

Abstract
Cognitive impairments are central to schizophrenia and may mark underlying biological dysfunction, but efforts to detect genetic associations for schizophrenia or cognitive phenotypes have been disappointing. Phenomics strategies emphasizing simultaneous study of multiple phenotypes across biological scales may help, particularly if the high heritabilities of schizophrenia and cognitive impairments are due to large numbers of genetic variants with small effect. Convergent evidence is reviewed, and a new collaborative knowledgebase – CogGene – is introduced to share data about genetic associations with cognitive phenotypes, and enable users to meta-analyze results interactively. CogGene data demonstrate the need for larger studies with broader representation of cognitive phenotypes. Given that meta-analyses will likely be necessary to detect the small association signals linking the genome and cognitive phenotypes, CogGene or similar applications will be needed to enable collaborative knowledge aggregation and specify true effects.
Cognitive Impairment in Schizophrenia: In Search of a Genomic Basis
Cognitive impairment has been seen as a hallmark of schizophrenia at least since Emil Kraepelin described the syndrome of dementia praecox in 1893 [7], but in the last few decades it has assumed new importance at least in part due to hopes that the cognitive functions might prove more tractable targets for genetic study than are the characteristic symptoms used to diagnose schizophrenia (see Box 1).
Box 1. Cognitive impairment in schizophrenia: why is it important?
The cognitive impairment associated with schizophrenia is severe, widespread, and apparent long before overt signs of psychosis emerge [1–3]. There were initially hopes that new antipsychotic drug treatments might ameliorate these deficits, but large scale trials with these agents have shown limited success, large-scale effectiveness studies suggest no significant cognitive benefit for new antipsychotic agents [4, 5], and so far no agent has been approved by the Food and Drug Administration for the indication of cognitive impairment associated with schizophrenia despite wide interest. There remains hope that in the future pharmacogenomic strategies may yield treatment benefits for schizophrenia [6], and innovative strategies are being advanced particularly to identify new molecular targets linked directly to cognitive dimensions rather than the traditional symptomatic dimensions of schizophrenia [8]. These findings highlight the importance of identifying genetic bases of cognitive dysfunction in schizophrenia both for increasing understanding of pathophysiology and for developing more effective treatments.
Despite heritability of the schizophrenia phenotype estimated at near 80%, initial family-based association studies, and then case-control genome-wide association studies (GWAS) have failed to identify any common genetic variants with large effects. A handful of reported associations surpass conventional levels of genome-wide significance (p<5*10−8)(see www.szgene.org for regularly updated meta-analytic findings), but none accounts for much of the morbidity associated with schizophrenia. It seems likely that many genetic variants (perhaps thousands) interact with each other and the environment to account for schizophrenia risk, and indeed this risk is likely shared at least with bipolar disorder and probably autism and other complex neurodevelopmental disorders [9, 10].
Investigations focused on “endophenotypes” or “intermediate phenotypes”, including cognitive phenotypes, are now emerging [11], with the hope that these might offer more traction in deciphering the complex genetics of brain dysfunction in schizophrenia and perhaps other neurodevelopmental syndromes. Meanwhile, most investigators are likely to agree that unraveling the genetic bases of schizophrenia and its associated cognitive impairment is proving more difficult than was hoped earlier [12].
Major questions remain as to how we may best gain traction on the elusive biological roots of schizophrenia. The new transdiscipline referred to as ‘phenomics’ (the systematic study of phenotypes on a genome-wide scale) may offer one perspective [13]. Most efforts so far have targeted the syndromal phenotype of schizophrenia in case-control studies in hopes of finding genetic association. In contrast, phenomics approaches consider multiple phenotypes, including those that may be measured on different biological scales, in order to better define biologically plausible traits. It is assumed that by combining information from multiple levels – from the level of gene expression through proteomic, metabolomic, cellular and systems levels – that we may better characterize the biological contributions of specific genetic variants and their interactions in a way that ultimately permits personalization of diagnosis and rational treatment. In research on schizophrenia, for example, it has been hoped that we might gain clearer insights from the simultaneous focus on the symptoms that mark the syndrome and the cognitive deficits that consistently accompany the syndrome.
