

Abstract
Papillomaviruses (PVs) comprise a large family of viruses infecting nearly all vertebrate species, with more than 100 human PVs identified. Our previous studies showed that a mutant chimera HPV18/16 genome, consisting of the upper regulatory region and early ORFs of HPV18 and the late ORFs of HPV16, was capable of producing infectious virus in organotypic raft cultures. We were interested in determining whether the ability of this chimeric genome to produce infectious virus was the result of HPV18 and HPV16 being similarly oncogenic, anogenital types and whether more disparate PV types could also interact functionally. To test this we created a series of HPV18 chimeric genomes where the ORFs for the HPV18 capsid genes were replaced with the capsid genes of HPV45, HPV39, HPV33, HPV31, HPV11, HPV6b, HPV1a, CRPV, and BPV1. All chimeras were able to produce infectious chimeric viral particles, although with lower infectivity than wild-type HPV18. Steps in the viral life cycle and characteristics of the viral particles were examined to identify potential causes for the decrease in infectivity.
1. INTRODUCTION
Papillomaviruses (PVs) comprise a family of small, nonenveloped, double-stranded DNA viruses that replicate only in differentiating and stratifying cutaneous or mucosal epithelia. More than 100 human PV (HPV) types and numerous animal PV types have been sequenced. HPVs have been classified into genera according to the sequence homology of the major capsid gene L1 (Bernard et al., 2006; de Villiers et al., 2004). Generally, membership in a genus also correlates with biological and pathological characteristics (Bernard et al., 2006; de Villiers et al., 2004).
The PV capsid is approximately 50–55 nm in diameter and has an icosahedral symmetry of T=7. The capsid is composed of 360 copies of the major capsid protein L1 organized into 72 capsomeres consisting of L1 pentamers. The position, structure and number of the minor capsid protein L2 in the capsid is unknown (Chen et al., 2000; Modis et al., 2002), however it has been suggested that there may be between 12 and 72 L2 proteins per capsid (Buck et al., 2008; Finnen et al., 2003; Trus et al., 1997; Volpers et al., 1994). Studies indicate that during infection the L2 protein is translocated into the nucleus independently of L1 and accumulates at the subnuclear structure, nuclear domain 10 (ND10) (Day et al., 1998; Florin et al., 2002a), where it induces a reorganization of the ND10 (Becker et al., 2003; Florin et al., 2002a; Florin et al., 2002b). Following the reorganization of ND10 by L2, the L1 and E2 viral proteins as well as viral genomes are targeted by L2 to relocate to these subnuclear regions (Day et al., 1998; Florin et al., 2002b). It has been suggested that this co-localization increases the local concentration of all of the necessary viral components involved in virion morphogenesis (Day et al., 1998). The targeting of L2 to ND10 may also facilitate the delivery of the viral genome to ND10 during primary infection to initiate viral transcription (Day et al., 2004). It is expected that additional interactions, direct and indirect, exist between viral nonstructural and structural genes. For example it has been shown that late viral life cycle functions are affected by the viral proteins E1^E4, E5 and E7 (Fang et al., 2006; Fehrmann et al., 2003; Flores et al., 2000; McLaughlin-Drubin et al., 2005; Nakahara et al., 2005; Peh et al., 2004; Wilson et al., 2005; Wilson et al., 2007). Little is known about what cellular players may contribute to the process of virion morphogenesis except that the chaperone protein Hsc70 transiently associates with the c-terminus of L2 at the ND10 subnuclear regions (Florin et al., 2004) and HSP70i promotes the nuclear localization of L1 (Song et al.), suggesting a function for these cellular proteins during viral assembly.
Roden, et al., using in vitro generated virus-like particles, demonstrated that HPV16 L2 and L1 capsid proteins expressed from a Semliki Forest viral vector could self-assemble capsid particles encapsidating BPV-1 genomes (Roden et al., 1996). This finding suggested that the capsid proteins of one PV type were capable of encapsidating the genome of another type. However, the potential for the capsid proteins of one PV type to package the genome of another PV type had not been tested in a physiologically relevant system where the mechanisms governing native PV replication are in control.
Chimeric genetic analysis systems have been used with other viruses to study viral infectivity, replication, transforming potential, immunity, and virulence factors. Often chimeric viruses are used to compare genes from one virus with homologous genes from a related virus to attempt to ascertain their similarities and differences. Using chimeric genetic systems one can assign a particular phenotype to a specific gene or sequence. The study of chimeric viruses may reveal relatedness between exchanged sequences via the ability of the foreign sequence to provide all the needed functions and interactions for the virus to replicate. Information gained from such studies can elucidate mechanisms of viral replication and pathology, and identify common mechanisms for therapeutic targeting. Chimeric viruses have been successfully used to genetically analyze the biology of many DNA viruses, including the JC virus, SV40, BK virus, herpesviruses, and adenoviruses (Bollag et al., 1989; Daniel et al., 1996; Eberle et al., 1997; Gall et al., 1996; Gall et al., 1998; Haggerty et al., 1989; Krasnykh et al., 1996; Lynch et al., 1994; Miyazawa et al., 1999; O’Neill et al., 1992; Roy et al., 1998; Sawada et al., 1994; Tavis et al., 1994; Telling and Williams, 1994; Trowbridge and Frisque, 1993; Vacante et al., 1989; Zabner et al., 1999).
Our laboratory has previously been successful in establishing in vitro organotypic raft culture systems capable of synthesizing native infectious HPV16 (McLaughlin-Drubin et al., 2004), HPV18 (Meyers et al., 1997), HPV31 (Meyers et al., 1992), HPV39 (McLaughlin-Drubin and Meyers, 2004), HPV45 (McLaughlin-Drubin et al., 2003), and a chimeric HPV18/16 virus (Chen et al.; Chen et al.; Meyers et al., 2002) in differentiating and stratifying host epithelial tissue. The extension of our raft culture system to studies on HPV18/16 capsid protein chimeras demonstrated for the first time the use of a viable chimeric virus system to study PV genetics under natural host replicating conditions (Meyers et al., 2002). Although few direct interactions between early and late PV gene products have been characterized, this study showed that the early proteins and their functions in the viral life cycle of HPV18 were functional when coupled with the late genes of HPV16 (Meyers et al., 2002).
Given that HPV16 capsid proteins can interact with HPV18 early genes to generate infectious virus, we address here whether such interactions are common among all PV types or whether productive interactions are based upon the relatedness of the two PV types, a phenomenon observed in other viral families. Relatedness could be defined in several ways including; genetic and evolutionary differences, disease association, or species and anatomic site tropism. To cover these different types of relatedness we created a series of intertypic, chimeric PV genomes. We generated cell lines from the intertypic, chimeric genomes and demonstrated that each genome was capable of immortalization. Furthermore, tissues grown from these cell lines differentiated and stratified appropriately. Surprisingly, each of the chimeric genomes generated infectious virus. These findings suggest the existence of conserved viral domains that could be targeted in the development of universal papillomavirus therapeutics.
2. MATERIALS AND METHODS
2.1. Plasmid construction
Using restriction digest and PCR technologies, the L2 and L1 ORFs from HPV18 were replaced with an unique BglII site creating the pHPV18L2/L1Δ construct (Meyers et al., 2002). PCR primers were designed to amplify the L2 and L1 ORFs of HPV45, 39, 33, 31, 11, 6b, 1a, CRPV, and BPV1, introducing BglII sites at both the 5′ and 3′ ends. Using the BglII sites, each L2/L1 PCR amplified sequence was ligated into the HPV18L2/L1Δ genome creating the following HPV18 L2/L1 chimeras; HPV18/45, HPV18/39, HPV18/33, HPV18/31, HPV18/11, HPV18/6b, HPV18/1a, HPV18/CRPV, HPV18/BPV1 (Fig. 1B). Full details of the methods used were described previously (Meyers et al., 2002). Primers used to amplify each L2/L1 ORF sequence are listed in the supplemental Table 1. Since the HPV18 genome sequence is cloned into the plasmid vector via an unique EcoRI site, and prior to electroporation the viral genomes are released from the vector sequences by digesting with EcoRI (McLaughlin-Drubin and Meyers, 2005; Meyers et al., 2002; Meyers et al., 1997), it was necessary to use site-directed mutagenesis to change EcoRI sites that existed within the L2/L1 ORFs of HPV39 (nt 6824) before they were cloned into HPV18. Additionally, three of the virus types contained BglII restriction sites within their L2/L1 ORFs, HPV1a (nt 5582 and 6358), BPV1 (nt 6945, 6249, and 6275) and CRPV (nt 5078), which were also changed using site directed mutagenesis to facilitate cloning into the unique BglII site of pHPV18L2/L1Δ. All restriction sites were removed by changing the restriction enzyme consensus sequence without changing the amino acid sequence of the virus. Primers used for site-directed mutagenesis are listed in supplemental Table 2. All constructs were analyzed by restriction enzyme digestion and sequenced to verify that they were correct.
