD44R43ARg, CD40Rg, or expression vector only. After five washes with 0.05 m Tris-HCl (pH 8.0), 0.15 m NaCl, (TBS) containing 0.1% Tween 20 and 0.1% Triton X-100, bound proteins were eluted with 2× SDS sample buffer, and loaded onto both 10% SDS-PAGE, and 10% SDS-PAGE containing 1 mg/ml gelatin for Western blot analysis of Rg protein expression and zymogram analysis of coprecipitated gelatinase activity. Alternatively, TA3 cells transfected with expression vector alone (TA3 c1 and TA3 c8) were lysed as above. The lysates were then precleared with agarose beads conjugated with goat anti-rat IgG (Sigma), and incubated with affinity-purified mAb KM201, KM81, or HB233 with fresh agarose beads conjugated with goat anti-rat IgG at 4°C overnight. Following extensive washing, immunoprecipitated proteins were subjected to zymogram analysis as above and to Western blot analysis with goat anti- MMP-9 antibody. TA3 cells transiently transfected with v5 epitope-tagged MMP-9 and sCD40 were lysed in RIPA buffer, and expression of the transfected cDNAs was assessed by Western blot analysis by use of a mouse mAb against the carboxy-terminal v5 peptide tag (Invitrogen). Lysates were precleared as above and incubated with anti-v5 mAb and fresh protein A beads at 4°C overnight. Following extensive washing, the immunoprecipitated proteins were eluted by 2× SDS sample buffer, and Western blots were performed with anti-CD44 mAb IM7.8.
Tumor growth and invasion assays
Transfected TA3 cells (2 × 106 in 0.2 ml of Hank’s balance solution, HBSS per mouse) were injected subcutaneously into male syngeneic A/Jax mice (Jackson Laboratory, Bar Harbor, ME). The animals were observed daily, sacrificed 3 weeks after injection, and the tumors were removed, weighed, fixed, and sectioned for further studies. At least six animals were injected with each transfectant and at least two independent isolates of each transfectant were used. G8 myoblast monolayers were prepared as described (Yu et al. 1997). Transfected TA3 and MC cells were seeded onto the fixed G8 monolayers at 5 × 103 cells/well in six-well plates with or without preincubation with the mAbs and the reagents described in type IV collagen degradation assays. After 7–10 days incubation with or without the presence of above reagents, the invasiveness of the cells was documented by microscopy. All experiments were done in triplicate and at least two independent isolates of each transfectant were used.
FL–HA uptake
A total of 5 × 105 of the transfected cells (TA3c, and TA3CD44H) was seeded onto 60-mm dishes in the presence of DMEM/10%FBS. On the following day, the culture medium was replaced by fresh DMEM/10%FBS containing 20 μg/ml Fl–HA. Twenty-four and 48 hr later, the cells were washed extensively with PBS, fixed in 4% paraformadehyde, and observed under a fluorescence microscope.