In this article we aim to: (a) provide a brief synopsis of current knowledge about the genetic basis of cognitive deficits in schizophrenia; (b) highlight selected conceptual issues that we believe will be important to make further progress in finding the genetic bases for cognitive phenotypes in schizophrenia; and (c) introduce a new, freely available resource – CogGene – that we hope can serve the field by helping to aggregate, visualize, and analyze relevant evidence for those interested in the genetic bases of cognitive function in schizophrenia and other disorders. An important point about a knowledgebase like CogGene is that it might help advance understanding of the genetic bases of cognitive deficits in schizophrenia even if the knowledgebase is focused selectively on findings in healthy people. In brief, we hope a tool like CogGene can help researchers “triangulate” genetic association findings: if a specific genetic variation is associated with the diagnosis of schizophrenia AND the same genetic variation is associated with cognitive impairment in otherwise healthy people, then it increases the likelihood that this variant may be related to BOTH schizophrenia and cognitive impairment through a common mechanism. Indeed this strategy may be more informative than examining association of a genetic variant with cognitive deficits within schizophrenia samples, because cognitive impairment is confounded with the diagnosis of schizophrenia.
What do we know so far about the genetics of cognitive impairment in schizophrenia?
It has long been known that both schizophrenia and cognitive impairment are highly heritable and it has long been assumed that some genetically mediated anomaly – probably a neurodevelopmental anomaly – underlies the vulnerability to both schizophrenia and the cognitive impairment that invariably accompanies the syndrome. The heritability of schizophrenia is estimated at greater than .8, while the heritability of cognitive phenotypes is most often found to be near .5, regardless of whether the estimate is derived from healthy or ill groups [14, 15]. Compelling evidence has also been provided to show that many different cognitive abilities may be linked, not only by their covariation within individuals, but further by their shared genetic correlations; indeed this has led to the “generalist gene” hypothesis that many presumably diverse cognitive functions are likely to be associated with a common set of genetic variations [16–18]. Recent work has begun to identify the shared genetic components of cognitive phenotypes and syndromal phenotypes like schizophrenia; in brief, the cognitive phenotypes and schizophrenia are significantly correlated, and the lion’s share of this covariance (72% to 92%) is due to shared genetic effects [19–21]. Despite the relatively high heritability of these phenotypes and their high genetic correlation, we so far have in hand no well-validated candidate genes that explain much of the variance in either schizophrenia or cognitive phenotypes (with the exception of selected rare genes in which mutations cause large effects on cognition, as described below). Meanwhile it increasingly looks as though the shared liability for schizophrenia and cognitive impairment is most likely to be identified through a relatively large number of genetic influences, some coming from larger impacts of rare variants, and some coming from larger numbers of more common variants with very small effects working in combination to undermine healthy brain development and signaling.
The search for cognitive phenotypes
Cognitive phenotypes as intermediate phenotypes or as paraphenotypes
One frequently asked question is whether cognitive deficits or symptoms are epiphenomenal: that is, do cognitive deficits cause schizophrenia or are cognitive deficits caused by schizophrenia? While this distinction might be seen as an irrelevant exercise in semantics, it is important for modeling. We see both symptomatic and cognitive measures as similar in level of explanation, given that both are behavioral manifestations of neural systems activity. This point of view casts skepticism on the likelihood that cognitive phenotypes will serve well as intermediate phenotypes because they are not really intermediate in the sense that this term is used in causal models (i.e., they are not likely mediating variables for symptoms, at least in schizophrenia). Cognitive phenotypes may nevertheless be of value as “paraphenotypes” (i.e., phenotypes that are at the same hierarchic level within a causal model, and are alongside each other), because they are better validated with respect to neural systems phenotypes (see Figure 1b: if path coefficient x > y). Adding cognitive measures to a multivariable phenotype may thus help constrain the neural system phenotypes to a subset of all neural systems that might be part of the mechanistic path from genome to syndrome, and thereby help increase statistical power for detecting associations with “lower” level biological processes including genetic variation. It should be recognized, however, that such arguments are at this point largely theoretical and there are few confirmatory or disconfirmatory examples in practice. So far we can say only that cognitive phenotypes have not shown “simpler” genetic architecture than complex syndromal phenotypes [22].
Figure 1. Relations of cognitive phenotypes to neural systems and diagnosis.

Path diagrams schematizing different points of view regarding the role of cognitive phenotypes as a) intermediate phenotypes or endophenotypes; or b) as different behavioral effects – paraphenotypes -- that differ primarily in the strength of relations to neural system activity.
The genetic bases of cognitive phenotypes
There are multiple potential windows on the genetic bases of cognitive phenotypes. Some of the earliest and most successful approaches found genetic associations with cognitive impairment syndromes, particularly mental retardation. Indeed mental retardation may be seen as a phenotype for which genetic studies have been particularly successful, with ~300 identified monogenic causes; but it should be recognized that these are rare (i.e., most account for only .01% of all cases) [23].
