

Abstract
Although a small decrease in survival and increase in tumor incidence was observed in ATR+/− mice, ATR−/− embryos die early in development, subsequent to the blastocyst stage and prior to 7.5 days p.c. In culture, ATR−/− blastocysts cells continue to cycle into mitosis for 2 days but subsequently fail to expand and die of caspase-dependent apoptosis. Importantly, caspase-independent chromosome breaks are observed in ATR−/− cells prior to widespread apoptosis, implying that apoptosis is caused by a loss of genomic integrity. These data show that ATR is essential for early embryonic development and must function in processes other than regulation of p53.
Keywords: ATR, ATM, p53, embryonic lethality, chromosome breaks
ATM and ATR are mammalian counterparts to a family of high molecular weight protein kinases conserved in a broad range of species including Schizosaccharomyces pombe, Saccharomyces cerevisiae, and Drosophila melanogaster (Keith and Schreiber 1995; Keegan et al. 1996; Cimprich et al. 1996; Canman et al. 1998). The genes most closely related to ATM and ATR are the MEC1 (S. cerevisiae) TEL1 (S. cerevisiae), RAD3 (S. pombe), and Mei-41 (D. melanogaster) genes. Each of these genes is involved in DNA damage responses and falls into two groups based both on homology and function. ATM is related most closely to TEL1, a gene that shares an overlapping role with MEC1 in checkpoint responses to γ-irradiation in S. cerevisiae (Morrow et al. 1995). ATR, on the other hand, is most closely related to RAD3, Mei-41, and MEC1 in descending order of similarity. The kinase domain of ATR is 61% and 53% identical to the kinase domains of RAD3 and Mei-41, respectively (Keith and Schreiber 1995; Cimprich et al. 1996; Keegan et al. 1996), whereas the ATM kinase domain shares only 39% identity with either RAD3 or Mei-41.
Recent studies indicate that ATR functions in DNA damage response pathways similar to those mediated by RAD3, Mei-41, and MEC1. RAD3, Mei-41, and MEC1 are required for checkpoint responses to pyrimidine dimer formation (UV radiation), DNA-alkylation (MMS), depletion of deoxyribonucleotides [hydroxyurea (HU)] and γ-irradiation (Weinert et al. 1994; Hari et al. 1995; Bentley et al. 1996). In addition to cell cycle checkpoint response, these genes have been implicated in the regulation of DNA repair (Weinert 1998). Consistent with the hypothesis that ATR is the functional mammalian homolog of RAD3, Mei-41, and MEC1, expression of a kinase inactive mutant of ATR (ATR–KI) sensitizes mammalian cells to these same forms of DNA damage and diminishes the G2/M checkpoint response induced by γ-irradiation (Cliby et al. 1998; Wright et al. 1998). Recently, this ATR–KI cell line has been shown to be deficient in the regulated phosphorylation of p53 in response to UV and γ-irradiation (Tibbetts et al. 1999). Because ATM-disrupted cells are deficient in regulating p53 levels in response to γ-irradiation but not in response to UV or MMS (Canman et al. 1994; Xu and Baltimore 1996), ATR and ATM may possess both overlapping and nonredundant roles in regulating p53 (Canman et al. 1998; Tibbetts et al. 1999).
Here, we describe the phenotype of mice deficient in ATR. Whereas ATR+/− mice display a small decrease in survival and increase in tumor incidence, ATR−/− embryos die early in development. Early embryonic lethality and observations of ATR−/− blastocysts cultured in vitro indicate that ATR has an essential role in the proliferation of early embryonic cells. In addition, we show that ATR−/− cultured blastocyst cells suffer chromosomal fragmentation, suggesting that that the early death of ATR−/− embryos is caused by a widespread loss of genomic integrity. Because ATM−/− and p53−/− mice do not display a similar phenotype, these data indicate that ATR must function in some manner that is not redundant with ATM and is independent of p53 regulation. Evidence in support of a role for ATR in regulation of the BRCA gene products and in S- to M-phase transition of early embryonic cells is discussed.
Results
ATR disruption
Targeted disruption of ATR was achieved by deletion of three exons encoding the translation initiating methionine and the following 90 amino acids (Fig. 1A). Homologous recombination of the targeting vector into the ATR gene introduces a neomycin resistance cassette containing a single EcoRV site (Fig. 1A). This EcoRV site was subsequently used for colony screening by Southern blot (Fig. 1B) and the genotype of positive clones was confirmed further by PCR (Fig. 1C). Chimeric mice originating from three different ES cell clones transmitted the ATR disruption to F1 offspring. Although quantitation of ATR mRNA in homozygous ATR-disrupted cells was not possible due to early embryonic lethality (below), ES cells and murine embryonic fibroblasts (MEFs) with a single ATR allele disrupted expressed 47% ± 7% (95% confidence interval) and 46% ± 10% less ATR mRNA, respectively (Fig. 1D). Truncated mRNA species in ATR+/− cells were not observed (Fig. 1D). Preliminary analysis of the effects of MMS, cisplatin, γ-irradiation, and HU on heterozygous ES cells and MEFs showed no significant differences in the survival of ATR+/+ and ATR+/− cells (data not shown).
