

Abstract
Mosquitoes in the Culex pipiens complex are important vectors of several disease-causing pathogens, including West Nile virus. In North America, the complex consists of Cx. pipiens pipiens form pipiens, Cx. pipiens pipiens form molestus, Cx. pipiens quinquefasciatus, and their hybrids that exhibit substantial diversity in physiology, behavior, and geographic range. Hybridization among these mosquitoes is of concern because of potential implications for disease transmission. Currently, several morphological and molecular markers exist for differentiating members of the Cx. pipiens complex; however, these markers have specific limitations. We report here two highly reliable ribosomal DNA-based single nucleotide polymorphism (SNP) markers, CxpG2T and CxpA2d, for detecting Cx. pipiens complex mosquitoes containing Cx. p. quinquefasciatus alleles. Both CxpG2T and CxpA2d contain one allele that is present in all members of the Cx. pipiens complex, and the other allele is specific to Cx. p. quinquefasciatus. Testing of field populations from the eastern United States showed that these two SNP markers are capable of identifying a south to north gradient of Cx. p. quinquefasciatus and hybrids. The northern limit of detection of Cx. p. quinquefasciatus alleles in this study was in Fort Totten, NY (40.79°N), whereas the southern boundary was determined between Atlanta, GA (33.81°N) and Gainesville, FL (29.64°N). CxpG2T and CxpA2d were more accurate than the ACE-2 marker, and they may conceivably provide comparable resolution with microsatellite markers for detecting Cx. p. quinquefasciatus alleles.
Introduction
Understanding population structure of arthropod vectors is critical for evaluating their roles in disease transmission and developing more effective control strategies. In North America, the Culex pipiens complex consists of Cx. pipiens pipiens form pipiens, Cx. p. pipiens f. molestus, Cx. p. quinquefasciatus, and their hybrids.1 Members of this species complex are principle vectors of several disease-causing pathogens, including West Nile virus (WNV),2,3 and exhibit substantial diversity in physiology, behavior, and geographic range of distribution.
Hybridization among members of the Cx. pipiens complex is of great concern because of the potential implications for disease transmission.4 It seems that limited hybridization occurs between Cx. p. pipiens f. pipiens and Cx. p. pipiens f. molestus.5–7 In contrast, Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus frequently hybridize within a sympatric zone between 36°N and 39°N latitudes in North America.1 Hybrid populations of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus within this zone had been traditionally identified by the relative positions of the dorsal (D) and ventral (V) arms (DV/D) of male phallosome as a morphological marker.1 Recently, Sanogo and others8 identified hybrid individuals beyond 39°N in Champaign, IL using DV/D ratio, whereas Kothera and others9 defined a much wider hybridization zone in the central United States by adopting molecular markers HotAce.2 and microsatellites. Molecular markers (i.e., ACE-210/Ace.211/HotAce.2,9,12 CQ11,13 and microsatellites14,15) have proven to be more accurate for detecting hybrids of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus than the morphological marker DV/D. Microsatellite markers provide more accurate information but are relatively costly compared with morphological markers, and they require specialized equipment. ACE-2/Ace.2/HotAce.2 and CQ11 are less costly but are not as accurate as microsatellites, and CQ11 is too polymorphic to be used as a diagnostic marker.7,8
In this study, we have developed two ribosomal DNA (rDNA) -based single nucleotide polymorphism (SNP) markers, denoted CxpG2T and CxpA2d, that are reliable and easy to use for detecting Cx. p. quinquefasciatus. rDNA has reliably been used as a source of genetic variation in phylogenetic studies.16,17 The internal transcribed spacer 1 (ITS1) and ITS2 regions of rDNA have been extensively explored for species differentiation in a variety of organisms because of their low to moderate rate of polymorphism.18 However, earlier attempts in using ITS1 and ITS2 to differentiate members of the Cx. pipiens complex were unsuccessful because of insufficient variation.19 In this study, we examined the intergenic spacer region (IGS), which exhibits higher rates of variation, contains subrepeats and important promoter, enhancer, and binding sites for protein factors regulating rDNA transcription,20 and may play an important role in adaptation of populations to local environments.21,22 This region has been successfully used in developing molecular markers for other complex mosquito species such as malaria vectors in the Anopheles gambiae species complex.23,24
Materials and Methods
Mosquito sampling and identification.