Despite the low frequency of these conditions, they may be informative about mechanisms important to brain development and cognition. For example, the study of Fragile × syndrome (a genetic condition involving changes in part of the × chromosome) has led to multiple insights about the genetics of trinucleotide repeats, X-linked genetic disorders, and the enormous pleiotropy of single-gene deficits on neural and other systems [24]. Similarly the study of neurofibromatosis (a genetic disorder of the nervous system, which mainly affects how nerve cells form and grow), and the NF1 gene, has yielded major insights into the molecular basis of these syndromes, yielded novel transgenic rodent models in which mutants have superior abilities, and may stimulate novel treatment development [25, 26].
It should be recognized that even when genetic studies reveal compelling associations that are considered significant at genome-wide levels and replicated, the identified variants may still account for only a small amount of the known heritability. Human height is a good example phenotype: despite a heritability near 80%, only about 5% of phenotypic variance is explained by more than 40 known loci [27]. This has been referred to as the problem of “missing heritability” or the “dark matter” of heritability, and may be due to many reasons, including: (1) variants that the GWAS arrays are missing (i.e., the SNP’s that have yielded association findings may not be the causative SNP’s, and the true causative SNP’s might have larger effects); (2) gene-gene interactions (epistasis) and/or gene-environment interaction effects too complicated to assess given current sample sizes and analytic strategies; (3) epigenetic effects; (4) much larger numbers of genetic variants with even smaller effects remaining to be found; and (5) inadequate accounting for shared environmental variance among relatives [28].
Although work so far using GWAS to detect associations with cognitive phenotypes has been unsuccessful at replicating results from some prior “candidate gene” studies, it is worth noting that the results remain consistent with the hypothesis that cognitive impairment may be associated with an increase in genetic variants each with small effect [29]. Sabb and colleagues summarized prior work on candidate genes for which investigators reported associations with cognitive phenotypes comprising “memory” (51 effects) and “intelligence” (42 effects) [30]. They found generally modest associations of candidate genes with varying cognitive phenotypes, with most effect sizes (Cohen’s d for the effect distinguishing alleles) ranging from .09 to .23. An interesting result of this survey was that among genes investigated, two had relations specifically with intelligence (CHRM2, DRD2), two had relations specifically with memory phenotypes (5-HTT, KIBRA), and four had reported links to both intelligence and memory phenotypes (DTNB1, COMT, BDNF, APOE). Others have highlighted the replication of selected findings related to rare variants in key genetic regions (such as PDE10A, CYSIP1, KCNE1/KCNE2, CHRNA7) and their possible connection to both schizophrenia and cognitive impairment phenotypes [31]. It must be recognized, however, that these findings may still reflect false positive reports. Sources of bias include the targeting of certain genes as candidates without very strong a priori evidence, considering different single-nucleotide polymorphisms (SNP’s) within a gene as replications of a specific gene finding, and as highlighted by Sabb and colleagues, dubious measurement of cognitive phenotypes (for example, one of the measures of “memory” was the Mini Mental State Exam, and another was “Fluency”, despite the fact that these would be questioned by most investigators). Sabb and colleagues established an open database to share knowledge about these associations (see www.Phenowiki.org; [14]). Hopefully, continued development of such resources will ultimately enable convergence on the meaningful associations and refinement of phenotype definitions, perhaps narrowing down to those that have the clearest relations for broader population studies.
CogGene: a collaborative knowledgebase for documenting genetic associations with cognitive phenotypes
With this background, we aimed to determine to what extent data regarding genetic associations with the schizophrenia phenotype might be enriched by examining correlations of the same genetic targets with cognitive phenotypes. This approach may be considered a triangulation of genes at the intersection of schizophrenia and cognitive impairment. In order to gather data relevant to this we elaborated on the Phenowiki database architecture and created a new knowledgebase and web service (UCLA CogGene; see www.CogGene.org) specifically to represent genetic association findings for cognitive phenotypes. We aimed to have an interface similar in some ways to those available in SZGene and ALZGene, which provide forest plots of effect sizes, but our data differ insofar as the phenotypes to be represented are quantitative trait scores (rather than categorical diagnoses). Further, due to the richness of the cognitive phenotype data (which includes both test names and then specific measurement variables or indicators within each test), we created features in CogGene to enable dynamic sorting and computation of weighted effect size statistics over groups of results that can be selected simply by clicking on the effect labels.