Figure 1.

Targeted disruption of ATR. (A) A schematic of the wild-type locus, targeting vector, and recombined locus are shown. In the targeting vector and recombined locus, the neomycin selection cassette replaces the first three coding exons of ATR, including the translation initiation codon (exon 1). Homologous recombination of the targeting vector into the ATR locus introduces an EcoRV site, shortening a 33-kb wild-type EcoRV fragment to 20 kb. (B) Southern blot of DNA from ES cell colonies confirms the projected truncation of the EcoRV fragment by homologous recombination. (Lanes 1,3) EcoRV-digested DNA from wild-type ES cells, (lanes 2,4) ES cells with a single recombined ATR allele. (C) DNA samples from wild-type (lanes 1,3) and ATR+/− (lanes 2,4) ES cells were subjected to PCR to confirm genotyping as performed by Southern analysis. PCR products derived from wild-type and disrupted alleles are 216 and 590 bp, respectively. (D) Northern blot of poly (A)+ mRNA from ATR+/+ and ATR+/− MEFs. Embryos used to generate MEFs (passage 2) were isolated from ATR-disrupted mouse lines 1 and 2 (see Materials and Methods). Autoradiographic detection of ATR, FRAP, and β-actin transcripts are shown.
Increase in tumor incidence in ATR+/− mice
Although p53+/− mice exhibit decreased longevity and increased tumor incidence (Jacks et al. 1994), ATM+/− mice survive similarly to wild-type mice (Barlow et al. 1999; C. Barlow, pers. comm.). Because ATR and ATM are speculated to have both overlapping and nonredundant roles in regulating p53, we asked whether the longevity of ATR+/− mice is compromised. ATR+/− and ATM+/− mice (Xu and Baltimore 1996) were crossed to produce populations of ATR+/−, ATR+/−ATM+/−, and ATM+/− mice. Although no difference was observed in the survival of ATR+/− mice in comparison to ATR+/− ATM+/− mice, a decrease in the survival of both ATR+/− and ATR+/−ATM+/− mice was observed in comparison to that of ATM+/− mice. By 18 months, 10 of 48 ATR+/− (21%) and 8 of 41 ATR+/−ATM+/− mice (20%) had died in comparison to 1 of 22 ATM+/− mice (4.5%). Of the 10 ATR+/− and ATR+/−ATM+/− mice autopsied postmortem, 6 had evident tumors. The types of tumors observed were histiocytic sarcoma (one in ATR+/−; two in ATR+/−ATM+/−), large follicular center cell lymphoma (ATR+/−), gastric adenoma (ATR+/−ATM+/−), and sebaceous gland adenoma (ATR+/−).
To further examine the tumor incidence in these mice, autopsies were performed on the remaining ATR+/−, ATR+/− ATM+/−, and ATM+/− mice at 79–89 weeks of age [mean age = 83 ± 3 (s.d.) weeks]. In comparison to ATM+/− mice, a 4- and 2.6-fold increase in tumor incidence was observed in ATR+/− and ATR+/−ATM+/− mice, respectively. In total, 5 of 25 ATR+/− mice (20%), 4 of 30 ATR+/−ATM+/− mice (13%), and 1 of 21 ATM+/− mice (4.8%) had obvious tumors ranging from 1 to 3 cm in diameter. Comparison of the tumor incidence in ATM+/− mice with that observed in ATR+/− mice and in ATR+/− and ATR+/−ATM+/− mice combined was significant to P values of 0.050 and 0.045, respectively. The types of tumors observed were plasma cell lymphoma (ATM+/−), mixed follicular center cell lymphoma (two in ATR+/−; two in ATR+/− ATM+/−), hepatocellular adenoma (one in ATR+/−; one in ATR+/−ATM+/−), fibrous histiocytoma (ATR+/−), ovarian cystadenoma (ATR+/−), and ovarian fibroma (ATR+/−ATM+/−). Although Southern blot analysis of DNA extracted from ATR+/− and ATR+/−ATM+/− tumors did not indicate a loss of heterozygosity at the targeted region of ATR (data not shown), it is possible that alterations outside the targeted region may have occurred in these tumors given the large size of the ATR genomic locus (>60 kb). Together, these results indicate that in contrast to heterozygosity of the ATM gene, ATR heterozygosity causes a small decrease in survival and increase in tumor incidence.