Populations of Cx. pipiens complex mosquitoes were sampled from 10 different geographic locations (Figure 1). Female mosquitoes were primarily sampled using CO2-baited Centers for Disease Control and Prevention (CDC) miniature light traps or gravid traps baited with hay infusion. Mosquitoes from New York City (NYC) were sampled with hand-held aspirators from above-ground hibernacula located at Fort Totten in the borough of Queens. An underground population of Cx. p. pipiens f. molestus was sampled from sewer catch basins located at 91st Street in the Borough of Manhattan, NYC using a battery-powered modified CDC backpack aspirator (John W. Hock Co., Gainesville, FL). This population of Cx. p. pipiens f. molestus has been found to be a pure Cx. p. pipiens f. molestus population in an earlier study.6 All mosquito specimens from Cambridge, MA, Hartford, CT, Fort Totten, NY, Manhattan, NYC, Trenton, NJ, and Houston, TX, have also been analyzed by microsatellite markers in the previous study.6 Mosquitoes were transported to the laboratory alive in coolers (4–8°C) with ice packs, inactivated on dry ice, or preserved in 95–100% ethanol. Mosquitoes were first identified based on morphological characters25 as members of the Cx. pipiens complex and subsequently, were subjected to a species-specific polymerase chain reaction (PCR) tests based on rDNA19 for confirmation. Identified specimens were either processed immediately for genomic DNA extraction or stored at −80°C.
Figure 1.
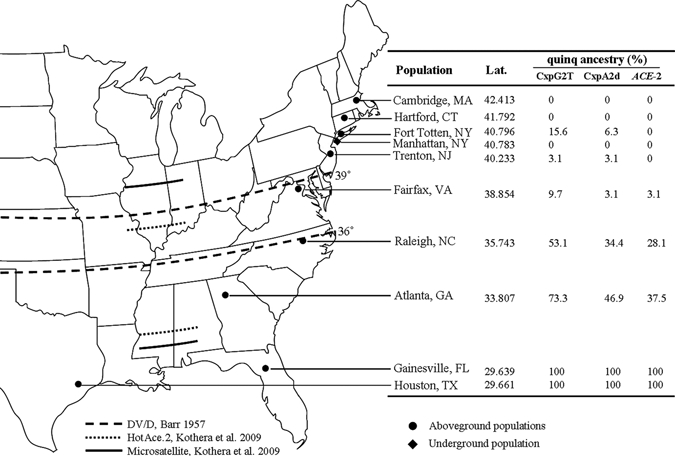
Map showing mosquito sampling sites along the eastern coast of the United States and results of hybrid detection by SNP markers, CxpG2T and CxpA2d, and ACE-2. Results were represented as percent of individuals that bear Cx. p. quinquefasciatus alleles. Dashed, dotted, and solid lines are the hybrid zone boundaries defined by DV/D,1 HotAce. 2,8 and microsatellite8 analyses, respectively.
Genomic DNA extraction.
The thorax of each female mosquito was used for genomic DNA extraction to avoid cross-contamination from sperm stored in the spermatheca. The thorax was homogenized with a disposable microtube pestle (USA Scientific, Enfield, CT) in a 1.5-mL tube containing 180 μL phosphate buffer saline (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4) and subjected to genomic DNA extraction using DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA) according to the manufacturer's recommended protocol for insect tissues. Isolated DNA from each mosquito was reconstituted in 50 μL AE buffer (10 mM Tris·Cl, 0.5 mM ethylenediaminetetraacetic acid [EDTA], pH 9.0; Qiagen) and stored at −20°C for later use.
Amplification, cloning, and sequencing of full-length IGS.
Eukaryotic rDNA is arranged in tandemly repeated units (Figure 2) containing the genes for the 18S, ITS1, 5.8S, ITS2, and 28S (in the order of 5′ end to 3′ end) separated by spacers.26 The sequence flanking the 3′ end of one array and the 5′ end of the next array is known as IGS. A pair of universal primers Cxp28SF and Cxp18SR located at the 3′ end of the 28S and the 5′ end of the 18S, respectively, were designed to amplify the full-length IGS region (Table 1 and Figure 2).