The data discussed in this paper and the CogGene system are now viewable at www.CogGene.org, and information is posted on the site regarding how to submit additional contributions. The findings described here were culled from publications identified through literature mining if they cited the names of at least one gene and related polymorphism, and at least one cognitive test (the names are from a lexicon developed in the Consortium for Neuropsychiatric Phenomics at UCLA; see www.phenomics.ucla.edu). From these publications we selected those with usable data (i.e., with data specifying at least one statistical association between a specific SNP and a specific cognitive test indicator), and extracted quantitative effect sizes for associations between SNP’s and cognitive test indicators. We highlight that these data were selected to represent results from healthy samples, in order to maximize the independence of findings from those in SZGene (and thus enabling us to inspect possible overlaps in the “top hits” free of the potential confounds between schizophrenia and cognitive impairment phenotypes). The results can now be browsed, sorted and reanalyzed using custom-designed software tools that permit visualization and execution of “meta-analysis” (sample size weighted averaging) over selected effects under user control. In brief, the CogGene system permits visualization of the effect sizes for specific allelic variants on the cognitive trait scores (expressed as Cohen’s d statistic, which is the standardized difference between group means), and the 95% confidence intervals around these difference scores, in an interactive Forest plot. This is similar to the representation of genetic association data in other widely used resources (SzGene, AlzGene) which use similar (but static) Forest plots to show allelic associations with case-control differences (but in these examples, the effect sizes are expressed as odds ratios rather than group differences). A typical screen-shot of CogGene is shown in Figure 2.
Figure 2. Screen-shot illustrating features of the CogGene web service.
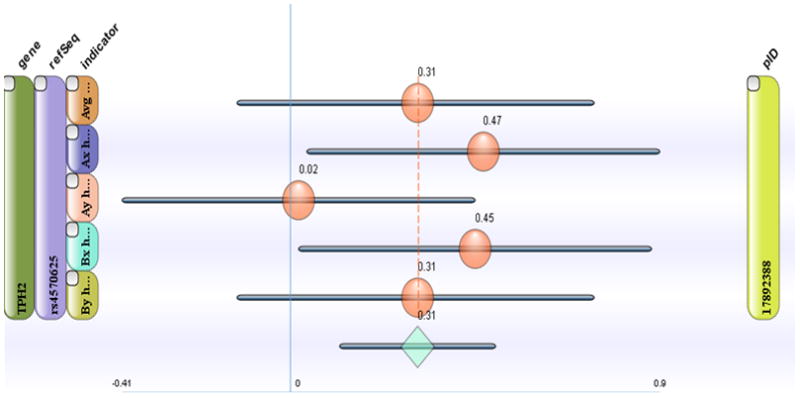
Users can filter records in the CogGene database either by “gene” or by “task”, and the system will then show all relevant effects in a dynamic forest plot. The example shows results selected for the task “AX-CPT” (the “AX” version of the Continuous Performance Test), filtered to show results only for the gene TPH2 from a single publication (see green bar on right side of figure, which gives the PubMedID (pID), and a single SNP (see purple bar on left side of figure, labeled “refSeq”). In this example, the publication had examined 5 different Indicators (see 5 bars stacked on the left side of the figure). If the user clicks on any of these bars the CogGene system sorts all results based on that column (i.e., by gene, task, indicator, refSeq, or PubMedID). Note also that each bar has a checkbox in its upper left corner; users can check these boxes and then the meta-analysis calculations will be executed over the checked results. The actual entries in the forest plot show the effect size d for the specific indicator and refSeq contrast for that study orange circles (not shown is that the mouse fly-over function reveals exactly which allelic contrast is being represented). At the bottom of the figure, the blue diamond represents the meta-analytic result (sample size weighted average of all selected effects). The gray bars represent 95% confidence intervals around the individual or meta-analytic effect size values. Further details are available at www.CogGene.org.
The SZGene (SchizophreniaGene) database contains information from 1727 studies, reporting data on 1008 genes, and 8,788 polymorphisms; this database has 287 meta-analyses (see www.szgene.org; accessed 5/31/2011)[32]. SZGene ranks its “Top Results” using the HuGENet interim guidelines published by Ioannidis and colleagues[33], which consider the amount of evidence (i.e., Grade A is given to studies where the total number of minor alleles exceeds 1,000); consistency of evidence (i.e., Grade A is given only when inconsistency is modest, for example I2 < 25); and bias (with Grade A given when there is probably no bias).