ATR is required for early embryonic cellular proliferation
To generate homozygous ATR-disrupted mice, ATR+/− mice were intercrossed. However, of 225 progeny analyzed, no homozygous ATR-disrupted pups were observed, implying that ATR is essential for embryonic development. The stage at which ATR−/− embryos arrest in development was then determined by isolation of embryos from time-mated heterozygous crosses (Table 1). Although an abnormally high number of decidua contained embryos in the final stages of resorbtion, no viable ATR−/− embryos were observed at day 7.5 postcoitum (p.c.) or beyond. (Table 1). The high number of resorbed embryos, however, suggested that ATR−/− embryos survive earlier stages of development and die following implantion. To test whether ATR−/− embryos survive to the blastocyst stage, embryos were isolated 3.5 days p.c. and genotyped. Of 83 embryos genotyped, 20 were ATR−/−. Thus, ATR−/− embryos successfully develop to the blastocysts stage but die subsequently, prior to E7.5.
Table 1.
Genotypic ratios of embryos from ATR heterozygous intercrosses
| Days p.c.
|
Viable embryos (litters)
|
+/+
|
+/−
|
−/−
|
Resorbed
|
P values
|
|---|---|---|---|---|---|---|
| 10.5–14.5 | 23 (5) | 9 | 14 | 0 | N.D. | 0.003 |
| 8.5–9.5 | 23 (4) | 6 | 17 | 0 | 4 | 0.003 |
| 7.5 | 35 (5) | 11 | 24 | 0 | 12 | 0.0003 |
P values were calculated by one tailed z-test. (N.D.) Not determined.
To determine if the developmental defect in ATR−/− mice was due to an inability of cells to proliferate subsequent to the blastocyst stage, day 3.5 embryos were isolated from ATR+/− intercrosses and grown in culture for 6 days. After 1 day in culture, ATR+/+, ATR+/−, and ATR−/− blastocysts hatched from the zona pellucida and implanted onto the tissue culture plastic. At isolation and during the first 2 days in culture, the inner cell mass (ICM) of ATR−/− blastocysts was indistinguishable from that of ATR+/+ and ATR+/− blastocysts. However, while the ICM cells of ATR+/+ and ATR+/− embryos continued to expand throughout the 6-day culture period, ATR−/− ICM cells failed to expand subsequent to day 2 and invariably died by day 4 in culture (Fig. 2A). Only the nondividing trophoblastic giant cells (TGC) of ATR−/− embryos remained after 6 days in culture (Fig. 2A). Identical results were observed with ATR−/− blastocysts (n = 8) derived from an independent ATR-disrupted line (line 2, data not shown).
Figure 2.
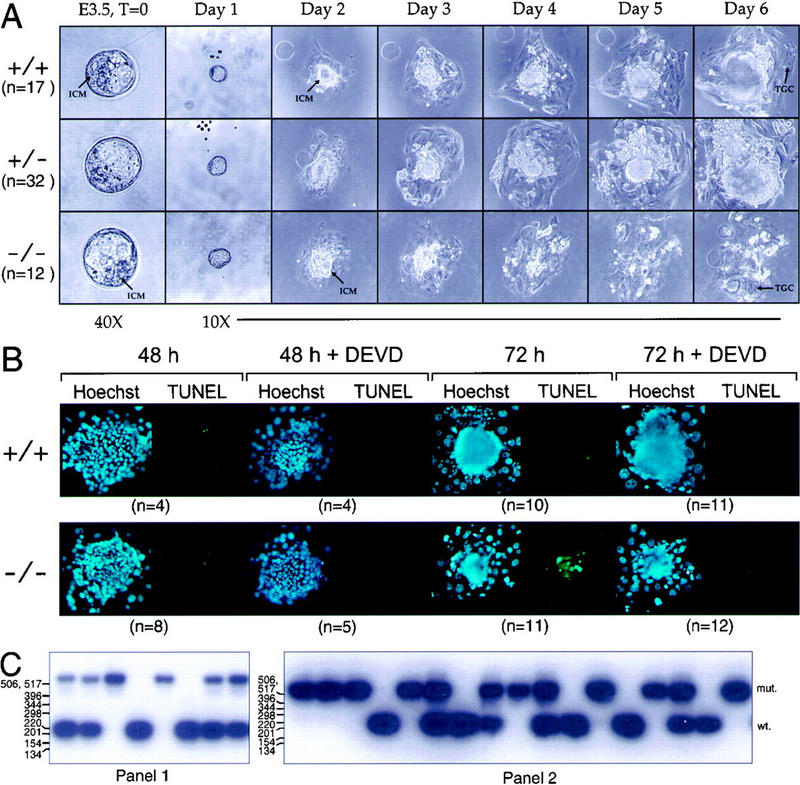
Deletion of ATR leads to apoptotic death of early embryonic cells. (A) Day 3.5 p.c. blastocysts were isolated from ATR+/− intercrosses and were cultured in 96-well plates for 6 days. ICMs and TGC are indicated. (B) TUNEL staining of ATR+/− and ATR−/− blastocysts cultured in the presence or absence of Z-DEVD-FMK. Blastocysts isolated from ATR+/− intercrosses were cultured in Terasaki-style microwell plates (Nunc) for 48 and 72 hr. Z-DEVD-FMK (200 μm) or vehicle was added after 24 hr of culture (for 48- and 72-hr time points) and added again after 48 hr (72-hr time point). Fluorescein (TUNEL) and Hoechst fluorescent images are shown. (C) Examples of PCR genotyping of blastocysts cultured as described in A and B are shown in panels 1 and 2, respectively.