FIGURE 1.

(A) Phylogenetic tree of PVs based on L1 sequence homology. PVs used in this study are bolded and boxed. The figure was adopted and modified (Bravo and Alonso, 2004). (B) Creation of chimeric PV recombinant plasmids. The areas representing the early genes (grey) and the late genes (black) of HPV18 are shown. The approximate positions of the ORFs are labeled. HPV18 was cloned into pBSSK(+) (dotted area) at the unique EcoRI site. The HPV18 late gene ORFs were removed and replaced with the corresponding sequences from the panel of PVs.
2.2. Keratinocytes and electroporation
Primary human foreskin keratinocytes (HFK) were isolated and maintained in culture as previously described (McLaughlin-Drubin et al., 2004; McLaughlin-Drubin et al., 2003). Keratinocyte lines were grown in 154 media (Cascade Biologics, Inc., Portland, OR). Each chimeric PV genome was electroporated into primary HFKs as previously described (McLaughlin-Drubin et al., 2003; Meyers et al., 2002; Meyers et al., 1997). Keratinocyte lines stably maintaining the chimeric PV genomes were grown in monolayer culture using E medium in the presence of mitomycin C-treated J2 3T3 feeder cells (McLaughlin-Drubin and Meyers, 2005; Meyers et al., 2002; Meyers et al., 1997). Multiple batches of HFKs were used for the electroporations of each PV chimeric mutant.
2.3. Organotypic raft cultures, histochemical analyses, and in situ hybridization
Organotypic raft cultures were grown as previously described (McLaughlin-Drubin and Meyers, 2005; Meyers et al., 1992; Meyers et al., 1997). Raft cultures were grown for 10 days, harvested, fixed in 10% neutral buffered formalin, and embedded in paraffin. Four-micrometer sections were cut and stained with hematoxylin and eosin as previously described (Mayer and Meyers, 1998; Meyers et al., 1992; Ozbun and Meyers, 1996; Visalli et al., 1997). Immunostaining was done using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. Keratin 10-specific monoclonal antibody (AM201-5M, BioGenex, San Ramon, CA) was used according to the manufacturer’s instructions. For in situ hybridizations, cloned HPV18 or HPV11 was removed from the vector sequences and labeled with Bio-11-dUTP by the random priming method using the Megaprime DNA Labeling System (Amersham, Piscataway, NJ). Probes were diluted at a concentration of 1 μg/ml in a 50% formamide hybridization cocktail containing 2.4 M NaCl, 0.04 M Tris {pH 7.4}, 0.002 M EDTA, 4 mg/ml BSA, 0.08% PVP, 0.08% ficoll, 0.6 mg/ml yeast tRNA and 0.08 M DTT. Paraffin sections were de-waxed, dehydrated and digested with 4 mg/ml pepsin in 0.1 N HCl. The tissues were then neutralized by washing in 95% ETOH with subsequent dehydration in 100% ETOH and allowed to air dry. The probe cocktail was applied and tissue and probe were simultaneously denatured at 95 °C for 6 min. Hybridization in a moist chamber was performed for 2 h at 37 °C. After a thorough wash in 2 X SSC, the hybridization probe was detected by incubation with an avidin-alkaline phosphatase conjugate, followed by colorimetric development in McGrady reagent. Slides were counterstained with nuclear fast red and coverslipped using permanent mounting media.
2.4. Southern blot hybridizations
Total cellular DNA was isolated from monolayer and raft cultures as previously described (Meyers et al., 2002; Meyers et al., 1992; Meyers et al., 1997; Ozbun and Meyers, 1998). Five micrograms of total cellular DNA was digested with either EcoRI, to linearize the chimeric viral genomes, or BglII, to separate the structural genes from the rest of the genome, or left undigested and then electroporated in a 0.8% agarose gel. DNA was transferred onto a GeneScreen Plus membrane (New England Nuclear Research Products, Boston, MA) and Southern blot hybridization performed as previously described (Meyers et al., 2002). The HPV-specific probes were prepared by gel purification of the 8 kb HPV cloned insert from recombinant vectors and labeled using the Random Primed DNA labeling kit (Roche Molecular Biochemicals, Indianapolis, IN). Labeled probe was purified with a Quick Spin Column for radiolabeled DNA purification (Roche Molecular Biochemicals). Blots were first probed with a complete HPV18 genomic probe and then they were stripped and reprobed with a complete HPV45, HPV39, HPV33, HPV31, HPV11, HPV6b, HPV1a, CRPV, or BPV1 genomic probe correlating to the PV chimera studied. Stripping of the membranes was done by placing the membranes in 0.1 × SSC−1% SDS and boiling for 1 h.
2.5. Infectivity and neutralization assays
Viral stocks were prepared as previously described (Alam et al., 2008; Bowser et al.; Chen et al.; Conway et al., 2009a; Conway et al., 2009b; McLaughlin-Drubin et al., 2005; McLaughlin-Drubin et al., 2003; Meyers et al., 2002).
The infectivity assay is based on an in vitro system described by Smith et al (Smith et al., 1993; Smith et al., 1995) and was performed as previously used in our lab (Alam et al., 2008; Bowser et al.; Chen et al.; Conway et al., 2009a; Conway et al., 2009b; McLaughlin-Drubin et al., 2005; McLaughlin-Drubin et al., 2003; Meyers et al., 2002) using the HaCaT cell line (kindly provided by Dr. Norbert Fusenig). Endpoint dilution titers were designated as the highest dilution of the wild-type/chimeric viral stock capable of infecting HaCaT cells and generating a detectable HPV18 early spliced E1^E4 mRNA species. A titer of 20 (dilution of the viral stock 1:20) is the lowest titer we are able to detect.
Infectivity neutralization assays were performed by infecting HaCaT cells with a 1:20 diluted viral sample that was preincubated with a panel of type-specific monoclonal antibodies (MAb) diluted 1:20 in HaCaT culture medium, as previously described (Chen et al.; Chen et al.; Conway et al., 2009a; Conway et al., 2009b; Conway et al.; McLaughlin-Drubin et al., 2005; McLaughlin-Drubin et al., 2004; McLaughlin-Drubin et al., 2003). The panel included HPV6b, 11, 16, 18, 31, 33, 45, CRPV, and BPV1 L1–reactive conformation-dependent monoclonal antibodies H6.N8, H11.B2, H16.V5, H18.J4, H31.A6, H33.B6, CRPV.4B, and B1.A1, respectively. All of these antibodies were previously characterized (Christensen et al., 1996a; Christensen and Kreider, 1991; Christensen and Kreider, 1993; Christensen et al., 1990; McLaughlin-Drubin et al., 2003). Neutralizing antibodies against HPV1a and HPV39 were not available. Following preincubation, HaCaT cells were infected with the virus-antibody mixture and incubated for 2 days as described (Smith et al., 1993; Smith et al., 1995). Total RNA was then extracted and the viral early spliced E1^E4 was detected as described above (Smith et al., 1993; Smith et al., 1995).
2.6. Virus titering
Three-raft/virus stocks were prepared by dounce homogenization in 500 μl Benzonase buffer (0.05 M Na-phosphate, 2 mM MgCl2) (Hogg et al., 1999). Titers were measured as previously described (Chen et al.; Chen et al.; Conway et al., 2009a; Conway et al., 2009b; Conway et al.; McLaughlin-Drubin et al., 2005; McLaughlin-Drubin et al., 2004; McLaughlin-Drubin et al., 2003).
2.7. ELISA
Stocks of chimeric viruses were used as antigens in ELISAs to assay for correct folding of the L1 capsid protein into conformationally correct structures. MAbs H45.L10, H33.B6, H31.A6, H18.J4, H16.V5, H11.B2, H6.N8, CRPV.4B, and B1.A1 were raised against L1-VLPs of HPV45, HPV33, HPV31, HPV18, HPV16, HPV11, HPV6b, CRPV, and BPV1, respectively (Christensen et al., 1996a; Christensen and Kreider, 1991; Christensen and Kreider, 1993; Christensen et al., 1990; McLaughlin-Drubin et al., 2003). Monoclonal antibodies directed against conformationally correct HPV1a and HPV39 were not available. Five μl of the viral stocks were put into 96-well plates, incubated overnight at 4 °C in neutral PBS buffer, washed and blocked for 2 h with 5% non-fat milk in PBS. Monoclonal antibody stocks were diluted 1:100 in blocking buffer and incubated for 1 h, followed by incubation with rabbit anti-mouse-AP (Pierce) at 1:1000 and developed with 1 mg/ml p-nitrophenyl phosphate substrate (Sigma). Wells were probed with an irrelevant MAb and this was used as a background measurement for comparison. Absorbance at 405 nm (A405) was measured using a microplate reader (ThermoLabsystems). The readings from wells probed with an irrelevant MAb were subtracted from readings from wells probed with capsid-specific MAbs to provide the final measurement for each chimeric PV.