Inspecting initial entries into the CogGene database, we note that the quality of genetic association studies for cognitive phenotypes so far is relatively low. For example, none of the studies meets the criteria to be considered Grade “A” following HuGENet criteria for amount of evidence, and only 3 studies would receive a Grade of “B” (i.e., with minor allele frequencies greater than 100; the rest would all be considered Grade “C”). Analysis of existing studies is further complicated by the lack of uniformity in phenotype definition, rendering replication of results difficult to determine because few studies use exactly the same indicators. Finally, the degree of bias in the cognitive studies may be considered relatively high, given the paucity of large effect sizes.
Examining the 45 “Top Results” of SZGene, we find that 10 of the same genes are listed in CogGene. Among these 10 genes, we find that only evidence supporting association for two of these genes (APOE, HTR2A) is considered Grade “A” in SZGene. APOE (e2/3/4; contrasting 4 versus 3 allele) is significantly associated with schizophrenia among Caucasians; and HTR2A (rs6311; contrasting A versus G allele) is associated with schizophrenia, also selectively in Caucasian samples.
Figure 3 provides a graphical summary of the “Top Results” from CogGene, considering only those individual SNP effects that had 95% confidence intervals not including zero difference between allelic variants. Among these, only the APOE genotype overlaps with those identified as a Top Result in the SZGene database, and as Figure 3 shows, the average effect size for APOE is small (d = .069, 95% confidence interval = .014 to .124). It should be recognized that this effect for APOE genotype is small in part because it is averaging together effects on different cognitive indicators. We have for APOE two studies with the same cognitive indicator (Buschke Selective Reminding Test, Long Term Recall), and the same contrast among alleles; these two studies [34, 35] have overlapping samples so probably the larger of the two studies should be relied on by itself. On a positive note, this single study [35] showed a medium effect (i.e., comparing the e2/2 and e2/3 to e3/3 had d = .29 and comparing e2/2 and e2/3 to e4 allele carries had d = .39), with total sample size of 912 and minor allele frequency of 76. On the other hand, this more detailed inspection of the findings indicates that this result stands as an isolated finding without replication.
Figure 3. Top results from CogGene.

Effect size statistics from CogGene (expressed in terms of Cohen’s d statistic) for genetic associations with cognitive indicators, based on sample size-weighted mean effect sizes for each SNP. Error bars provide 95% confidence intervals around each mean effect, only effects not including zero are shown. DRD2 and SNAP25 each had two SNP’s satisfying these criteria.
Among the Top results in CogGene, none of the effects so far would be considered significant at conventional genome-wide levels, at least in part because the sample sizes are so low. For example, only the APOE and DTNB1 findings are supported by a study with a sample size exceeding 500 cases, and the largest effect (CACNA1C) is supported by a study of only 80 people, only 10 of whom possessed the minor allele at the investigated locus (rs1006737). These results are consistent with those reported by Sabb and colleagues [30], where as noted above, all effect sizes were in the range of d = .09 to d = .23, with the single exception (d = .44) being a study for which total sample size was only 201. This highlights the possibility of publication bias and so far small sample studies, which poses a major challenge to cognitive genomics, and the likelihood that many of the reported associations will turn out to be false positive results. Currently, the literature remains rife with findings that center on selected “candidate” genes that have been investigated at least in part due to inertia from earlier positive reports (for example, the study of APOE genotype in schizophrenia reflects more the “smoke” from positive findings in Alzheimer’s disease than the likely “fire” in schizophrenia). This bias may soon be overcome as more GWAS results and then genome sequencing findings are disseminated. At that point the biggest priorities will be to obtain unbiased sampling of the “cognitive phenome,” else we will run similar risk of biases from studying the wrong candidate phenotypes that we currently face in studying false positive candidate genotypes [36]. This will be an interesting challenge for future investigations, which will need to balance consistency and standardization of phenotyping that are critical for replication, with sufficiently broad sampling to help reduce phenotyping bias. We hope that further development of CogGene will help aggregate findings across investigations, increase our understanding of where relevant signals may lie, and shed light on the design of future studies and collaborative research programs. The most recent findings regarding the genetics of schizophrenia and cognitive impairment phenotypes suggest we are likely to face a deluge of associations with very small effects, and a smaller number of rare variants possibly with larger effects, along with likely complex gene-gene and gene-environment interactions. These observations make the availability of a collaborative knowledge-building tool like CogGene particularly valuable, because sifting through the findings, and aggregation of results across diverse studies, may ultimately be more important than results from any single study. By structuring knowledge in CogGene we hope also to facilitate links to other knowledgebases (such as the Entrez systems supported by the National Library of Medicine) to promote biological discovery and better constrain our models of the causal paths that connect the human genome to complex disorders of brain and behavior.