Histology and immunocytochemistry
Tumors from the experimental animals were dissected and fixed in 4% paraformaldehyde (Fisher) in PBS, washed with PBS, dehydrated through 30%, 70%, 95%, and 100% ethanol and xylene, and embedded in paraffin wax (Fisher). 5- to 10-μm sections were cut, mounted onto slides, and stained with Gill-2 hematoxylin (Shandon) for histologic analysis. In immunocytochemical analyses, cells were seeded on 35-mm dishes, cultured overnight, and treated with or without 0.5 mg/ml HA (Anika Research) or 10 U/dish of Streptomyces hyaluronidase (ICN, Costa Mesa, CA) for 2 hr at 37°C. After three washes with PBS, the cells were fixed with 4% paraformaldehyde (Fisher), washed with PBS, blocked with 2% nonfat milk in PBS, and incubated with either 5 μg/ml of goat anti-mouse MMP-9 or MMP-2 polyclonal antibody (Santa Cruz), in the case of TA3 cells, anti-human MMP-2, and MMP-9 mAb (Neomarkers, Fremont, CA), in the case of MC cells, or mouse anti-TIMP-1 mAb (Chemicon International, Temecula, CA), which recognizes both mouse and human TIMP-1 and 5 μg/ml IM7.8 or KM201 mAb at 4°C overnight. After extensive washing with PBS, cells were incubated with secondary antibodies for 30 min at 4°C, washed, and examined by fluorescence microscopy. The secondary antibodies were rhodamine (TRITC)-conjugated AffiniPure donkey anti-goat IgG (Accurate Co., Westbury, NY) to detect goat anti-MMP-2 and MMP-9 antibodies, rhodamine-conjugated AffiniPure donkey anti-mouse IgG (Accurate Co.) to detect mAbs against human MMP-2, MMP-9, and TIMP1, and FITC-conjugated rabbit anti-rat IgG (Sigma) to detect IM7.8, and KM201 mAb. As controls, the tumor cells were incubated with secondary antibodies alone, or with the primary and unmatched secondary antibody. To induce CD44 capping, 5 μg/ml KM201 mAb was added to TA3 cells that had been seeded onto 35-mm plates, cultured overnight, and incubated for 2 hr at 37°C. FITC-conjugated rabbit anti-rat antibody (1 μg/ml) was then added to the cell culture medium for an additional hour at 37°C. Cells were then washed extensively, fixed with 3.7% paraformaldehyde, and examined by fluorescence microscopy.
Type IV collagen degradation assay
Collagen type IV degradation assays were performed as described previously (Garbisa et al. 1987). Briefly, 24-well plates were coated with 3H-labeled type-IV collagen (5000 cpm/well, Dupont NEN, Boston, MA), the transfected TA3 cells and MC melanoma cells were seeded at 2 × 105 cells/well onto the coated plates, and incubated at 37°C for 16 hr. Cell culture supernatants were then removed and centrifuged; released radiolabel, corresponding to degraded collagen IV, was measured in 100 μl of each supernatant (one-fifth of the total volume) in a β-counter (Beckman Instruments). Tumor cells were untreated or preincubated for 1 hr on ice with 20 μg/ml anti-human MMP-2, or MMP-9 mAb (NeoMarkers Inc, Fremont, CA), KM201, IM7.8, or HB-233 mAb; or 1 mm MMP inhibitor 1,10-phenanthroline (Sigma), 10 μg/ml serine proteinase inhibitor, aprotinin (Sigma), or 10 μm MMP-3 inhibitor peptide (Calbiochem, San Diego, CA), or 60 μm MMP peptide inhibitor I (Calbiochem). All experiments were done in triplicate.
Acknowledgments
We thank Luisa Cironi for preparing the MC cell transfectants. This work was supported by National Institutes of Health (NIH) grants CA55735, GM48614, and GM54176. Q.Y. was supported by NIH training grant CA09216. I.S. is a Scholar of the Leukemia Society of America.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL stamenko@helix.mhg.harvard.edu; FAX (617) 726-5684.