All experiments have been reviewed by the Pennsylvania State University College of Medicine Biological Safety and Recombinant DNA Committee and comply with NIH Guidelines for Research Involving Recombinant DNA Molecules, and adhere to Biosafety Level practices and containment institutional policies. The studies were performed under biosafety assurances number CMM09-01p-2.5.
3. RESULTS
3.1. Construction of papillomavirus chimeras
The structural genes of HPV16 and nonstructural genes of HPV18 are able to interact to produce infectious chimeric viruses (Chen et al.; Chen et al.; Meyers et al., 2002). This ability to interact may correlate with the sequence homology of HPV16 and HPV18 structural genes. The amino acid sequences of HPV16 L1 and L2 are 73.7% and 64.0% similar, respectively, to those of HPV18 (Table 1A). HPV16 and HPV18 are also both mucosotropic viruses and have a similar pathological spectrum. Since the Family Papillomaviridae contains a large group of biologically and pathologically diverse viruses, we tested the hypothesis that there may be incompatibility between structural and nonstructural genes of more evolutionarily diverse PV types. We constructed a panel of PV chimeras to test this hypothesis (Figure 1B). This panel of PV chimeras was generated in the context of an HPV18 backbone to facilitate comparisons with our original HPV18/16 chimera. We replaced the HPV18 structural genes with the structural genes of a wide range of evolutionarily diverse PVs.
TABLE 1A.
L1 homology
| HPV type | Identity (nt) to HPV18 | Similarity (aa) to HPV18 |
|---|---|---|
| 18 | 100% | 100% |
| 16 | 60.1% | 73.7% |
| 31 | 57.5% | 71.5% |
| 33 | 57.5% | 69.3% |
| 6b | 55.7% | 69.9% |
| 11 | 54.3% | 68.5% |
| 39 | 68.0% | 76.4% |
| 45 | 77.4% | 82.8% |
| 1a | 43.9% | 59.4% |
| CRPV | 45.2% | 59.0% |
| BPV1 | 42.2% | 58.5% |
The PV types chosen for inclusion in our panel of chimeras were picked because they permitted comparisons between high-risk and low-risk types, mucosal and cutaneous types, and human versus animal tropic papillomaviruses. The PVs chosen for analysis here included HPV39 and HPV45 since they are both high-risk, mucosal viruses that belong to the same species and are pathologically closely related to HPV18. We also tested HPV31 and HPV33 because they are both high-risk, mucosal viruses from the same species and are pathologically more closely related to HPV16. In contrast with these high-risk viruses, we also tested HPV6b and HPV11 which are both low-risk, mucosal viruses that induce benign lesions and have different biological and pathological activities than those of HPV16 and HPV18. We also analyzed HPV1a which is a low-risk, cutaneous HPV that causes benign plantar and other common warts. Finally, we tested two animal PVs, cottontail rabbit papillomavirus (CRPV) and bovine papillomavirus (BPV1). The evolutionary diversity of these viruses is represented in the phylogenetic tree presented in Figure 1A. We designated the resulting chimeric mutants as HPV18/39, HPV18/45, HPV18/31, HPV18/33, HPV18/6b, HPV18/11, HPV18/1a, HPV18/CRPV, and HPV18/BPV1. These mutant constructs are diagrammed in Figure 1B.
3.2. Development of cell lines maintaining episomal copies of chimeric genomic PV DNA
Each of the chimeric genomes (Figure 1B) was introduced into primary human foreskin keratinocytes (HFKs) by electroporation, using standard protocols (McLaughlin-Drubin et al., 2005; McLaughlin-Drubin et al., 2004; McLaughlin-Drubin and Meyers, 2004; McLaughlin-Drubin and Meyers, 2005; McLaughlin-Drubin et al., 2003; Meyers et al., 2002; Meyers et al., 1997). To account for potential host differences, the mutant PV genomes were electroporated into multiple batches of HFKs and multiple cell lines for each chimera were selected by immortalization, as measured by greater than 20 population doublings. HFKs lacking PV genomes could not be immortalized and senesced after 6–8 population doublings. Southern blots were performed for each of the cell lines and were first probed with HPV18-specific radiolabeled probes. The blots were then stripped and reprobed with a probe specific for the PV from which the L2/L1 open reading frames came. All cell lines stably maintained chimeric genomes episomally (Figure 2, lane 1). Samples digested with EcoRI and probed with HPV18 or chimeric PV demonstrated a linear band consistent with the PV genome length of ~8kb (Figure 2, lane 2). Samples digested with BglII and probed with HPV18 revealed a band consistent with the size of the URR and early region of HPV18 (4,887 nt) (Figure 2, lane 3, top panels). The samples that were digested with BglII and probed with the chimeric PV sequence generated a band consistent with the length of the capsid open reading frames (Figure 2, lane 3, bottom panels). This analysis demonstrated that each chimeric viral DNA genome was maintained episomally and that the chimeric structure was preserved. We were unable to detect integrated PV DNA in these cell lines; the nicked band (Form II) migrated well below where the chromosomal band appears on the ethidium bromide stained agarose gel (data not shown). However, integration may still occur below our sensitivity of detection. The efficiency of the chimeric PV genomes for developing cell lines capable of stable episomal maintenance was 100%, the same as we achieve with wild-type HPV18 (Table 2).
FIGURE 2.

Southern blot hybridizations of DNA from chimeric PV infected cell lines grown in undifferentiated monolayer cultures. Each chimeric PV cell line was analyzed for the episomal maintenance, copy number, and integrity of the chimeric PV genome. The blot was first probed with an HPV18-specific probe (top panels), stripped and reprobed with the chimeric PV-specific probe (bottom panels). Lanes 1, undigested DNA form I supercoiled (FI) and form II nicked (FII) DNA. Lanes 2, DNA digested with EcoRI to linearize the viral genome (FIII). Lanes 3, DNA digested with BglII to separate the HPV18 sequences (early ORFs) from the chimeric PV sequences (late ORFs). HPV18 genomic 100- and 10-copy number standards (left side) and the chimeric PV genomic 100- and 10-copy number standards (right side) are shown. Note extensive nicking of the supercoiled DNA during DNA isolation from tissues has converted the majority of the supercoiled viral genomes to the nicked form.
TABLE 2.
| Independently derived immortalized cell linesa | Genome amplification b | |
|---|---|---|
| HPV18/45 | 6/6 | NO |
| HPV18/39 | 3/3 | YES |
| HPV18/33 | 3/3 | YES |
| HPV18/31 | 3/3 | YES |
| HPV18/11 | 3/3 | NO |
| HPV18/6b | 3/3 | NO |
| HPV18/1a | 7/7 | NO |
| HPV18/CRPV | 3/3 | NO |
| HPV18/BPV1 | 4/4 | NO |
The number of established chimeric PV cell lines over the number of attempts we electroporated primary HFK to establish a cell line with a particular chimeric PV.
Genome amplification determined by comparing undifferentiated monolayer and differentiated raft cultures side-by-side by using Southern blot analysis to identify any increase in the amount of viral DNA seen in differentiated host tissue.
Multiple replicates of each of the chimeric cell lines were made, grown in organotypic raft cultures, and analyzed. Unless otherwise stated, the results of the replicates were reproducible.
3.3. Morphology and differentiation of chimeric PV cell lines grown in organotypic raft culture
Having established that each chimeric cell line was made, we then examined the effects, if any, of the chimeric PVs on tissue differentiation and morphology. Each of the established chimeric PV cell lines and wild-type HPV18 was grown as a stratified and differentiated epithelial tissue in organotypic raft culture. These organotypic raft culture tissues were sectioned and stained with hematoxylin and eosin to observe tissue morphology. Figure 3A shows representative results for wild-type HPV18 and each of the chimeric mutants. All of the chimeric mutants displayed morphologies that were similar to the wild-type HPV18 tissue and were consistent with what we typically observe for tissues infected with wild-type HPVs (McLaughlin-Drubin et al., 2004; McLaughlin-Drubin et al., 2003; Meyers et al., 2002; Meyers et al., 1992; Meyers et al., 1997). Tissue sections from each of these rafts were also stained with the differentiation marker keratin 10 (Figure 3B). The expression of keratin 10 indicated that the differentiation program of the chimeric tissues resembled that of wild-type HPV18 tissue and was consistent with what we have observed with other wild-type HPVs (McLaughlin-Drubin et al., 2003; Meyers et al., 1992; Meyers et al., 1997).
FIGURE 3.


Immunohistochemical analyses of chimeric PV-infected tissues. Chimeric infected, fully stratified and differentiated raft culture tissue sections were stained with (A) hematoxylin and eosin or (B) immunostained with a keratin 10-specific MAb (AM201-5M, BioGenex).