A related challenge pertains to developing standards for cognitive phenotyping and refinement of ontologies that can help formalize knowledge within this scientific domain. Sabb and colleagues showed how fickle investigators can be, introducing new concept labels despite lack of change in the actual measurement methods [14]. We have suggested frameworks for developing cognitive ontologies elsewhere [13, 37, 38], and the Cognitive Atlas project (www.CognitiveAtlas.org) is dedicated specifically to development of a consensus ontology about cognitive concepts and their measurement. This work will be essential to help determine which specific findings can be meaningfully averaged in meta-analytic studies that will ultimately help us identify and understand what are likely to be myriad small signals relating cognitive phenotypes to the genome.
Finally, the development of tools like CogGene can help represent quantitative trait data for genetic associations and thus offer a means for collaboration, storage, and reuse of knowledge that is important to the dimensional representation of phenotypes. This is compatible with the National Institute of Mental Health Strategic Plan, and specifically of potential value to the new Research Domain Criteria (RDoC) initiative [39, 40], which aims to support research on phenotypic dimensions that may be more informative than traditional diagnostic phenotypes.
Concluding remarks
In summary, we considered the existing literature on genetic associations with the syndromal phenotype of schizophrenia and its conjunction with findings from the study of genetic associations with cognitive phenotypes in healthy people. The work on schizophrenia is more advanced and contains a few leads, albeit we are seeing at most the tip of the iceberg in understanding the contributions to this genetic risk, and much “dark matter” (missing heritability) remains to be defined. The work on cognitive genomics requires significant advances in methods and study quality to yield more credible findings, and to add substantively to understanding genetic risks for cognitive impairments in schizophrenia and other neurodevelopmental disorders. Among the challenges are: (a) increasing sample sizes, probably at least by an order of magnitude relative to the published work available now; (b) increasing standardization in cognitive phenotyping to enable more direct replication; (c) increasing coverage of cognitive domains within each study to help attenuate bias in sampling from the cognitive phenome (see also Box 2). These goals are particularly daunting to achieve given constraints on time and budgets. Major progress may be fostered by routine aggregation of genome-wide sequencing data (which we consider likely within a decade) together with widely distributed (internet- and mobile application-based) cognitive phenotyping, and the refinement of methods to represent cognitive concepts and the specific measurements use to define these. We introduced here the CogGene knowledgebase, a freely available online collaborative tool to help aggregate and meta-analyze relevant evidence, which we hope will advance understanding of the genetic bases of complex syndromes involving brain and behavior.
Box 2. Questions for Future Research.
How can we best investigate those cognitive phenotypes that reflect the most discrete, biologically meaningful targets (i.e., what are the best measures to increase understanding of the biological bases of cognitive phenotypes and how do we validate these)?
How can we best aggregate information from different measures that we think may be measuring the same basic dimension of cognitive function (i.e., when can we successfully “mate” findings from different studies in meta-analyses)?
How can we best translate cognitive phenotype studies across species, and particularly how can we best leverage the ability to develop strong genetic models in the mouse that will be informative about cognitive phenotypes in humans?
How can we best target genetic analyses based on prior knowledge about cognitively-relevant biology (i.e., can we effectively constrain analysis models based on prior knowledge of selected neurodevelopmental processes, or signaling pathways, that we believe are critical for cognitive function)?
Assuming we can identify some genetic variants that confer shared vulnerability to schizophrenia and cognitive impairment, can we leverage this knowledge to better understand those genetic features that are unique to schizophrenia and not associated with cognitive impairment (and those that are associated with cognitive impairment but not schizophrenia)?
Acknowledgments
This work was supported by the Consortium for Neuropsychiatric Phenomics (NIH Roadmap for Medical Research grants UL1-DE019580, RL1LM009833, PL1MH083271) and the Tennenbaum Family Center for the Biology of Creativity.