The timing and cause of ATR−/− ICM cell death was then explored by TUNEL staining of blastocysts placed in culture for 48 or 72 hr. TUNEL staining detects extensive DNA fragmentation that results from apoptotic cell death. Although few TUNEL-stained cells were observed in ATR−/− blastocysts cultured for 48 hr, widespread TUNEL staining was observed after 72 hr (Fig. 2B). A majority of the TUNEL-stained ATR−/− cells was confirmed to be due to apoptosis through the use of the caspase 3-inhibitor Z-DEVD-FMK (Longthorne and Williams 1997). Caspase-3 is a protease required for initiation of apoptotic chromosome fragmentation following exposure of ES cells and other cell types to a vast array of DNA-damaging agents (Woo et al. 1998). The potent inhibitor of caspase-3, Z-DEVD-FMK, has been shown to inhibit Fas-induced apoptosis in T cells (Longthorne and Williams 1997). As shown, preincubation of ATR−/− blastocyst cells with Z-DEVD-FMK inhibited the TUNEL staining observed after 48 and 72 hr of culture (Fig. 2B). Overall, the number of TUNEL-stained cells in ATR−/− blastocysts was reduced 80% by preincubation with Z-DEVD-FMK. Consistent with caspase-3 knockout studies (Woo et al. 1998), Z-DEVD-FMK did not rescue ATR−/− cells from other morphological effects of apoptosis such as changes in nuclear morphology (Fig. 2B, day 3). These results demonstrate that ATR is essential for expansion of early embryonic cells in culture and that loss of ATR ultimately results in apoptotic cell death.
Chromosome fragmentation in ATR−/− cells
Given the potential role of ATR in DNA replication and damage checkpoints and DNA repair, we speculated that the apoptotic signal in ATR−/− ICM cells may be initiated by damaged DNA resulting from the lack of ATR. If so, one would expect that such damage would occur equally in the presence and absence of Z-DEVD-FMK. To examine this possibility, mitotic spreads were prepared from blastocysts grown for 48 hr in culture and then treated with nocodazole or left untreated for 6 hr. The number of mitotic cells in ATR−/− blastocyst cultures increased significantly upon treatment with nocodazole, indicating that ATR−/− cells attempt proliferation at day 2 in culture (Table 2). However, consistent with the hypothesis that disruption of ATR causes a loss of genomic integrity, chromosomal fragmentation was apparent in >60% of the mitotic spreads from ATR−/− cells and appeared equally with or without Z-DEVD-FMK pre-incubation (Table 2). Thus, the Z-DEVD-FMK-resistant chromosomal fragmentation observed in ATR−/− blastocysts at day 2 in culture (Fig. 3) correlates to and is the likely cause of the widespread caspase-dependent apoptosis observed at day 3 (Fig. 2B). As shown, chromosome fragmentation ranged from mild (Fig. 3A) to extensive (Fig. 3B). This extensive mitotic DNA fragmentation (Fig. 3B) may contribute to the residual caspase-independent TUNEL staining observed in the presence of Z-DEVD-FMK (discussed above); however, the degree of fragmentation observed in a majority of mitotic spreads was apparently beyond the limits of detection by TUNEL. Although we cannot rule out the possibility that the observed chromosome fragmentation in ATR−/− cells may result from early stages of a caspase-independent apoptotic process, these data are consistent with the hypothesis that the aborted development of ATR−/− embryos results from a loss of genomic integrity.
Table 2.