3.4. Amplification of chimeric viral genomic DNA in cell lines grown in organotypic raft cultures
Once we had established that each of the chimeric cell lines could be grown in organotypic raft cultures, we analyzed them for amplification of their genomes. Viral genome amplification is an important step in the PV life cycle and is believed to occur in differentiating host tissues in preparation for encapsidation. We have previously shown that the HPV18/16 chimeric virus is capable of genome amplification in the context of organotypic raft cultures (Meyers et al., 2002). We therefore asked whether the panel of diverse chimeras generated here would also be capable of genome amplification. Total DNA was isolated from monolayer cultures and organotypic raft cultures of each chimeric cell line and Southern blot analysis was performed as before (Meyers et al., 2002). Genome amplification was described as an increase in the amount of viral DNA between undifferentiated monolayers and differentiated raft cultures. We observed that HPV18/39, HPV18/33 and HPV18/31 were able to amplify their chimeric genomes in differentiated organotypic raft cultures while the HPV18/45, HPV18/11, HPV18/6b, HPV18/1a, HPV18/BPV1, and HPV18/CRPV cell lines did not amplify their genomes (Figure 4 and Table 2).
FIGURE 4.
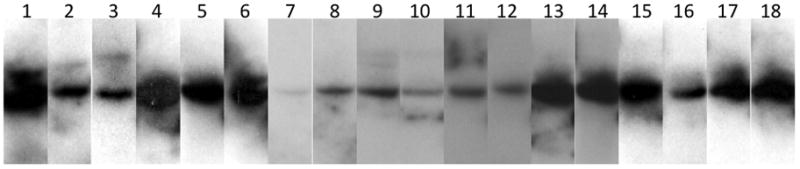
Southern blot analyses of chimeric PV genome amplification. Each chimeric PV cell line was analyzed for genome amplification during differentiation of the host tissue. The blot was probed with an HPV18-specific probe. Odd numbered lanes: DNA isolated from undifferentiated monolayer cells and digested with EcoRI to linearize the viral genome. Even numbered lanes: DNA isolated from differentiated raft culture tissues and digested with EcoRI to linearize the viral genome. Lanes 1–2, HPV18/45; Lanes 3–4, HPV18/39; Lanes 5–6, HPV18/33; Lanes 7–8, HPV18/31; Lanes 9–10, HPV18/11; Lanes 11–12, HPV18/6b; Lanes 13–14, HPV18/1a; Lanes 15–16, HPV18/CRPV; Lanes 17–18, HPV18/BPV1.
To confirm that some chimeric PV genomes did not amplify in the context of differentiated organotypic raft cultures, we selected a few of the mutants to perform in-situ hybridization to support the Southern blot findings. Positive control hybridizations were performed with wild-type HPV11 infected xenograft tissue (Unger et al., 1986) and wild-type HPV18 raft tissue. Both controls demonstrated hybridization, indicative of genome amplification (Figure 5). In contrast, HPV18/11, HPV18/CRPV, and HPV18/45 chimeric PV genomes showed no signs of genome amplification (Figure 5). HPV18/BPV1 showed only rare evidence of genome amplification by in-situ hybridization. The results of our Southern analysis and in-situ hybridization indicated that the genome amplification of chimeric HPV18/45, HPV18/11, HPV18/6b, HPV18/1a, HPV18/BPV1, and HPV18/CRPV was affected by the introduction of the corresponding structural open reading frames.
Figure 5.

In situ hybridization analysis for the presence of chimeric PV DNA amplification in differentiating raft tissues harboring chimeric PV genomes. (A) wild-type HPV11 xenograft tissue, (B) wild-type HPV18, (C) HPV18/45, (D) HPV18/11, (E) HPV18/CRPV, (F) HPV18/BPV1. Arrows indicate cells stained positive for genome amplification. HPV18/45, HPV18/11, and HPV18/CRPV showed no positive staining, whereas HPV18/BPV1 showed only one positively stained cell.
3.5. Analysis of viral titers and infectivity
Knowing that the introduction of foreign PV structural genes into the HPV18 backbone could impede genome amplification, we reasoned that the chimeric genomes could also vary in their ability to perform other aspects of the PV life cycle. For that reason, we assessed the ability of the chimeric genomes to assemble capsids and infect host cells. Virus stocks of wild-type HPV18 and each chimeric PV were treated with the endonuclease Benzonase to remove exogenous DNA. After Benzonase treatment, the encapsidated PV genomes were extracted from their capsids according to the protocol described in the Materials and Methods. The number of protected genomes was measured by qPCR based upon amplification of a fragment within the HPV18 E2 open reading frame. A standard curve was also generated by amplifying serially-diluted pBSHPV18 copy number controls. The exact number of endonuclease-resistant viral genomes was determined by comparing experimental values to the copy control standard curve and is presented in Table 3. Viral titers were measured for three Benzonase-resistant viral genome preparations obtained from three independent cell lines to confirm reproducibility.
Table 3.
Viral infectivity and titer.
| Virus | Infectivitya | Titera,b |
|---|---|---|
| HPV18 | 1,000 | 3.1 |
| HPV18/45 | 20 | 2.7 |
| HPV18/39 | 100 | 3.8 |
| HPV18/16 | 100 | 13.7 |
| HPV18/33 | 20 | 4.4 |
| HPV18/31 | 20 | 330.2 |
| HPV18/11 | 20 | 4.9 |
| HPV18/6b | 100 | 29.2 |
| HPV18/1a | 20 | 6.02 |
| HPV18/CRPV | 20 | 3.6 |
| HPV18/BPV-1 | 20 | 67.9 |
Endpoint Dilution titers measured from three raft tissues.
Encapsidated viral genomes (×106)
The capsid chimera titers were compared with wild-type HPV18 to assess the relative effects of mutation on the ability to encapsidate the viral genomes (Figure 6). Interestingly, five of the capsid chimeras (HPV18/45, HPV18/39, HPV18/33, HPV18/11, and HPV18/CRPV) encapsidated their genomes with nearly the same efficiency as wild-type HPV18, while the other five capsid chimeras (HPV18/1a (2-fold), HPV18/16 (4-fold), HPV18/6b (9-fold), HPV18/BPV-1 (21-fold), and HPV18/31 (105-fold)) encapsidated their genomes more efficiently than wild-type HPV18 (Figure 6). When compared with the ability of the capsid chimeras to amplify their genomes (Table 2 and Figures 4,5), these data indicated that the efficiency of genome encapsidation could not be correlated with genome amplification. The data demonstrated a vast difference in the ability of capsids from the different PV types to encapsidate the viral genomes.
FIGURE 6.
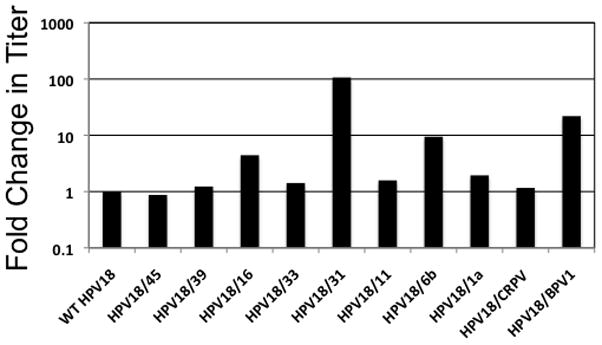
Relative virus titers. Quantification of the amounts of encapsidated chimeric PV genomes by qPCR was performed as described in the Materials and Methods. Each endonuclease-resistant viral genome preparation was analyzed in triplicate. In addition, three endonuclease-resistant viral genome preparations obtained from three independent cell lines were analyzed for each mutant virus. Data are presented as a fold change in titer compared with wild-type HPV18.
To measure infectivity, we performed endpoint dilution infectivity assays. We grew wild-type HPV18 and each chimeric cell line in organotypic raft cultures and generated virus stocks. HaCaT cells were incubated with 1:20, 1:100, and 1:1000 dilutions of wild-type HPV18 or chimeric virus stock for 48 hours. Surprisingly, each capsid chimera generated infectious virus (Table 3). Wild-type HPV18 was infectious at a dilution of 1:1000; HPV18/6b, HPV18/39, and HPV18/16 were infectious at a dilution of 1:100; and HPV18/33, HPV18/31, HPV18/11, HPV18/45, HPV18/1a, HPV18/CRPV, and HPV18/BPV1 were infectious at a dilution of 1:20 (Table 3). We had hypothesized that one or more of the chimeric PV generated here would be unable to propagate infectious virus due to incompatibilities between the structural and nonstructural genes of diverse types, especially between a human PV type (HPV18) and the two animal-specific types (CRPV and BPV1). However, our results clearly showed that regardless of this diversity, all PV chimeras were able to produce infectious virus.