Glossary
- Phenomics
the systematic study of phenotypes on a genome-wide scale, emphasizing the simultaneous analysis of multiple phenotypes
- Endophenotype/Intermediate phenotype
The term “endophenotype” as it is now widely used in psychiatry stems from original usage by Gottesman and Shields who referred to “internal features”, “only knowable after aid to the naked eye”, such as the help provided by “a biochemical test or a microscopic examination” ([41]p. 19). More recently, Gottesman and Gould defined endophenotypes as “measurable components unseen by the unaided eye along the pathway between disease and distal genotype” ([42]p. 636). We believe this added specification is important because it implicates endophenotypes as being the product of genetic variation that is part of the causal path between genotype and some other phenotype of interest (usually one that is either the diagnosis or part of the diagnosis). The terms “endophenotype” and “intermediate phenotype” are now widely used interchangeably to refer to any phenotype that is presumed to be “closer” to the level of gene action than is the observable phenotype of interest (for example, “molecular expression” is considered closer to gene action than behavioral “symptoms” of schizophrenia). We believe the term “intermediate phenotype” better reflects the putative causal attributes of phenotypes that actually are part of the causal path from genotype to some higher-level phenotype; but so far few such intermediate phenotypes have been proven
- Paraphenotype
a phenotype at the same level of expression with respect to causal models of the relation between a genotype and some other phenotype of interest; a phenotype that shares causal precedents with another phenotype (see Figure 1)
- Candidate gene study
a study of association between genotype and phenotype(s) in which the genetic variation is selected based on prior evidence of association
- Genome-wide association study (GWAS)
a study of association between genotypes and phenotype(s) in which genetic variations are sampled widely over the entire genome, theoretically in a manner that provides “coverage” of the entire genome
- Single-nucleotide polymorphism (SNP)
a variation between individuals of the same species in the structural sequence of nucleotides in the DNA molecule at a single location involving the substitution of one nucleotide for another (i.e., given that there are four nucleotides, adenosine (A), thymine (T), cytosine (C), and guanine (G); a SNP is said to exist when one of these nucleotides is replaced by another)
- Copy number variation (CNV)
a variation between individuals of the same species in the structural sequence of nucleotides due to repetition or deletion of part of the sequence (e.g., a “trinucleotide repeat” is the repetition of three nucleotides; “CAG repeats” are one example already identified as responsible for multiple neurological disorders including Huntington’s disease).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allott K, et al. Cognition at illness onset as a predictor of later functional outcome in early psychosis: Systematic review and methodological critique. Schizophrenia research. 2011;125:221–235. doi: 10.1016/j.schres.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Bora E, et al. Cognitive functioning in schizophrenia, schizoaffective disorder and affective psychoses: meta-analytic study. The British Journal of Psychiatry. 2009;195:475–482. doi: 10.1192/bjp.bp.108.055731. [DOI] [PubMed] [Google Scholar]
- 3.Mesholam-Gately RI, et al. Neurocognition in first-episode schizophrenia: A meta-analytic review. Neuropsychology. 2009;23:315. doi: 10.1037/a0014708. [DOI] [PubMed] [Google Scholar]
- 4.Keefe RS, et al. Neurocognitive Effects of Antipsychotic Medications in Patients With Chronic Schizophrenia in the CATIE Trial. Archives of general psychiatry. 2007;64:633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- 5.Hill SK, et al. Effect of second-generation antipsychotics on cognition: current issues and future challenges. Expert review of neurotherapeutics. 2010;10:43. doi: 10.1586/ern.09.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cacabelos R, et al. Pharmacogenomics of antipsychotics efficacy for schizophrenia. Psychiatry and clinical neurosciences. 2011;65:3–19. doi: 10.1111/j.1440-1819.2010.02168.x. [DOI] [PubMed] [Google Scholar]
- 7.Kraepelin E. Dementia praecox and paraphrenia. E and S Livingstone; 1919. [Google Scholar]
- 8.Krystal JH, et al. Neuroplasticity as a target for the pharmacotherapy of anxiety disorders, mood disorders, and schizophrenia. Drug discovery today. 2009;14:690–697. doi: 10.1016/j.drudis.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009 doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rzhetsky A, et al. Probing genetic overlap among complex human phenotypes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11694–11699. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenwood TA, et al. Analysis of 94 Candidate Genes and 12 Endophenotypes for Schizophrenia From the Consortium on the Genetics of Schizophrenia. Am J Psychiatry. 2011 doi: 10.1176/appi.ajp.2011.10050723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bilder RM. The neuropsychology of schizophrenia circa 2009. Neuropsychol Rev. 2009;19:277–279. doi: 10.1007/s11065-009-9112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bilder RM, et al. Phenomics: the systematic study of phenotypes on a genome-wide scale. Neuroscience. 