Figure 2.
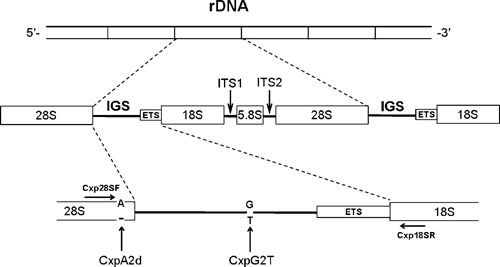
Schematic drawing of the typical organization of eukaryotic rDNA and strategy to amplify intergenic spacer sequences by primers Cxp28sF and Cxp18sR.
Table 1.
Primers used to amplify full-length IGS sequences and perform SNP PCR restriction assays
| Primer name | Primer sequence | Amplicon size |
|---|---|---|
| Cxp28sF | 5′-AGG GAA CGT GAG CTG GGT TTA G-3′ | 2.8 kb |
| Cxp18sR | 5′-TGC CTT CCT TGG ATG TGG TAG C-3′ | |
| CxpG2TF | 5′-AGG ACC CGC AGA GCA TGC CTA CCT TCG G-3′ | 239 bp |
| CxpG2TApaLI | 5′-GTA CGA GAA AGC AGT GCG TCG CCC AAC AG-3′ | |
| CxpA2dF | 5′-TTC GAT GTC GGC TCT TCC TAT CAT TGT G-3′ | 158 bp |
| CxpA2dEcoRI | 5′-TCC GTT AAG ATA GCG TTC GCT GTT GAA-3′ |
F = forward primer; R = reverse primer.
The sequences of the conserved regions used for alignments and design of universal primers were obtained from GenBank. Sequences for the 28S were from insects (GenBank accession numbers M21017 for Drosophila melanogaster, L22060 for Aedes albopictus, and L78065 for An. albimanus), tick (AF291874), copepod (DQ180347), nematodes (AY580057 and AJ512837), and flatworm (EF095264). Sequences for the 18S were from insects (M21017 for D. melanogaster, EU057177 for Helicoverpa assulta, AY216700 for Aphelinus gossypii, and EU179519 for Anastrepha ludens), frog (X04025), bird (TURRR18S), scorpion (AF005442), human (X03205), and rat (X01117).
PCR amplification products from three individuals of each pure population of Cx. p. pipiens f. pipiens (Hartford, CT), Cx. p. pipiens f. molestus (Manhattan, NYC), and Cx. p. quinquefasciatus (Houston, TX) were cloned for full-length IGS identification. The GeneAmp XL PCR Kit (Applied Biosystems, Foster City, CA) that contains rTth DNA polymerase, XL for increased fidelity, and long products was used for PCR amplifications. The 50-μL PCR reactions contained 0.2 μL (~1 ng) genomic DNA, 1× XL Buffer II (Applied Biosystems), 200 μM each dNTP, 0.2 μM each primer, 1.1 mM Mg (OAc)2, and 1.5 units rTth DNA Polymerase XL (Applied Biosystems). PCR reactions were initially denatured at 94°C for 1 minute followed by 16 cycles of amplification at 94°C for 15 seconds and 68°C for 3 minutes and an additional 14 cycles of amplification at 94°C for 15 seconds and 68°C for 3 minutes with 15-second increments per cycle; they were finally extended at 72°C for 10 minutes.
PCR products were analyzed using a 0.8% low-melting agarose gel where bands corresponding to 1.5–5 kb were excised and purified using S.N.A.P. UV Free Gel Purification Kit (Invitrogen, Carlsbad, CA). Purified DNA was cloned into pCR-XL-TOPO vector and subsequently transformed into One Shot TOP10 chemically competent Escherichia coli cells using TOPO XL PCR Cloning Kit (Invitrogen). E. coli cells were spread onto Luria–Bertani (LB) agar plates containing 50 μg/mL kanamycin and cultured overnight. For each clone, five well-isolated colonies were randomly selected and cultured overnight in LB liquid medium containing 50 μg/mL kanamycin. Plasmids from all bacterial colonies were isolated using the QIAprep Miniprep kit (Qiagen, Santa Clara, CA). Plasmid DNAs were then subjected to EcoRI restriction assay to ensure that the cloned fragment was the correct size. Finally, the cloned full-length IGS was sequenced by primer walking.