3.6. Neutralization assays
None of the capsid chimeras that we have generated have yielded equivalent infectivity with wild-type HPV18 and yet half of these chimeras generated more encapsidated genome particles than wild-type HPV18. One possible explanation for this discrepancy could be that the combination of HPV18 early gene backbone with foreign structural genes induced structural changes that affected infectivity. To determine whether structural changes took place, we performed neutralization assays using available PV-specific antibodies. Neutralizing antibodies against HPV1a and HPV39 were unavailable. The conformation-specific MAbs H11.B2, H31.A6, H45.L10, CRPV.4B, H33.B6, H6.N8, and B1.A1, specific for the L1 proteins of HPV11, HPV31, HPV45, CRPV, HPV33, HPV6b, and BPV1, respectively, were used for neutralization assays (Christensen et al., 1996a; Christensen and Kreider, 1991; Christensen and Kreider, 1993; Christensen et al., 1990). Virus stocks were generated for each of these PV chimeras and were preincubated with the corresponding type-specific MAbs at 37 °C for one hour at a 1:1 volume ratio (viral stock:MAb). HaCaT cells were then incubated with the virus-antibody mixture for 48 hours. Total RNA was harvested and nested RT-PCR was performed as previously described (Meyers et al., 2002). The chimeric viruses of HPV18/31, HPV18/11, HPV18/45, HPV18/CRPV, and HPV18/BPV1 were effectively neutralized by their corresponding MAbs (Figure 7). Neither HPV18/33 nor HPV18/6b could be neutralized by the type-specific MAbs used here (Figure 7). The MAbs H6.N8 (HPV6b) and H33.B6 (HPV33) have not been tested previously with native viruses. Our results indicate that they may not be neutralizing antibodies in an infectious assay. Alternatively, minor changes in the structure of these chimeras may have precluded neutralization by the MAbs used.
FIGURE 7.

Neutralization analyses of chimeric PV. Neutralization of chimeric HPV18/31 (lanes 1 and 2), HPV18/11 (lane 3), HPV18/45 (lane 4), HPV18/CRPV (lane 5), HPV18/BPV1 (lane 6), HPV18/33 (lane 7) and HPV18/6b (lane 8) with MAbs H31.A6, H11.B2, H45.L10, CRPV.4B, B1.A1, H33.B6 and H6.N8, respectively. Shown is a 2% agarose gel of nested RT-PCR-amplified HPV 18 E1^E4 and β-actin. The arrowhead indicates β-actin and the arrow indicates HPV18 E1^E4 amplified products. The chimeric PV dilution was 1:20 and each MAb was diluted 1:20.
3.7. ELISA
Infectivity, genome encapsidation, and virus neutralization assays each suggested possible changes in capsid structure for some of the PV chimeras. To further examine the capsid structures of each chimera, an ELISA was performed with the conformation-dependent MAbs H45.L10, H33.B6, H31.A6, H18.J4, H16.V5, H11.B2, H6.N8, CRPV.4B, and B1.A1 which are specific for the L1 proteins of HPV45, HPV33, HPV31, HPV18, HPV16, HPV11, HPV6b, CRPV, and BPV1, respectively (Christensen et al., 1996a; Christensen and Kreider, 1991; Christensen and Kreider, 1993; Christensen et al., 1990). Each chimeric PV was used as an antigen and wild-type HPV18 served as a positive control. Wells probed with an irrelevant MAb were included and used to subtract background signal. For HPV18, HPV18/45, HPV18/31, HPV18/16, HPV18/33, HPV18/CRPV, and HPV18/BPV1, the HPV11-reactive MAb H11.B2 was used as the irrelevant background control. For HPV18/6b and HPV18/11, the HPV16-reactive antibody H16.V5 was used as the irrelevant background control. H16.V5 was chosen for a second background control to eliminate specific reactivity between the MAb H11.B2 and HPV18/11 and was also used to eliminate cross-reactivity with HPV18/6b (Christensen et al., 1996b). The ELISA measurement for each PV was defined as the difference between wells probed with a PV-specific MAb and the irrelevant MAb control. All chimeric PV particles reacted with their PV-specific MAb (Figure 8). The abundance of each PV sample loaded per well and the fact that antibodies with different specificities were used to probe the samples are both inherent limitations of this ELISA, making quantitative comparisons among the samples difficult. However, these data demonstrated that each MAb could recognize its virus-specific target, indicating that any capsid structural changes that existed were insufficient to prevent antibody/antigen recognition in this assay.
FIGURE 8.

Relative binding of chimeric PV-specific MAbs to chimeric PV particles by ELISA assay. Reading for each chimeric PV was defined as the difference between wells probed with a chimera-specific and an irrelevant MAb. Values are means + standard error bars of the means (S.E.M.)
4. DISCUSSION
The study of protein chimeras can provide important information on protein structure and function, particularly as a tool to study viral infection and replication (Belnap et al., 1996; Daniel et al., 1996; Gall et al., 1996; Gall et al., 1998). For example, using chimeric JC viruses in which the JC virus early region was replaced with the corresponding sequence from the SV40 genome helped to identify functional sequences required by JC virus for host cell transformation (Bollag et al., 1989). In addition, a JCV-SV40 chimeric viral genome that contains the regulatory region and the early genes of SV40 and the late structural genes of JCV was studied (Chen and Atwood, 2002). The resulting chimeric virus induced an SV40-like cytopathic effect in human glial cells while maintaining the host range of JCV, suggesting that the interactions between the virus capsid and host cell receptors contribute to JCV tropism (Chen and Atwood, 2002). Here, we show that a chimeric approach can be applied to investigate the functional, biological and evolutionary similarities and dissimilarities among PVs.
PVs only replicate in terminally differentiating keratinocytes, therefore our lab has developed a raft culture system to mimic in vivo differentiating host tissues which we have used to produce infectious PV in vitro (Bodily and Meyers, 2005; McLaughlin-Drubin et al., 2004; Meyers et al., 2002; Meyers et al., 1997; Ozbun and Meyers, 1997; Ozbun and Meyers, 1998; Sen et al., 2004; Sen et al., 2002). Using this raft culture system, we have previously shown that the HPV18 L2/L1 sequence could be functionally replaced with the HPV16 L2/L1 sequence as measured by the generation of infectious chimeric virus (Meyers et al., 2002). The amino acid sequence of the HPV16 L2/L1 region is 64–74% similar to the HPV18 L2/L1 region and this similarity may explain why the structural genes appear to be interchangeable between the two types (Meyers et al., 2002). However, we hypothesized that there would be incompatibility between structural and nonstructural genes of more evolutionarily diverse PV types. To test our hypothesis, we investigated the L2 and L1 genes from a group of PVs displaying different degrees of relatedness to HPV18, including PVs from four genera, six species and with tropism for three animal hosts (de Villiers et al., 2004). HPV18, HPV39 and HPV45 are human viruses of the Alpha genus, species 7. HPV16, HPV33 and HPV31 are human viruses of the Alpha genus, species 9 (de Villiers et al., 2004). HPV6b and HPV11 are human viruses of the Alpha genus, species 10 (de Villiers et al., 2004). HPV1a is a human virus of the Mu genus, species 1 (de Villiers et al., 2004). CRPV is a rabbit virus of the Kappa genus (de Villiers et al., 2004). BPV1 is a virus of cattle, belonging to the Delta genus, species 4 (de Villiers et al., 2004).
We created a panel of PV chimeras and established cell lines for each mutant construct. Studying these PV chimeras, we found that the late genes did not appear to affect immortalization or episomal genome maintenance. In addition, tissue morphology of cell lines containing each PV chimera was similar to that of wild-type HPV18. When we examined viral genome amplification, HPV18/45, HPV18/11, HPV18/6b, HPV18/1a, HPV18/BPV1 and HPV18/CRPV did not amplify their viral genomes (Figures 4,5 and Table 2). Therefore, viral genome amplification may be affected by the structural genes, as others have previously suggested (Terhune et al., 2001). How viral genome amplification actually relates to virion morphogenesis is currently unknown.
In addition to viral genome amplification, the PV chimeras also displayed altered infectivity and/or DNA encapsidation. Each of the capsid chimeric viruses displayed reduced viral infectivity compared with the wild-type HPV18 virus (Table 3) while five of the chimeric viruses (HPV18/1a (2-fold), HPV18/16 (4-fold), HPV18/6b (9-fold), HPV18/BPV-1 (21-fold), and HPV18/31 (105-fold)) demonstrated increased DNA encapsidation compared with the wild-type HPV18 virus (Figure 6). Others have reported the generation of chimeric viruses that are more infectious (Delgrange et al., 2007; Kawakami et al., 2003) or less infectious (Osman et al., 1998) than wild-type controls. One possible explanation for the reduced virus infectivity that we have observed with our panel of chimeric viruses is that the viral particles may not be folded properly. Minor changes in the capsid’s three-dimensional structure may reduce its infectivity without affecting its ability to encapsidate genomes. This is reflected in our neutralization and ELISA results. We found that all but HPV18/33 and HPV18/6b could be neutralized (Figure 7) and each of the chimeric viruses for which a conformation-dependent antibody was available could be detected by ELISA (Figure 8). These results suggest that minor changes may exist in the viral capsids outside of the epitope recognized by the conformation-dependent antibodies. These changes might permit genome encapsidation, but interfere with infection. The use of additional conformation-dependent antibodies against different regions of the viral capsids may identify and localize these minor changes in the capsid structure.