References
- Alexander CM, Hansell EJ, Behrendtsen O, Flannery ML, Kishnani NS, Hawkes SP, Werb Z. Expression and function of matrix metalloproteinases and their inhibitors at maternal-embryonic boundary during mouse embryo implantation. Development. 1996;122:1723–1736. doi: 10.1242/dev.122.6.1723. [DOI] [PubMed] [Google Scholar]
- Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- Bartolazzi A, Peach R, Aruffo A, Stamenkovic I. Interaction between CD44 and hyaluronan is directly implicated in the regulation of tumor development. J Exp Med. 1994;180:53–66. doi: 10.1084/jem.180.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum C, Werb Z. Focalized proteolysis: Spatial and temporal regulation of extracellular matrix degradation at the cell surface. Curr Opin Cell Biol. 1996;8:731–738. doi: 10.1016/s0955-0674(96)80116-5. [DOI] [PubMed] [Google Scholar]
- Bennett KL, Modrell B, Greenfield B, Bartolazzi A, Stamenkovic I, Peach R, Jackson DG, Spring F, Aruffo A. Regulation of CD44 binding to hyaluronan by glycosylation of variably spliced exons. J Cell Biol. 1995a;131:1623–1633. doi: 10.1083/jcb.131.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett KL, Jackson DG, Simon JC, Tanczos E, Peach R, Modrell B, Stamenkovic I, Plowman G, Aruffo A. CD44 isoforms containing exon v3 are responsible for the presentation of heparin-binding growth factors. J Cell Biol. 1995b;128:687–698. doi: 10.1083/jcb.128.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkedal-Hansen H. Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol. 1995;7:728–735. doi: 10.1016/0955-0674(95)80116-2. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA. Localization of matrix metalloproteinase mmp-2 to the surface of invasive cells by interaction with integrin αv β3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Silletti S, von Schalscha TL, Friedlander M, Cheresh DA. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell. 1998;92:391–400. doi: 10.1016/s0092-8674(00)80931-9. [DOI] [PubMed] [Google Scholar]
- Clement MV, Stamenkovic I. Fas and tumor necrosis factor receptor-mediated cell death: similarities and distinctions. J Exp Med. 1994;180:557–567. doi: 10.1084/jem.180.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culty M, Shizari M, Thompson EW, Underhill CB. Binding and degradation of hyaluronan by human breast cancer cell lines expressing different forms of CD44. J Cell Physiol. 1994;160:275–286. doi: 10.1002/jcp.1041600209. [DOI] [PubMed] [Google Scholar]
- Fotouhi N, Lugo A, Visnick M, Lusch L, Walsky R, Coffey JW, Hanglow AC. Potent peptide inhibitors of stromelysin based on the prodomain region of matrix metallopreinases. J Biol Chem. 1994;269:30227–30231. [PubMed] [Google Scholar]
- Garbisa S, Pozzatti R, Muschel RJ, Saffiotti U, Ballin M, Goldfarb RH, Khoury G, Liotta LA. Secretion of type IV collagenolytic protease and metastatic phenotype: Induction by transfection with c-Ha-ras but not c-Has-ras plus Ad2-Ela. Cancer Res. 1987;47:1523–1528. [PubMed] [Google Scholar]
- Gomis-Ruth F-X, Maskos K, Betz M, Bergner A, Huber R, Suzukl K, Yoshida N, Nagase H, Brew K, Bourenkov GP, Bartunik H, Bode W. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature. 1997;388:77–81. doi: 10.1038/37995. [DOI] [PubMed] [Google Scholar]
- Gunthert U, Hofmann M, Rudy W, Reber S, Zoller M, Haussmann I, Matzku S, Wenzel A, Ponta H, Herrlich P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991;65:13–24. doi: 10.1016/0092-8674(91)90403-l. [DOI] [PubMed] [Google Scholar]
- Herron GS, Banda MJ, Clark EJ, Gavrilovic J, Werb Z. Secretion of metalloproteinases by stimulated capillary epithelial cells. J Biol Chem. 1986;261:2814–2818. [PubMed] [Google Scholar]