Sequences were assembled using SeqMan II, Lasergene 7.0 (DNASTAR Inc., Madison, WI). All sequences were verified as rDNA fragments, and putative boundaries were annotated by searching against GenBank and Cx. pipiens genome database (Broad Institute, Cambridge, MA) using nucleotide blast program. Putative full-length IGS sequences were submitted to GenBank (accession numbers GU911305 to GU911348). Finally, all sequences were aligned in SeqMan II to identify SNP loci.
Design of SNP genotyping by restriction assay.
An SNP containing a G–T transversion, designated CxpG2T, was located downstream of IGS (Figure 2). Another SNP containing A to deletion (hereafter called −) mutation, designated CxpA2d, was identified at the 3′ end of the 28S (Figure 2).
For CxpG2T, allele G is specific to Cx. p. quinquefasciatus, and T is present in all members of the Cx. pipiens complex, indicating that the genotypes for Cx. p. pipiens f. pipiens, Cx. p. pipiens f. molestus, and Cx. p. quinquefasciatus and hybrids of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus would be TT, TT, GT, and GT. When T is present, the site forms a restriction site GTGCAC for enzyme ApaLI. A pair of primers, CxpG2TF and CxpG2TApaLI (Table 1), flanking the restriction site were designed to amplify a 239-bp fragment (Figure 3A). Digestion of this fragment with ApaLI generates two fragments of 119 and 120 bp when T is present and one intact 239-bp fragment when G is present (Figure 3B). Thus, one band of 119/120 bp would be present for Cx. p. pipiens f. pipiens and Cx. p. pipiens f. molestus, and two individual bands containing 239-bp and 119/120-bp fragments, respectively, are present for Cx. p. quinquefasciatus and hybrids of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus (Figure 3B).
Figure 3.
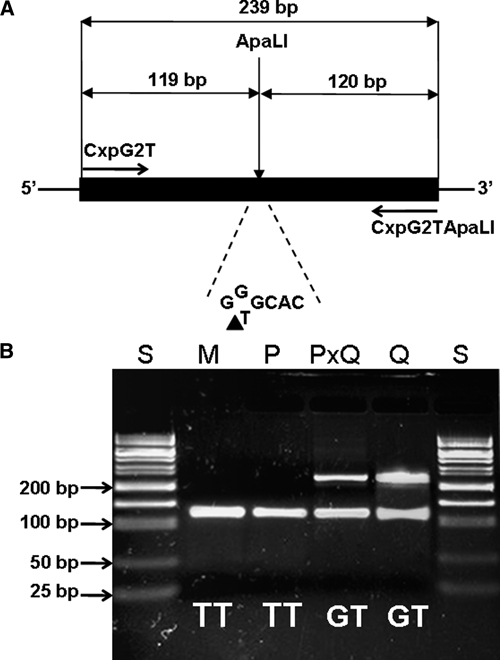
Design of CxpG2T SNP genotyping by PCR-ApaLI restriction assay. (A) Strategy of PCR amplification for G to T mutation. When T is present, the site contains an ApaLI sequence (GTGCAC). The solid black bar indicates the region corresponding to amplicon. The black triangle indicates the cutting position of restriction enzyme ApaLI. (B) Electrophoretic patterns of digested PCR products amplified by primers CxpG2TF and CxpG2TApaLI. S = 50 bp DNA size standard; M = pure Cx. p. pipiens f. molestus; P = pure Cx. p. pipiens f. pipiens; PxQ = hybrid of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus; Q = pure Cx. p. quinquefasciatus.