Other possible explanations for the reduced chimera infectivity include defects in the ability of the virus particles to dissociate and improper virion trafficking. An example of the inability of a virus particle to dissociate properly can be seen with the SV40 VP1 capsid protein mutant E330K. The E330K mutant virus is unable to uncoat its virus particles due to altered interactions at the pentamer-pentamer interface of its capsid (Kawano et al., 2009). An example of altered infectivity through improper virion trafficking is provided by adenovirus fiber swap. Fiber swap between adenovirus subgroup B (Ad serotype 7) and C (Ad serotype 5) was shown to alter the intracellular trafficking of adenovirus gene transfer vectors (Miyazawa et al., 1999). While Ad5 virions rapidly translocated to the nucleus and Ad7 virions remained cytoplasmic, a chimeric vector containing an Ad5 capsid and Ad7 fiber remained mostly cytoplasmic, similar to Ad7 (Miyazawa et al., 1999).
The increased encapsidation that we observed with the mutants HPV18/1a, HPV18/16, HPV18/6b, HPV18/BPV-1, and HPV18/31 (Figure 6) may reflect differences in the ability of the capsid proteins to interact with the viral genome. Interestingly, a putative packaging signal has been identified within the BPV1 E1 ORF (Zhao et al., 1999). This sequence was reported to be recognized by the capsid proteins of BPV1 and HPV6b. Therefore, it is possible that the capsid proteins of some of the foreign PVs were better able to recognize an HPV18 packaging sequence, leading to increased genome encapsidation. This interaction could be demonstrated genetically by mutating the candidate packaging sequences.
We show that each of the chimeric viruses was able to generate infectious virus. A comparison of amino acid sequence homology between L2 and L1 of HPV18 and the foreign PV types (Table 1) showed that HPV18 and HPV45 are the most homologous, suggesting that the HPV18/45 chimera would have the closest phenotype to wild-type HPV18. However, HPV18/45 is unable to amplify its genome and has reduced infectivity compared with the chimeras HPV18/16, HPV18/39, and HPV18/6b, which have lower sequence homology to HPV18 (Table 2 and Table 3). Conversely, HPV18/6b is infectious at a dilution of 100 and encapsidates its genome better then wild-type HPV18, and yet HPV18/6b has lower homology to HPV18 than HPV45 (Table 3). These data indicate that the sequence homology of PV structural genes is not a predictor of biological compatibility.
Our results bring to light some important points concerning PV biology. We demonstrate that interactions between the HPV18 nonstructural genes and the structural genes of a very diverse array of PV types can produce infectious virus. This result implies that all PVs share a similar set of mechanisms for viral replication. If this is true, then all PV may share a therapeutic target in the pathway of virion morphogenesis. This result also supports the possibility of phenotypic mixing in vivo. Several studies have demonstrated the existence of multiple HPV infections in a single host (Chaturvedi et al., 2005; Franco et al., 1999; Levi et al., 2002; Liaw et al., 2001). Our data, coupled with the prevalence of multiple infections, suggests the possibility that phenotypic mixing could take place in a human host, similar to other virus families (Chang et al., 1994; Hong et al., 2006).
Supplementary Material
TABLE 1B.
L2 homology
| HPV type | Identity (nt) to HPV18 | Similarity (aa) to HPV18 |
|---|---|---|
| 18 | 100% | 100% |
| 16 | 48.7% | 64.0% |
| 31 | 49.5% | 65.2% |
| 33 | 49.0% | 64.2% |
| 6b | 50.7% | 65.9% |
| 11 | 51.3% | 67.1% |
| 39 | 70.4% | 82.0% |
| 45 | 82.1% | 90.1% |
| 1a | 32.5% | 48.0% |
| CRPV | 32.8% | 44.4% |
| BPV1 | 28.0% | 40.5% |
L1 and L2 sequence comparisons were done using the website: http://www.ebi.ac.uk/clustalw/.
Highlights.
We constructed HPV18 chimeric genomes in which the HPV18 capsid genes were replaced with those of evolutionarily diverse PV types
Cell lines were generated from these chimeric genomes and each was capable of immortalization
Each of the chimeric genomes generated infectious virus
Acknowledgments
We thank members of the Meyers’ laboratory for many helpful discussions and Lynn R. Budgeon for histological assistance. This work was supported by grant RO1 AI 057988 (C.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- Alam S, Conway MJ, Chen HS, Meyers C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J Virol. 2008;82(2):1053–8. doi: 10.1128/JVI.01813-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KA, Florin L, Sapp C, Sapp M. Dissection of human papillomavirus type 33 L2 domains involved in nuclear domains (ND) 10 homing and reorganization. Virology. 2003;314(1):161–7. doi: 10.1016/s0042-6822(03)00447-1. [DOI] [PubMed] [Google Scholar]
- Belnap DM, Olson NH, Cladel NM, Newcomb WW, Brown JC, Kreider JW, Christensen ND, Baker TS. Conserved features in papillomavirus and polyomavirus capsids. J Mol Biol. 1996;259(2):249–63. doi: 10.1006/jmbi.1996.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard HU, Calleja-Macias IE, Dunn ST. Genome variation of human papillomavirus types: phylogenetic and medical implications. Int J Cancer. 2006;118(5):1071–6. doi: 10.1002/ijc.21655. [DOI] [PubMed] [Google Scholar]
- Bodily JM, Meyers C. Genetic analysis of the human papillomavirus type 31 differentiation-dependent late promoter. J Virol. 2005;79(6):3309–21. doi: 10.1128/JVI.79.6.3309-3321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag B, Chuke WF, Frisque RJ. Hybrid genomes of the polyomaviruses JC virus, BK virus, and simian virus 40: identification of sequences important for efficient transformation. J Virol. 1989;63(2):863–72. doi: 10.1128/jvi.63.2.863-872.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser BS, Alam S, Meyers C. Treatment of a human papillomavirus type 31b-positive cell line with benzo[a]pyrene increases viral titer through activation of the Erk1/2 signaling pathway. J Virol. 85(10):4982–92. doi: 10.1128/JVI.00133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo IG, Alonso A. Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. J Virol. 2004;78(24):13613–26. doi: 10.1128/JVI.78.24.13613-13626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. Arrangement of L2 within the papillomavirus capsid. J Virol. 2008;82(11):5190–7. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Zhou S, Ganem D, Standring DN. Phenotypic mixing between different hepadnavirus nucleocapsid proteins reveals C protein dimerization to be cis preferential. J Virol. 1994;68(8):5225–31. doi: 10.1128/jvi.68.8.5225-5231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi AK, Myers L, Hammons AF, Clark RA, Dunlap K, Kissinger PJ, Hagensee ME. Prevalence and clustering patterns of human papillomavirus genotypes in multiple infections. Cancer Epidemiol Biomarkers Prev. 2005;14(10):2439–45. doi: 10.1158/1055-9965.EPI-05-0465. [DOI] [PubMed] [Google Scholar]
- Chen BJ, Atwood WJ. Construction of a novel JCV/SV40 hybrid virus (JCSV) reveals a role for the JCV capsid in viral tropism. Virology. 2002;300(2):282–90. doi: 10.1006/viro.2002.1522. [DOI] [PubMed] [Google Scholar]
- Chen HS, Bromberg-White J, Conway MJ, Alam S, Meyers C. Study of infectious virus production from HPV18/16 capsid chimeras. Virology. 405(2):289–99. doi: 10.1016/j.virol.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Conway MJ, Christensen ND, Alam S, Meyers C. Papillomavirus capsid proteins mutually impact structure. Virology. 412(2):378–83. doi: 10.1016/j.virol.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XS, Garcea RL, Goldberg I, Casini G, Harrison SC. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol Cell. 2000;5(3):557–67. doi: 10.1016/s1097-2765(00)80449-9. [DOI] [PubMed] [Google Scholar]
- Christensen ND, Dillner J, Eklund C, Carter JJ, Wipf GC, Reed CA, Cladel NM, Galloway DA. Surface conformational and linear epitopes on HPV-16 and HPV-18 L1 virus-like particles as defined by monoclonal antibodies. Virology. 1996a;223(1):174–84. doi: 10.1006/viro.1996.0466. [DOI] [PubMed] [Google Scholar]
- Christensen ND, Kreider JW. Neutralization of CRPV infectivity by monoclonal antibodies that identify conformational epitopes on intact virions. Virus Res. 1991;21(3):169–79. doi: 10.1016/0168-1702(91)90031-p. [DOI] [PubMed] [Google Scholar]