- Jalkanen S, Jalkanen M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J Cell Biol. 1992;116:817–825. doi: 10.1083/jcb.116.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalkanen S, Joensuu H, Soderstrom KO, Klemi P. Lymphocyte homing and clinical behavior of non-Hodgkin’s lymphoma. J Clin Invest. 1991;87:1835–1840. doi: 10.1172/JCI115205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya G, Rodriguez I, Jorcano L, Vassalli P, Stamenkovic I. Selective suppression of CD44 in keratinocytes of mice bearing an antisense CD44 transgene driven by a tissue-specific promoter disrupts hyaluronate metabolism in the skin and impairs keratinocyte proliferation. Genes & Dev. 1997;11:996–1007. doi: 10.1101/gad.11.8.996. [DOI] [PubMed] [Google Scholar]
- Kitson RP, Appasamy PM, Nannmark U, Albertsson P, Gabauer MK, Goldfarb RH. Matrix metalloproteinases produced by rat IL-2-activated NK cells. J Immunol. 1998;160:4248–4253. [PubMed] [Google Scholar]
- Kleiner DE, Stetler-Stevenson WG. Structural biochemistry and activation of matrix metalloproteinase. Curr Opin Cell Biol. 1993;5:891–897. doi: 10.1016/0955-0674(93)90040-w. [DOI] [PubMed] [Google Scholar]
- Lesley J, Hyman R, Kincade P. CD44 and its interaction with extracellular matrix. Adv Immunol. 1993;54:271–335. doi: 10.1016/s0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Steeg PS, Stetler-Stevenson W. Cancer metastasis and angiogenesis: An imbalance of positive and negative regulation. Cell. 1991;64:327–336. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- Mignatti P, Rifkin D. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev. 1993;73:161–195. doi: 10.1152/physrev.1993.73.1.161. [DOI] [PubMed] [Google Scholar]
- Odake S, Morita Y, Morikawa T, Yoshida N, Hori H, Nagal Y. Inhibition of matrix metalloproteinase by peptidyl hydroxamic acids. Biochem Biophys Res Commun. 1994;199:1442–1446. doi: 10.1006/bbrc.1994.1392. [DOI] [PubMed] [Google Scholar]
- Ogata Y, Itoh Y, Nagase H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinase-1 complex by 4-aminophenylmercuric acetate and proteinases. J Biol Chem. 1995;270:18506–18511. doi: 10.1074/jbc.270.31.18506. [DOI] [PubMed] [Google Scholar]
- Okada Y, Naka K, Kawamura K, Matsumoto T, Nakanishi I, Fujimoto M, Sato H, Seiki M. Localization of matrix metalloproteinase-9 in osteoclasts: implication for bone resporption. Lab Invest. 1995;72:311–322. [PubMed] [Google Scholar]
- Pals S, Horst E, Ossekoppele G, Figdor C, Scheper R, Meijer C. Expression of lymphocyte homing receptor as a mechanism of dissemination in non-Hodgkin’s lymphoma. Blood. 1989;37:885–888. [PubMed] [Google Scholar]
- Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death-domain-containing receptor for TRAIL. Science. 1997;277:815–818. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- Peach RJ, Hollenbaugh D, Stamenkovic I, Aruffo A. Identification of hyaluronic acid binding sites in the extracellular domain of CD44. J Cell Biol. 1993;122:257–264. doi: 10.1083/jcb.122.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaper HW, Grant DS, Stetler-Stevenson WG, Fridman R, D’Orazi G, Murphy AN, Bird RE, Hoythya M, Fuerst TR, Frence DL, Quigley JP, Kleinman HK. Type IV collagenase(s) and TIMPs modulate endothelial cell morphogenesis in vitro. J Cell Physiol. 1993;156:235–246. doi: 10.1002/jcp.1041560204. [DOI] [PubMed] [Google Scholar]
- Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI. Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc Natl Acad Sci. 1992;89:12160–12164. doi: 10.1073/pnas.89.24.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton GR, Bell MV, Bell JI, Jackson DG. The identification of a new alternative exon with highly restricted tissue expression in transcripts encoding the mouse Pgp-1 (CD44) homing receptor: Comparison of all 10 variable exons between mouse, human, and rat. J Biol Chem. 1993;268:12235–12238. [PubMed] [Google Scholar]