For CxpA2d, allele A is specific to Cx. p. quinquefasciatus, and deletion (d) is present in all members of the Cx. pipiens complex, indicating that the genotypes of Cx. p. pipiens f. pipiens and Cx. p. pipiens form molestus, Cx. p. quinquefasciatus, and hybrids of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus would be −, −, A−, and A−, respectively. To detect allele A, primer set CxpA2dF and CxpA2dEcoRI (Table 1) amplifying a 158-bp fragment was designed so that the reverse primer (CxpA2dEcoRI) contained a T to A mismatch at the second position at the 3′ end (Figure 4A). Introduction of this mismatch creates a recognition site GAATTC for the restriction enzyme EcoRI when A is present (Figure 4A). On EcoRI digestion, the amplicon would be cut into 131- and 27-bp fragments if only A is present and would be intact if only deletion is present (Figure 4B). Thus, one band containing a 158-bp fragment would be present for Cx. p. pipiens f. pipiens and Cx. p. pipiens f. molestus, and three bands containing 158, 131, and 27 bp, respectively, would be present for Cx. p. quinquefasciatus and its hybrids with Cx. p. pipiens f. pipiens (Figure 4B).
Figure 4.
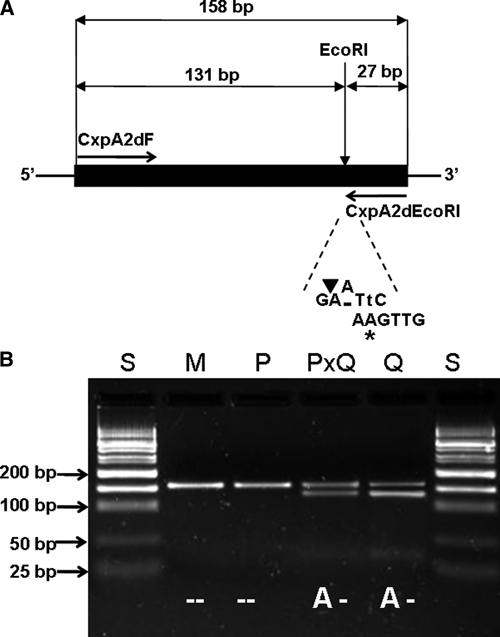
Design of CxpA2d SNP genotyping by PCR-EcoRI restriction assay. (A) Strategy of introducing mismatched PCR for A to deletion mutation. The nucleotide (A) marked with an asterisk indicates the mismatch (originally T) incorporated into the CxpA2dEcoRI primer to create a new EcoRI site (GAATTC) at the mutation location. The nucleotide in lower case indicates the complementary mismatch incorporated into the amplicon after PCR amplification. The solid black bar indicates the region corresponding to the amplicon. The black triangle indicates the cutting position of restriction enzyme EcoRI. (B) Electrophoretic patterns of digested PCR products amplified by primers CxpA2dF and CxpA2dEcoRI. S = 50 bp DNA size standard; M = pure Cx. p. pipiens f. molestus; P = pure Cx. p. pipiens f. pipiens; PXQ = hybrid of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus; Q = Cx. p. quinquefasciatus.
Both CxpG2T and CxpA2d contain one allele that is present in all members of the Cx. pipiens complex, whereas the other allele is specific to Cx. p. quinquefasciatus. This polarity determines that CxpG2T and CxpA2d exclusively detect individuals that contain Cx. p. quinquefasciatus genetic signature, but they do not differentiate pure Cx. p. quinquefasciatus and its hybrids with Cx. p. pipiens f. pipiens; this is because both contain the two alleles. These two SNP markers also do not differentiate Cx. p. pipiens f. pipiens, Cx. p. pipiens f. molestus, or their hybrids.
PCR reactions for both SNPs were performed in 20 μL reaction mix containing 0.4 μL genomic DNA, 2 μL 10× PCR buffer II (Applied Biosystems), 2 μL 25 mM MgCl2, 0.4 μL 10 mM 2′-deoxynucleoside 5′-triphosphate (dNTP) mix, 0.4 μL 10 mg/μL bovine serum albumin (BSA), 0.2 uM each primer, and 0.5 units AmpliTaq Gold DNA polymerase (Applied Biosystems). PCR reactions were initially denatured at 95°C for 10 minutes followed by 30 cycles of amplification at 95°C for 15 seconds, 65°C for CxpG2T or 61°C for CxpA2d for 20 seconds, and 72°C for 20 seconds, and finally extended at 72°C for 7 minutes. ApaLI digestion assays were conducted in 30 μL containing 10 μL PCR product, 2 μL 10× buffer Tango (Fermentas, Ontario, Canada), and 10 units ApaLI (Alw44I) and incubated at 37°C for 4 hours. EcoRI digestion assays were carried out in 30 μL containing 10 μL PCR product, 2 μL 10× NEBuffer EcoRI (New England Biolabs, Ipswich, MA), 0.2 μL 10 μg/μL BSA, and 15 units EcoRI (New England Biolabs) and incubated at 37°C for 4 hours. Digested PCR products were visualized under ultraviolet (UV) light on ethidium bromide-stained 2.5% agarose gel.