- Christensen ND, Kreider JW. Monoclonal antibody neutralization of BPV-1. Virus Res. 1993;28(2):195–202. doi: 10.1016/0168-1702(93)90136-b. [DOI] [PubMed] [Google Scholar]
- Christensen ND, Kreider JW, Cladel NM, Patrick SD, Welsh PA. Monoclonal antibody-mediated neutralization of infectious human papillomavirus type 11. J Virol. 1990;64(11):5678–81. doi: 10.1128/jvi.64.11.5678-5681.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen ND, Reed CA, Cladel NM, Hall K, Leiserowitz GS. Monoclonal antibodies to HPV-6 L1 virus-like particles identify conformational and linear neutralizing epitopes on HPV-11 in addition to type-specific epitopes on HPV-6. Virology. 1996b;224(2):477–86. doi: 10.1006/viro.1996.0554. [DOI] [PubMed] [Google Scholar]
- Conway MJ, Alam S, Christensen ND, Meyers C. Overlapping and independent structural roles for human papillomavirus type 16 L2 conserved cysteines. Virology. 2009a;393(2):295–303. doi: 10.1016/j.virol.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway MJ, Alam S, Ryndock EJ, Cruz L, Christensen ND, Roden RB, Meyers C. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J Virol. 2009b;83(20):10515–26. doi: 10.1128/JVI.00731-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway MJ, Cruz L, Alam S, Christensen ND, Meyers C. Cross-neutralization potential of native human papillomavirus N-terminal L2 epitopes. PLoS One. 6(2):e16405. doi: 10.1371/journal.pone.0016405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel AM, Swenson JJ, Mayreddy RP, Khalili K, Frisque RJ. Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology. 1996;216(1):90–101. doi: 10.1006/viro.1996.0037. [DOI] [PubMed] [Google Scholar]
- Day PM, Baker CC, Lowy DR, Schiller JT. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A. 2004;101(39):14252–7. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day PM, Roden RB, Lowy DR, Schiller JT. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J Virol. 1998;72(1):142–50. doi: 10.1128/jvi.72.1.142-150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324(1):17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouille Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol. 2007;88(Pt 9):2495–503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- Eberle R, Tanamachi B, Black D, Blewett EL, Ali M, Openshaw H, Cantin EM. Genetic and functional complementation of the HSV1 UL27 gene and gB glycoprotein by simian alpha-herpesvirus homologs. Arch Virol. 1997;142(4):721–36. doi: 10.1007/s007050050114. [DOI] [PubMed] [Google Scholar]
- Fang L, Budgeon LR, Doorbar J, Briggs ER, Howett MK. The human papillomavirus type 11 E1/\E4 protein is not essential for viral genome amplification. Virology. 2006;351(2):271–9. doi: 10.1016/j.virol.2006.01.051. [DOI] [PubMed] [Google Scholar]
- Fehrmann F, Klumpp DJ, Laimins LA. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol. 2003;77(5):2819–31. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnen RL, Erickson KD, Chen XS, Garcea RL. Interactions between papillomavirus L1 and L2 capsid proteins. J Virol. 2003;77(8):4818–26. doi: 10.1128/JVI.77.8.4818-4826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000;74(14):6622–31. doi: 10.1128/jvi.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L, Becker KA, Sapp C, Lambert C, Sirma H, Muller M, Streeck RE, Sapp M. Nuclear translocation of papillomavirus minor capsid protein L2 requires Hsc70. J Virol. 2004;78(11):5546–53. doi: 10.1128/JVI.78.11.5546-5553.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L, Sapp C, Streeck RE, Sapp M. Assembly and translocation of papillomavirus capsid proteins. J Virol. 2002a;76(19):10009–14. doi: 10.1128/JVI.76.19.10009-10014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L, Schafer F, Sotlar K, Streeck RE, Sapp M. Reorganization of nuclear domain 10 induced by papillomavirus capsid protein l2. Virology. 2002b;295(1):97–107. doi: 10.1006/viro.2002.1360. [DOI] [PubMed] [Google Scholar]
- Franco EL, Villa LL, Sobrinho JP, Prado JM, Rousseau MC, Desy M, Rohan TE. Epidemiology of acquisition and clearance of cervical human papillomavirus infection in women from a high-risk area for cervical cancer. J Infect Dis. 1999;180(5):1415–23. doi: 10.1086/315086. [DOI] [PubMed] [Google Scholar]
- Gall J, Kass-Eisler A, Leinwand L, Falck-Pedersen E. Adenovirus type 5 and 7 capsid chimera: fiber replacement alters receptor tropism without affecting primary immune neutralization epitopes. J Virol. 1996;70(4):2116–23. doi: 10.1128/jvi.70.4.2116-2123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall JG, Crystal RG, Falck-Pedersen E. Construction and characterization of hexon-chimeric adenoviruses: specification of adenovirus serotype. J Virol. 1998;72(12):10260–4. doi: 10.1128/jvi.72.12.10260-10264.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggerty S, Walker DL, Frisque RJ. JC virus-simian virus 40 genomes containing heterologous regulatory signals and chimeric early regions: identification of regions restricting transformation by JC virus. J Virol. 1989;63(5):2180–90. doi: 10.1128/jvi.63.5.2180-2190.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg RS, Yip B, Kully C, Craib KJ, O’Shaughnessy MV, Schechter MT, Montaner JS. Improved survival among HIV-infected patients after initiation of triple-drug antiretroviral regimens. CMAJ. 1999;160(5):659–65. [PMC free article] [PubMed] [Google Scholar]
- Hong EM, Perera R, Kuhn RJ. Alphavirus capsid protein helix I controls a checkpoint in nucleocapsid core assembly. J Virol. 2006;80(18):8848–55. doi: 10.1128/JVI.00619-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Li H, Lam JT, Krasnykh V, Curiel DT, Blackwell JL. Substitution of the adenovirus serotype 5 knob with a serotype 3 knob enhances multiple steps in virus replication. Cancer Res. 2003;63(6):1262–9. [PubMed] [Google Scholar]
- Kawano MA, Xing L, Tsukamoto H, Inoue T, Handa H, Cheng RH. Calcium bridge triggers capsid disassembly in the cell entry process of simian virus 40. J Biol Chem. 2009;284(50):34703–12. doi: 10.1074/jbc.M109.015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnykh VN, Mikheeva GV, Douglas JT, Curiel DT. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J Virol. 1996;70(10):6839–46. doi: 10.1128/jvi.70.10.6839-6846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi JE, Kleter B, Quint WG, Fink MC, Canto CL, Matsubara R, Linhares I, Segurado A, Vanderborght B, Neto JE, Van Doorn LJ. High prevalence of human papillomavirus (HPV) infections and high frequency of multiple HPV genotypes in human immunodeficiency virus-infected women in Brazil. J Clin Microbiol. 2002;40(9):3341–5. doi: 10.1128/JCM.40.9.3341-3345.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw KL, Hildesheim A, Burk RD, Gravitt P, Wacholder S, Manos MM, Scott DR, Sherman ME, Kurman RJ, Glass AG, Anderson SM, Schiffman M. A prospective study of human papillomavirus (HPV) type 16 DNA detection by polymerase chain reaction and its association with acquisition and persistence of other HPV types. J Infect Dis. 2001;183(1):8–15. doi: 10.1086/317638. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Haggerty S, Frisque RJ. DNA replication of chimeric JC virus-simian virus 40 genomes. Virology. 1994;204(2):819–22. doi: 10.1006/viro.1994.1600. [DOI] [PubMed] [Google Scholar]
- Mayer TJ, Meyers C. Temporal and spatial expression of the E5a protein during the differentiation-dependent life cycle of human papillomavirus type 31b. Virology. 1998;248(2):208–17. doi: 10.1006/viro.1998.9262. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Bromberg-White JL, Meyers C. The role of the human papillomavirus type 18 E7 oncoprotein during the complete viral life cycle. Virology. 2005;338(1):61–8. doi: 10.1016/j.virol.2005.04.036. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Christensen ND, Meyers C. Propagation, infection, and neutralization of authentic HPV16 virus. Virology. 2004;322(2):213–9. doi: 10.1016/j.virol.2004.02.011. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Meyers C. Evidence for the coexistence of two genital HPV types within the same host cell in vitro. Virology. 2004;321(2):173–80. doi: 10.1016/j.virol.2003.12.019. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Meyers C. Propagation of infectious, high-risk HPV in organotypic “raft” culture. Methods Mol Med. 2005;119:171–86. doi: 10.1385/1-59259-982-6:171. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Wilson S, Mullikin B, Suzich J, Meyers C. Human papillomavirus type 45 propagation, infection, and neutralization. Virology. 2003;312(1):1–7. doi: 10.1016/s0042-6822(03)00312-x. [DOI] [PubMed] [Google Scholar]