Mitotic index and analysis of mitotic spreads from blastocysts cultured for 2 days in vitro
| Genotypea
|
Nb
|
DEVD
|
Total cells/blastocystc
|
Percentc
|
|
|---|---|---|---|---|---|
| mitotic
|
fragmented
|
||||
| +/+ (4) | − | − | 164 ± 95 | 1 ± 1 | 0 |
| +/+ (6) | + | − | 111 ± 67 | 25 ± 10 | 0 |
| +/+ (5) | + | + | 100 ± 14 | 21 ± 8 | 0 |
| +/− (7) | − | − | 158 ± 57 | 1 ± 0.8 | 0 |
| +/− (7) | + | − | 115 ± 41 | 23 ± 11 | 2 ± 3 |
| +/− (6) | + | + | 142 ± 47 | 20 ± 8 | 0 |
| −/− (7) | − | − | 133 ± 41 | 3 ± 2 | 60 ± 45 |
| −/− (7) | + | − | 79 ± 24 | 12 ± 5 | 64 ± 22 |
| −/− (6) | + | + | 81 ± 47 | 13 ± 7 | 65 ± 21 |
The number of blastocysts analyzed per condition are indicated in parentheses.
Nocodazole.
Confidence intervals (95%) are shown following the ± symbol.
Figure 3.
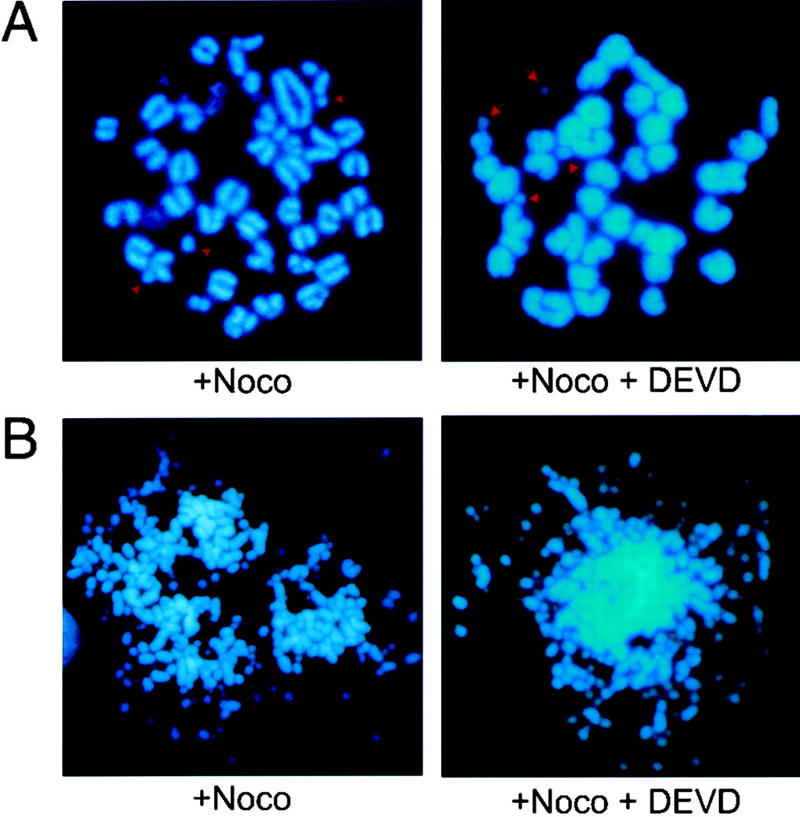
Chromosomal fragmentation in ATR−/− cells at day 2 in culture. Chromosome fragmentation was observed ranging from few (A) to many (B) breaks. This range of fragmentation was observed similarly in ATR−/− blastocysts cultured in the presence or absence of 200 μm Z-DEVD-FMK added after 24 hr of culture.
Discussion
We have found that disruption of the ATR gene leads to a small increase in tumorigenesis in heterozygotes and very early embryonic lethality in homozygotes. Although the total incidence of tumors in ATR heterozygotes was not as great as those observed in p53 heterozygotes (Jacks et al. 1994), the effect of ATR heterozygosity was statistically significant. It is interesting to note that the incidence of large benign tumors was particularly increased in ATR heterozygotes. This increase in benign tumors may indicate that deficiency in ATR has a greater effect on the rate of tumor initiation than on the rate of progression to malignancy. It is currently not known whether ATR defects exist in humans; however the region to which ATR maps in humans is a site of genetic alteration in lung cell carcinomas (Cimprich et al. 1996)
As described here, our data indicate that ATR has an essential role in preventing the occurrence of DNA damage early in embryogenesis. Although recent studies suggest that ATR regulates p53, defective regulation of p53 is unlikely to be the sole cause of such DNA damage, as p53−/− mice do not suffer a block in early embryonic development similar to that observed in ATR−/− mice. Based on previous studies and data provided in this paper, two plausible essential roles for ATR are apparent and are discussed here: regulation of the BRCA gene products and control of the S- to M-phase transition.