Field testing.
To validate the two SNP markers, 32 adult female mosquitoes for each population were subjected to PCR restriction assays. To ensure that the two SNPs were not created by polymerase errors and/or cloning artifacts, one specimen for each genotype was randomly selected from each field population and subjected to PCR amplification by using a proofreading enzyme, AccuPrim Pfx DNA Polymerase (Invitrogen). PCR products were then directly sequenced to examine the presences of each allele. For comparison purposes, hybrid detection by ACE-2 marker was also performed in parallel on all populations.
Results
We obtained 44 cloned full-length IGS sequences (accession numbers GU911305 to GU911348), all of which had approximately the same size of 1.8 kb because of the lack of 60 bp, ~200 bp, or other size subrepeats commonly found in other Dipteran insects. The IGS region in the present study contained many microsatellite sequences (simple sequence repeats) of mostly 2–4 bp. When all microsatellite sequences were removed, the IGS sequences shared greater than 90% identity (data not shown). There was greater sequence identity in the IGS region among individuals within each group of Cx. pipiens complex mosquitoes compared with that of individuals from different groups (Figure 5). However, a few individuals from the Cx. p. pipiens f. pipiens group exhibited higher sequence identity to individuals from Cx. p. quinquefasciatus in the IGS region than those individuals within each group. Cx. p. pipiens f. molestus mosquitoes showed closer relatedness to Cx. p. pipiens f. pipiens mosquitoes in the IGS sequences than they did to Cx. p. quinquefasciatus (Figure 5). These full-length IGS sequences also contained several SNPs. By thorough screening of these SNPs, we identified two markers, designated CxpG2T and CxpA2d, that consistently detected the Cx. p. quinquefasciatus genetic signature among Cx. pipiens complex mosquitoes.
Figure 5.
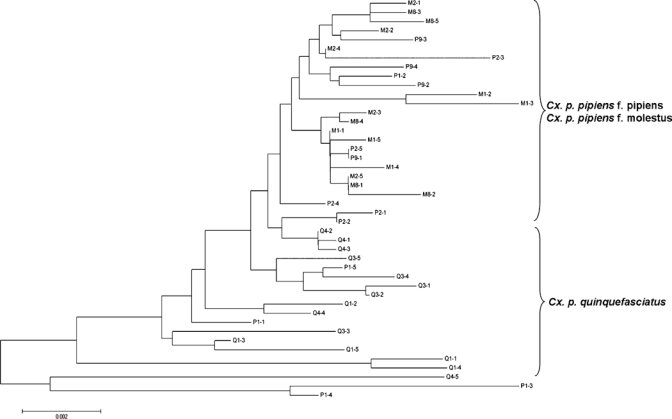
Neighbor-joining phylogenetic tree of the cloned IGS sequences from Cx. pipiens complex mosquitoes. The optimal tree with the sum of the branch length equal to 0.10589842 is shown. The tree is drawn to scale, with branch lengths in the same units as those units of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the maximum composite likelihood method, and analyses were conducted in software MEGA5.43 Letters in the sequence labels were P, M, and Q, representing Cx. p. pipiens f. pipiens, Cx. p. pipiens f. molestus, and Cx. p. quinquefasciatus, respectively. Numbers after the letters P, M, and Q depict individual mosquito identification followed by the clone number.