- Meyers C, Bromberg-White JL, Zhang J, Kaupas ME, Bryan JT, Lowe RS, Jansen KU. Infectious virions produced from a human papillomavirus type 18/16 genomic DNA chimera. J Virol. 2002;76(10):4723–33. doi: 10.1128/JVI.76.10.4723-4733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers C, Frattini MG, Hudson JB, Laimins LA. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science. 1992;257(5072):971–3. doi: 10.1126/science.1323879. [DOI] [PubMed] [Google Scholar]
- Meyers C, Mayer TJ, Ozbun MA. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J Virol. 1997;71(10):7381–6. doi: 10.1128/jvi.71.10.7381-7386.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa N, Leopold PL, Hackett NR, Ferris B, Worgall S, Falck-Pedersen E, Crystal RG. Fiber swap between adenovirus subgroups B and C alters intracellular trafficking of adenovirus gene transfer vectors. J Virol. 1999;73(7):6056–65. doi: 10.1128/jvi.73.7.6056-6065.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis Y, Trus BL, Harrison SC. Atomic model of the papillomavirus capsid. EMBO J. 2002;21(18):4754–62. doi: 10.1093/emboj/cdf494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara T, Peh WL, Doorbar J, Lee D, Lambert PF. Human papillomavirus type 16 E1circumflexE4 contributes to multiple facets of the papillomavirus life cycle. J Virol. 2005;79(20):13150–65. doi: 10.1128/JVI.79.20.13150-13165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill FJ, Xu X, Gao Y. Host range analysis of a chimeric simian virus 40 genome containing the BKV capsid genes. Virus Res. 1992;25(3):169–87. doi: 10.1016/0168-1702(92)90132-s. [DOI] [PubMed] [Google Scholar]
- Osman F, Choi YG, Grantham GL, Rao AL. Molecular studies on bromovirus capsid protein. Virology. 1998;251(2):438–48. doi: 10.1006/viro.1998.9421. [DOI] [PubMed] [Google Scholar]
- Ozbun MA, Meyers C. Transforming growth factor beta1 induces differentiation in human papillomavirus-positive keratinocytes. J Virol. 1996;70(8):5437–46. doi: 10.1128/jvi.70.8.5437-5446.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbun MA, Meyers C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J Virol. 1997;71(7):5161–72. doi: 10.1128/jvi.71.7.5161-5172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbun MA, Meyers C. Human papillomavirus type 31b E1 and E2 transcript expression correlates with vegetative viral genome amplification. Virology. 1998;248(2):218–30. doi: 10.1006/viro.1998.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh WL, Brandsma JL, Christensen ND, Cladel NM, Wu X, Doorbar J. The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J Virol. 2004;78(4):2142–51. doi: 10.1128/JVI.78.4.2142-2151.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden RB, Greenstone HL, Kirnbauer R, Booy FP, Jessie J, Lowy DR, Schiller JT. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J Virol. 1996;70(9):5875–83. doi: 10.1128/jvi.70.9.5875-5883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Shirley PS, McClelland A, Kaleko M. Circumvention of immunity to the adenovirus major coat protein hexon. J Virol. 1998;72(8):6875–9. doi: 10.1128/jvi.72.8.6875-6879.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Raskova J, Fujinaga K, Raska K., Jr Identification of functional domains of adenovirus tumor-specific transplantation antigen in types 5 and 12 by viable viruses carrying chimeric E1A genes. Int J Cancer. 1994;57(4):598–603. doi: 10.1002/ijc.2910570426. [DOI] [PubMed] [Google Scholar]
- Sen E, Alam S, Meyers C. Genetic and biochemical analysis of cis regulatory elements within the keratinocyte enhancer region of the human papillomavirus type 31 upstream regulatory region during different stages of the viral life cycle. J Virol. 2004;78(2):612–29. doi: 10.1128/JVI.78.2.612-629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen E, Bromberg-White JL, Meyers C. Genetic analysis of cis regulatory elements within the 5′ region of the human papillomavirus type 31 upstream regulatory region during different stages of the viral life cycle. J Virol. 2002;76(10):4798–809. doi: 10.1128/JVI.76.10.4798-4809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LH, Foster C, Hitchcock ME, Isseroff R. In vitro HPV-11 infection of human foreskin. J Invest Dermatol. 1993;101(3):292–5. doi: 10.1111/1523-1747.ep12365409. [DOI] [PubMed] [Google Scholar]
- Smith LH, Foster C, Hitchcock ME, Leiserowitz GS, Hall K, Isseroff R, Christensen ND, Kreider JW. Titration of HPV-11 infectivity and antibody neutralization can be measured in vitro. J Invest Dermatol. 1995;105(3):438–44. doi: 10.1111/1523-1747.ep12321173. [DOI] [PubMed] [Google Scholar]
- Song H, Moseley PL, Lowe SL, Ozbun MA. Inducible heat shock protein 70 enhances HPV31 viral genome replication and virion production during the differentiation-dependent life cycle in human keratinocytes. Virus Res. 147(1):113–22. doi: 10.1016/j.virusres.2009.10.019. [DOI] [PubMed] [Google Scholar]
- Tavis JE, Trowbridge PW, Frisque RJ. Converting the JCV T antigen Rb binding domain to that of SV40 does not alter JCV’s limited transforming activity but does eliminate viral viability. Virology. 1994;199(2):384–92. doi: 10.1006/viro.1994.1136. [DOI] [PubMed] [Google Scholar]
- Telling GC, Williams J. Constructing chimeric type 12/type 5 adenovirus E1A genes and using them to identify an oncogenic determinant of adenovirus type 12. J Virol. 1994;68(2):877–87. doi: 10.1128/jvi.68.2.877-887.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhune SS, Hubert WG, Thomas JT, Laimins LA. Early polyadenylation signals of human papillomavirus type 31 negatively regulate capsid gene expression. J Virol. 2001;75(17):8147–57. doi: 10.1128/JVI.75.17.8147-8157.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge PW, Frisque RJ. Analysis of G418-selected Rat2 cells containing prototype, variant, mutant, and chimeric JC virus and SV40 genomes. Virology. 1993;196(2):458–74. doi: 10.1006/viro.1993.1502. [DOI] [PubMed] [Google Scholar]
- Trus BL, Roden RB, Greenstone HL, Vrhel M, Schiller JT, Booy FP. Novel structural features of bovine papillomavirus capsid revealed by a three-dimensional reconstruction to 9 A resolution. Nat Struct Biol. 1997;4(5):413–20. doi: 10.1038/nsb0597-413. [DOI] [PubMed] [Google Scholar]
- Unger ER, Budgeon LR, Myerson D, Brigati DJ. Viral diagnosis by in situ hybridization. Description of a rapid simplified colorimetric method. Am J Surg Pathol. 1986;10(1):1–8. doi: 10.1097/00000478-198601000-00001. [DOI] [PubMed] [Google Scholar]
- Vacante DA, Traub R, Major EO. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology. 1989;170(2):353–61. doi: 10.1016/0042-6822(89)90425-x. [DOI] [PubMed] [Google Scholar]
- Visalli RJ, Courtney RJ, Meyers C. Infection and replication of herpes simplex virus type 1 in an organotypic epithelial culture system. Virology. 1997;230(2):236–43. doi: 10.1006/viro.1997.8484. [DOI] [PubMed] [Google Scholar]
- Volpers C, Schirmacher P, Streeck RE, Sapp M. Assembly of the major and the minor capsid protein of human papillomavirus type 33 into virus-like particles and tubular structures in insect cells. Virology. 1994;200(2):504–12. doi: 10.1006/viro.1994.1213. [DOI] [PubMed] [Google Scholar]
- Wilson R, Fehrmann F, Laimins LA. Role of the E1--E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J Virol. 2005;79(11):6732–40. doi: 10.1128/JVI.79.11.6732-6740.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Ryan GB, Knight GL, Laimins LA, Roberts S. The full-length E1E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology. 2007;362(2):453–60. doi: 10.1016/j.virol.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Zabner J, Chillon M, Grunst T, Moninger TO, Davidson BL, Gregory R, Armentano D. A chimeric type 2 adenovirus vector with a type 17 fiber enhances gene transfer to human airway epithelia. J Virol. 1999;73(10):8689–95. doi: 10.1128/jvi.73.10.8689-8695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao KN, Frazer IH, Jun Liu W, Williams M, Zhou J. Nucleotides 1506–1625 of bovine papillomavirus type 1 genome can enhance DNA packaging by L1/L2 capsids. Virology. 1999;259(1):211–8. doi: 10.1006/viro.1999.9714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.