BRCA1 and BRCA2 regulation
Several lines of evidence have substantiated a link between ATR and the functions of BRCA1 and BRCA2. BRCA1−/−, BRCA2−/−, and RAD51−/− mice die early in development and have cellular proliferation defects similar to that observed in ATR−/− cultured blastocysts (Hakem et al. 1996; Lim and Hasty 1996; Sharan et al. 1997; Suzuki et al. 1997). Secondly, like ATR−/− cells, chromosome breaks are observed in RAD51−/−, BRCA1 exon 11-deleted, and BRCA2-truncated cells (Lim and Hasty 1996; Patel et al. 1998; Xu et al. 1999). Finally, the ATR homolog ATM recently has been shown to be required for the phosphorylation of BRCA1 in response to γ-irradiation but is dispensable for BRCA1 phosphorylation in response to HU, MMS, and UV (Scully et al. 1997; Cortez et al. 1999). Because ATR–KI expression renders cells sensitive to these later reagents (Cliby et al. 1998; Wright et al. 1998), it is possible that ATR and ATM both regulate BRCA1 phosphorylation, albeit in a DNA damage-specific manner. These genetics and biochemical similarities provide correlative evidence that the BRCA gene products may be dependent on ATR function.
S- to M-phase transition
A second plausible essential function for ATR is suggested by the similarity between the extensive chromosomal fragmentation in ATR−/− cells (Fig. 3B) and that observed in cells undergoing mitotic catastrophe. Mitotic catastrophe is caused by premature entry of cells into mitosis prior to completion of DNA synthesis and is characterized by a high degree of chromosomal fragmentation (Heald et al. 1993; Schlegel and Pardee 1986). The treatments and mutations that cause mitotic catastrophe are now recognized to influence a pathway that regulates DNA damage and replication checkpoints in mammalian cells and S. pombe (Heald et al. 1993; Russell 1998; Chan et al. 1999; Pines 1999). Importantly, in S. pombe this pathway requires RAD3, the closest known relative of ATR (Cimprich et al. 1996; Keegan et al. 1996). Although recent studies indicate ATR may have a redundant role with ATM in regulating this checkpoint pathway in response to γ-irradiation (Cliby et al. 1998; Matsuoka et al. 1998; Chaturvedi et al. 1999), an essential role for ATR in DNA replication checkpoint responses has been implied (Cliby et al. 1998; Chaturvedi et al. 1999; Sarkaria et al. 1999).
If ATR regulates a DNA replication checkpoint in mammalian cells, mitotic catastrophe in ATR−/− cells might be due to the lack of such a checkpoint combined with the extremely rapid cellular proliferation that occurs normally in the course of early embryonic development. Early embryonic cells proliferate with doubling times as short as 2 hr at day 6.5 p.c. (Snow 1977) and can completely lack a G2 phase (Aladjem et al. 1998). Such rapid proliferation indicates a high degree of precision in transition from S to M phase. Thus, the chromosomal fragmentation in ATR−/− cells might be due to an inability to coordinate this transition accurately, resulting in premature entry into mitosis. According to this analysis, ATR may be dispensable in cells that cycle through a more extensive G2 phase, but might be particularly essential in the early embryo to sense incomplete DNA replication and prevent mitotic catastrophe.
Materials and methods
Generation of the ATR targeted allele
A 17-kb genomic fragment encoding the amino terminus of murine ATR was cloned from a 129SVJ genomic library (Stratagene). The ATR targeting vector was constructed by subcloning the 2.9-kb SalI and 7.5-kb XbaI genomic fragments flanking the 6.5-kb ATR amino terminal encoding fragment into the pPNT vector (Fig. 1A). ATR-disrupted ES cells cells were generated in both D3 and J1 lines. A single targeted allele was observed in 9 of 310 isolated ES cell colonies. Isolated colonies were expanded and DNA was prepared by digestion in PK buffer (100 mm Tris at pH 8.0, 400 mm NaCl, 5 mm EDTA, 0.8% SDS), followed by extraction with one-third volume of saturated NaCl and precipitation in an equal volume of isopropanol. EcoRV fragments were separated on a 0.66% agarose gel and detected by Southern analysis using the indicated probe (Fig. 1A). ATR-disrupted lines 1 and 2 are derived from J1 and D3 ES cell lines, respectively. PCR genotyping was performed using a common primer, 121 bp 5′ of the initiating methionyl codon in exon 1 (5′-ttccgggaggagaattttggac-3′) in combination with primers discriminating wild-type exon 1-containing alleles (5′-cggcgactcgaagctggcgacgacgc-3′) and knockout alleles encoding the 3′ end of the neomycin resistance gene (5′-cagcgcatcgccttctatcgccttcttgac-3′). PCR was performed in 25-μl reactions with 1× PCR buffer (Boehringer Mannheim), 1.25 units of Taq polymerase, and 2% DMSO. Temperature cycling conditions were (1) 94°C for 4 min, (2) 94°C for 1 min, (3) 62°C for 2.5 min, and (4) 72°C for 2.5 min, cycling 33 times to step 2. Poly (A)+ mRNA from ES cells and day 14.5 MEFs was isolated, subjected to Northern blot analysis, and probed with the full-length human ATR cDNA (Cimprich et al. 1996). Normalization was performed upon subsequent probing with the full-length cDNAs for FRAP (Brown et al. 1994) and β-actin (Clontech). Levels of mRNA were quantitated by PhosphorImager readout (Molecular Dynamics), and 95% confidence intervals for the average reduction of normalized ATR mRNA levels were calculated by Student's t-test.