CxpG2T and CxpA2d successfully amplified corresponding sequences from all field-sampled specimens. The restriction banding patterns were consistent. Testing of all field populations enabled us to identify a south to north gradient of Cx. p. quinquefasciatus and hybrids. The southernmost and northernmost ends contained pure Cx. p. quinquefasciatus and Cx. p. pipiens f. pipiens, respectively. Varying degrees of individuals with Cx. p. quinquefasciatus genetic signature were identified at both the northernmost and southernmost end of the hybridization zone (Figure 1). The northern detection limit of Cx. p. quinquefasciatus genetic signature by CxpG2T and CxpA2d was at Fort Totten, NY.
CxpG2T showed greater resolution than CxpA2d in detecting individuals bearing Cx. p. quinquefasciatus alleles within the hybridization zone except in Trenton, NJ (Figure 1). Likewise, both CxpG2T and CxpA2d were more reliable than ACE-2 except in Fairfax, VA. The northern detection limit of Cx. p. quinquefasciatus genetic signature by ACE-2 was also much farther south (Fairfax, VA; 38.854°N) compared with Fort Totten, NY (40.796°N) as identified by CxpG2T and CxpA2d.
All mosquitoes sampled from the southernmost locations (Gainesville, FL and Houston, TX) contained genetic signatures of Cx. p. quinquefasciatus, which were examined by CxpG2T, CxpA2d, and ACE-2, suggesting that the southern boundary of the hybridization zone of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus is likely located between Atlanta, GA and Gainesville FL, approximately 32°N. None of the markers used in the present study identified Cx. p. quinquefasciatus alleles in any individuals in the Cx. p. pipiens f. molestus population sampled from an underground habitat in Manhattan, NYC.
Discussion
Understanding genetic variation and species distribution boundaries is vital for evaluating the respective role of local vector populations in disease transmission. Among molecular markers for population genetic studies, SNPs offer rapid and cost-efficient technology to resolve some of the outstanding questions regarding species differentiation and hybrid groups. Based on the rDNA IGS region, we developed two SNP markers, CxpG2T and CxpA2d, to evaluate the extent of introgression of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus in Cx. pipiens complex and redefine the hybridization zone in the eastern United States that extends beyond the currently recognized boundaries.
Although the core domains of the rDNA coding regions have been highly conserved, the IGS region intervening between the 28S and 18S rRNA subunits often displays sequence divergence and length variation among members of closely related species21,27 or among local populations of the same species,28,29 thus allowing differentiation of morphologically inseparable cryptic species and characterization of genetic structure in mosquito vector populations.17,28–30 The full-length IGS sequences obtained in our study had approximately the same size because of the lack of 60 bp, ~200 bp, or other size subrepeats that are found in other insect species, including Drosophila spp. (IGS length = 3.3–11.6 kb),21 Ae. albopictus (2–10 kb),28 and An. sinensis (3.2–4.3 kb)31 (Figure 6). The lack of subrepeats has also been reported in mosquitoes Ae. aegypti (1.8 kb)32 and An. gambiae (1.4 kb)30 and in the copepod Tigriopus californicus (2.8 kb)33 (Figure 6).
Figure 6.
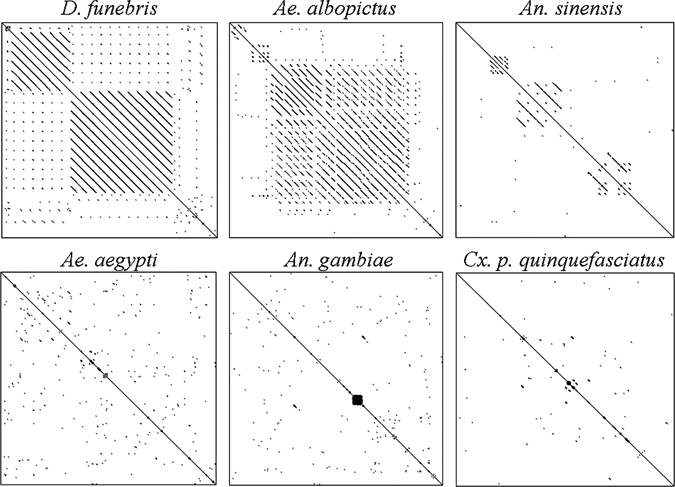
Dot plot matrices of IGS sequences of six Dipteran insect species revealing different sizes of subrepeats. Symmetric lines forming a square box along the central diagonal indicate presence of a subrepeat. The size of the square dictates the size of the subrepeat. Dot plots were generated using the Dotlet program44 with an 11-bp sliding window. Symmetric dots or lines along the diagonal represent the repeat number and length. GenBank accession numbers for IGS sequences used for the plot were AF004986 (Ae. aegypti), AGU10135 (An. gambiae), AF498237 (An. sinensis), M65063 (Ae. albopictus), L17048 (D. funebris), and GU911335 (Cx. p. quinquefasciatus).