- Seiter S, Arch R, Reber S, Komitowski D, Hofmann M, Ponta, H. H, Herrlich P, Matzku S, Zoller M. Prevention of tumor metastasis formation by anti-variant CD44. J Exp Med. 1993;177:443–455. doi: 10.1084/jem.177.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman L, Wainwright D, Ponta H, Herrlich P. A splice variant of CD44 expressed in the apical ectoderm ridge presents fibroblast growth factors to limb mesenchyme and is required for limb outgrowth. Genes & Dev. 1998;12:1058–1071. doi: 10.1101/gad.12.7.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman JP, Rudy W, Hofamnn M, Moll J, Herrlich P, Ponta H. Regulated clustering of variant CD44 proteins increases their hyaluronan binding capacity. J Cell Biol. 1996a;135:1139–1150. doi: 10.1083/jcb.135.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman JP, Arming S, Moll JF, Hekele A, Rudy W, Sherman LS, Kreil G, Ponta H, Herrlich P. Hyaluronan-independent metastatic behavior of CD44 variant-expressing pancreatic carcinoma cells. Cancer Res. 1996b;56:3134–3141. [PubMed] [Google Scholar]
- Southern JA, Young DF, Heaney F, Baumgartner W, Randall RE. Identification of an epitope on the P and V proteins of Simian virus 5 that distinguishes between two isolates with different biological characteristics. J Gen Virol. 1991;72:1551–1557. doi: 10.1099/0022-1317-72-7-1551. [DOI] [PubMed] [Google Scholar]
- Stamenkovic I, Aruffo A, Amiot M, Seed B. The hematopoietic and epithelial forms of CD44 are distinct polypeptide with different adhesion potentials for hyaluronan bearing cells. EMBO J. 1991;10:343–348. doi: 10.1002/j.1460-2075.1991.tb07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol. 1993;9:541–573. doi: 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- Sy MS, Guo Y-J, Stamenkovic I. Distinct effects of two CD44 isoforms on tumor growth in vivo. J Exp Med. 1991;174:859–866. doi: 10.1084/jem.174.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudbeck BD, Pilcher BK, Welgus HG, Parks WC. Induction and repression of collagenase-1 by keratinocytes is controlled by distinct components of different extracellular matrix compartments. J Biol Chem. 1997;272:22103–22110. doi: 10.1074/jbc.272.35.22103. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Adams DH, Hubscher S, Hirano H, Siebenlist U, Shaw S. T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1β. Nature. 1993;361:79–82. doi: 10.1038/361079a0. [DOI] [PubMed] [Google Scholar]
- Thomas L, Byers HR, Vink J, Stamenkovic I. CD44H regulates tumor cell migration on hyaluronan-coated substrate. J Cell Biol. 1992;118:971–977. doi: 10.1083/jcb.118.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TH, Werb Z. Gelatinase B: Structure, regulation, and function. In: Parks WC, Mecham RP, editors. Matrix metalloproteinases. San Diego, CA: Academic Press; 1998. pp. 115–148. [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber GF, Ashkar S, Glimcher MJ, Cantor H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1) Science. 1996;270:7437–7444. doi: 10.1126/science.271.5248.509. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Collier IE, Marmer BL, Eisen AZ, Grant GA, Goldberg G. SV40-transformed human lung fibroblasts secrete a 92kDa type IV collagenase which is identical to that secreted by normal macrophages. J Biol Chem. 1989;264:17213–17221. [PubMed] [Google Scholar]
- Yu Q, Toole BP. Biotinylated hyaluronan as a probe for detection of binding proteins in cells and tissues. Biotechniques. 1995;19:122–129. [PubMed] [Google Scholar]
- Yu Q, Toole BP, Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. J Exp Med. 1997;186:1985–1196. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Katoh S, He Q, Oritani K, Miyake K, Lesley J, Hyman R, Hamik A, Parkhouse RME, Farr AG, Kincade PW. Monoclonal antibodies to CD44 and their influence on hyaluronan recognition. J Cell Biol. 1995;130:485–495. doi: 10.1083/jcb.130.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]