Survival of and tumors in ATR+/− mice
ATR+/− and ATM+/− of 129Sv and C57BL/6 mixed background were intercrossed to produce populations of ATR+/−, ATR+/−ATM+/−, and ATM+/− mice. Mice suffering from severe fighting wounds (∼10%) were excluded from further study. The percentages of deceased and euthanized mice from ATR+/− and ATR+/−ATM+/− populations were compared to that of ATM+/− by Kaplan–Meier analysis, and log-rank P values were calculated (Biostat 2000, Cupertino, CA). Individual comparison of ATR+/− and ATR+/−ATM+/− survival with ATM+/− survival was significant to P values of 0.067 and 0.097, respectively. Histopathology on formalin-fixed tissues was performed by the Research Animal Diagnostic and Investigative Laboratory (Columbia, MO). Test significance of difference in tumor incidence for animals autopsied 79–89 weeks of age was calculated by one tailed z-test.
Cultured blastocysts and TUNEL assays
Blastocysts were flushed from the uterus of ATR+/− females 3.5 days p.c. and washed five times in M2 media (Sigma) or PBS containing 5% FBS. Blastocysts were cultured for the times indicated in 50 μl (Fig. 2A) or 15 μl (Fig. 2B) of DMEM containing 15% FBS, 100 μm β-mercaptoethanol, 2 mm glutamine, 100 μm nonessential amino acids, and 1× penicillin/streptomycin (GIBCO–BRL). Z-DEVD-FMK (200 μm) or vehicle was added after 24 and 48 hr (72-hr time point) of culture. At 200 μm, Z-DEVD-FMK has been shown to completely prevent Fas-induced apoptosis in human cells without affecting cellular proliferation (Longthorne and Williams 1997). For TUNEL assay, blastocysts were fixed at the indicated times in 3% paraformaldyhyde/PBS for 30 min. Permeabilization and TUNEL assays were performed using the In Situ Cell Death Detection Kit, Fluorescein (Boehringer Mannheim) as per the manufacturer's instructions. For PCR genotyping, DNA was prepared by incubation of individual blastocysts with 1.5 μl of NSPK buffer [300 μg/ml proteinase K (Boehringer Mannheim), 100 mm KCl, 20 mm Tris at pH 8.0, 4 mm MgCl2, 0.9% NP-40, 0.9% Triton X-100] for 4 hr at 60°C, followed by incubation at 90°C for 30 min. The entire DNA isolate was used directly for PCR, which was performed as described for Figure 1C. PCR products were then Southern blotted and probed with a 32P end-labeled internal primer common to both wild-type and mutant PCR products (5′-gacctcgcggggctccgtcga-3′). Hybridized products were detected by autoradiography.
Preparation of mitotic spreads and genotyping
Blastocysts were grown in 96-well round-bottom plates as described (Fig. 2A) and treated with 200 μm Z-DEVD-FMK or vehicle (0.1% DMSO) after 24 and 48 hr in culture. At 48 hr in culture, blastocysts were treated with 2.5 μm nocodazole for 6 hr and subsequently processed for preparation of mitotic spreads. To suspend cells, blastocysts were washed twice in PBS and trypsinized in 30 μl of 0.25% trypsin/1 mm EDTA (GIBCO–BRL). Trypsin was then neutralized with 150 μl of culture media and, cells were tritriated and transferred to 200-μl micocentrifuge tubes. At this step, 60 μl of the cell suspension was transferred to a separate tube for PCR analysis (below). The remaining 120-μl cell suspension was washed once in PBS (250 g centrifugation), resuspended in 75 mm KCl, and incubated at room temperature for 20 min. Cells were then centrifuged at 500g, resuspensed in ice-cold 3:1 (vol/vol) methanol/acetic acid fixative, incubated at 4°C for 10 min, and centrifuged at 650g. The fixation steps were repeated once, and cells were resuspended in 15 μl of fixative and dropped onto prewarmed (37°C) glass slides. Slides were stained with Hoechst or Giemsa, and mitotic and interphase cells were counted. Confidence intervals (95%) were calculated by Student's t-test. For genotyping, cells (60 μl, above) were washed five times with PBS (250g centrifugation). All centrifugation steps in these procedures were for 5 min. The final cell pellet was resupended in 1.5 μl of NSPK buffer, and DNA was isolated and PCR genotyped as described above.