2009;164:30–42. doi: 10.1016/j.neuroscience.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabb FW, et al. A collaborative knowledge base for cognitive phenomics. Molecular psychiatry. 2008;13:350–360. doi: 10.1038/sj.mp.4002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenwood TA, et al. Initial heritability analyses of endophenotypic measures for schizophrenia: the consortium on the genetics of schizophrenia. Archives of general psychiatry. 2007;64:1242–1250. doi: 10.1001/archpsyc.64.11.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plomin R, et al. Generalist Genes: Genetic Links Between Brain, Mind, and Education. Mind Brain Educ. 2007;1:11–19. doi: 10.1111/j.1751-228X.2007.00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haworth CM, et al. Generalist genes and high cognitive abilities. Behavior Genetics. 2009;39:437–445. doi: 10.1007/s10519-009-9271-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plomin R, Haworth CM. Genetics of high cognitive abilities. Behavior Genetics. 2009;39:347–349. doi: 10.1007/s10519-009-9277-9. [DOI] [PubMed] [Google Scholar]
- 19.Toulopoulou T, et al. Substantial genetic overlap between neurocognition and schizophrenia: genetic modeling in twin samples. Archives of general psychiatry. 2007;64:1348–1355. doi: 10.1001/archpsyc.64.12.1348. [DOI] [PubMed] [Google Scholar]
- 20.Toulopoulou T, et al. Impaired intellect and memory: a missing link between genetic risk and schizophrenia? Archives of general psychiatry. 2010;67:905–913. doi: 10.1001/archgenpsychiatry.2010.99. [DOI] [PubMed] [Google Scholar]
- 21.Owens SF, et al. Genetic overlap between episodic memory deficits and schizophrenia: results from the Maudsley Twin Study. Psychological Medicine. 2011;41:521–532. doi: 10.1017/S0033291710000942. [DOI] [PubMed] [Google Scholar]
- 22.Flint J, Munafo MR. The endophenotype concept in psychiatric genetics. Psychol Med. 2007;37:163–180. doi: 10.1017/S0033291706008750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butcher LM, et al. Generalist genes and cognitive neuroscience. Curr Opin Neurobiol. 2006;16:145–151. doi: 10.1016/j.conb.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Heulens I, Kooy F. Fragile X syndrome: from gene discovery to therapy. Frontiers in bioscience: a journal and virtual library. 2011;16:1211. doi: 10.2741/3785. [DOI] [PubMed] [Google Scholar]
- 25.Silva AJ, et al. Molecular and cellular approaches to memory allocation in neural circuits. Science. 2009;326:391–395. doi: 10.1126/science.1174519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nature reviews. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Visscher PM. Sizing up human height variation. Nature genetics. 2008;40:489–490. doi: 10.1038/ng0508-489. [DOI] [PubMed] [Google Scholar]
- 28.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Need AC, et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Human molecular genetics. 2009;18:4650–4661. doi: 10.1093/hmg/ddp413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabb FW, et al. Challenges in phenotype definition in the whole-genome era: multivariate models of memory and intelligence. Neuroscience. 2009;164:88–107. doi: 10.1016/j.neuroscience.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tam GW, et al. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochemical Society Transactions. 2010;38:445–451. doi: 10.1042/BST0380445. [DOI] [PubMed] [Google Scholar]
- 32.Allen NC, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 33.Ioannidis JP, et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol. 2008;37:120–132. doi: 10.1093/ije/dym159. [DOI] [PubMed] [Google Scholar]
- 34.Helkala EL, et al. Memory functions in human subjects with different apolipoprotein E phenotypes during a 3-year population-based follow-up study. Neurosci Lett. 1996;204:177–180. doi: 10.1016/0304-3940(96)12348-x. [DOI] [PubMed] [Google Scholar]
- 35.Helkala EL, et al. The association of apolipoprotein E polymorphism with memory: a population based study. Neurosci Lett. 1995;191:141–144. doi: 10.1016/0304-3940(95)11575-h. [DOI] [PubMed] [Google Scholar]
- 36.Yarkoni T, et al. Cognitive neuroscience 2.0: building a cumulative science of human brain function. Trends Cogn Sci. 2010;14:489–496. doi: 10.1016/j.tics.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilder RM. Neuropsychology 3.0: evidence-based science and practice. J Int Neuropsychol Soc. 2011;17:7–13. doi: 10.1017/S1355617710001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bilder RM, et al. Cognitive ontologies for neuropsychiatric phenomics research. Cogn Neuropsychiatry. 2009;14:419–450. doi: 10.1080/13546800902787180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Insel TR, Cuthbert BN. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biological psychiatry. 2009;66:988–989. doi: 10.1016/j.biopsych.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 40.Insel T, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 41.Gottesman II, Shields J. Genetic theorizing and schizophrenia. Br J Psychiatry. 1973;122:15–30. doi: 10.1192/bjp.122.1.15. [DOI] [PubMed] [Google Scholar]
- 42.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]