Acknowledgments
We are indebted to M. Scott and J. Harrison for invaluable assistance in the generation of knockout cells and chimeric ATR+/− mice. We also thank the following people for reagents, technical training, and helpful discussions: K. Cimprich, S. Schreiber, A. Koleske, C. Lois, J. Pomerantz, Y. Yamanashi, J. Baer, I. Stancovski, H. Chang, Y. Xu, S. Cherry, M. Meffert, and M. Porteus. We are grateful to P. Svec and L. Newman for laboratory assistance and L. Anonuevo, K. Owler, B. Kennedy, and S. Pease. E.J.B. was supported by a fellowship from the Cancer Research Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL baltimo@caltech.edu; FAX (626) 585-9495.
References
- Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R, Wahl GM. ES cells do not activate p53-dependent stress responses and undergo p53- independent apoptosis in response to DNA damage. Curr Biol. 1998;8:145–155. doi: 10.1016/s0960-9822(98)70061-2. [DOI] [PubMed] [Google Scholar]
- Barlow C, Eckhaus MA, Schaffer AA, Wynshaw-Boris A. Atm haploinsufficiency results in increased sensitivity to sublethal doses of ionizing radiation in mice. Nat Genet. 1999;21:359–360. doi: 10.1038/7684. [DOI] [PubMed] [Google Scholar]
- Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Canman CE, Wolff AC, Chen CY, Fornace AJ, Jr, Kastan MB. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54:5054–5058. [PubMed] [Google Scholar]
- Canman C E, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3 Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- Chaturvedi P, Eng WK, Zhu Y, Mattern MR, Mishra R, Hurle MR, Zhang X, Annan RS, Lu Q, Faucette LF, et al. Mammalian Chk2 is a downstream effector of the ATM-dependent DNA damage checkpoint pathway. Oncogene. 1999;18:4047–4054. doi: 10.1038/sj.onc.1202925. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of Brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- Hari KL, Santerre A, Sekelsky JJ, McKim KS, Boyd JB, Hawley RS. The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell. 1995;82:815–821. doi: 10.1016/0092-8674(95)90478-6. [DOI] [PubMed] [Google Scholar]
- Heald R, McLoughlin M, McKeon F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell. 1993;74:463–474. doi: 10.1016/0092-8674(93)80048-j. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bornson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Keegan KS, Holtzman DA, Plug AW, Christenson ER, Brainerd EE, Flaggs G, Bentley NJ, Taylor EM, Meyn MS, Moss SB, et al. The Atr and Atm protein kinases associate with different sites along meiotically pairing chromosomes. Genes & Dev. 1996;10:2423–2437. doi: 10.1101/gad.10.19.2423. [DOI] [PubMed] [Google Scholar]
- Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- Lim D-S, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longthorne VL, Williams GT. Caspase activity is required for commitment to Fas-mediated apoptosis. EMBO J. 1997;16:3805–3812. doi: 10.1093/emboj/16.13.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge S J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell. 1995;82:831–840. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- Patel KJ, Vu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- Pines J. Cell cycle. Checkpoint on the nuclear frontier. Nature. 1999;397:104–105. doi: 10.1038/16344. [DOI] [PubMed] [Google Scholar]
- Russell P. Checkpoints on the road to mitosis. Trends Biochem Sci. 1998;23:399–402. doi: 10.1016/s0968-0004(98)01291-2. [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- Schlegel R, Pardee AB. Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science. 1986;232:1264–1266. doi: 10.1126/science.2422760. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- Snow MHL. Gastrulation in the mouse: Growth and regionalization of the epiblast. J Embryol Exp Morphol. 1977;42:293–303. [Google Scholar]
- Suzuki A, de la Pompa JL, Hakem R, Elia A, Yoshida R, Mo R, Nishina H, Chuang T, Wakeham A, Itie A, et al. Brca2 is required for embryonic cellular proliferation in the mouse. Genes & Dev. 1997;11:1242–1252. doi: 10.1101/gad.11.10.1242. [DOI] [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes & Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T. DNA damage and checkpoint pathways: Molecular anatomy and interactions with repair. Cell. 1998;94:555–558. doi: 10.1016/s0092-8674(00)81597-4. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes & Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes & Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JA, Keegan KS, Herendeen DR, Bentley NJ, Carr AM, Hoekstra MF, Concannon P. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc Natl Acad Sci. 1998;95:7445–7450. doi: 10.1073/pnas.95.13.7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- Xu Y, Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes & Dev. 1996;10:2401–2410. doi: 10.1101/gad.10.19.2401. [DOI] [PubMed] [Google Scholar]