CxpG2T and CxpA2d showed better resolution than ACE-2 in detecting individuals containing Cx. p. quinquefasciatus alleles. rDNA-based molecular markers do not usually follow Mendelian mode of inheritance in eukaryotes because of the presence of multiple copies of rDNA arranged in tandems.26 Sequence variation among multiple copies in the same rDNA may compensate for the Mendelian dilution effect of the genotype frequency in hybrids after Fn (N > 1) generations and lead to greater resolution of CxpG2T and CxpA2d in contrast to ACE-2, which follows a simple Mendelian inheritance pattern. In the northern end of hybridization in Fort Totten, NY, CxpG2T and CxpA2d surprisingly identified 15.6% and 6.3% presumable hybrids, respectively, that had not been previously recognized. A plausible interpretation would be that, by using a single marker, it is not possible to calculate ancestry coefficient to determine if all individuals are true hybrids, because some individuals might have contained a very low level of Cx. p. quinquefasciatus signature but were still scored as hybrids. It is also noteworthy that, in our previous study using 12 microsatellite markers, we identified 2.1% hybrids of Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus in the aforementioned population (data not shown).6 In general, a higher proportion of hybrids can be expected if fewer microsatellite markers are used. The SNP marker CxpG2T had better resolution than CxpA2d in detecting individuals bearing Cx. p. quinquefasciatus alleles. This finding is most likely because of the location of CxpG2T at the highly polymorphic IGS region, whereas CxpA2d is located at the conserved region of the 3′ end of 28S.
Neither CxpG2T nor CxpA2d detected Cx. p. quinquefasciatus alleles in individuals of the Cx. p. pipiens f. molestus population sampled from the underground habitat in Manhattan, NYC. This finding is consistent with the results of other studies indicating that introgression between Cx. p. pipiens f. molestus and Cx. p. quinquefasciatus does not exist, most likely because of distribution and/or habitat isolation.4,6,9 CxpG2T and CxpA2d did not differentiate Cx. p. pipiens f. pipiens and Cx. p. pipiens f. molestus. Based on recent genetic studies and increasing evidence that Cx. p. pipiens f. molestus might have originated from the nearby aboveground populations of Cx. p. pipiens f. pipiens,6,7,34,35 it is likely that Cx. p. pipiens f. molestus inherited the same polymorphism at CxpG2T and CxpA2d loci from Cx. p. pipiens f. pipiens.
Two major hypotheses have been proposed on the formation of current Cx. pipiens complex in North America. Barr36 postulated that an ancestral form of Cx. p. pipiens f. pipiens in tropical Africa (Ethiopian region) invaded northern temperate regions in North America. Some of the Cx. p. pipiens f. pipiens thrived in this region, giving rise to today's Cx. p. pipiens f. pipiens, and others subsequently spread slowly to the southern portion of the region to form the current Cx. p. quinquefasciatus population. Microsatellite studies have shown that Cx. p. quinquefasciatus populations in the United States have much lower genetic diversity compared with Cx. p. pipiens f. pipiens, which seems to support the hypothesis that Cx. p. quinquefasciatus was derived from Cx. p. pipiens f. pipiens.6,9,37 However, Ross38 stated that Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus were independently introduced to North America, with Cx. p. pipiens f. pipiens coming from Europe by sailing ships and Cx. p. quinquefasciatus coming from Africa by slave ships. Testing of these hypotheses could be a challenging task further complicated by multiple mosquito introductions because of increasing global trade and transportation. Recent global genetic analyses of Cx. pipiens complex mosquitoes have not permitted clear identification of the origin of Cx. p. quinquefasciatus in North America,37,39 and at least a second introduction of Cx. p. quinquefasciatus into Hawaii has been suggested.39,40 In our study, both CxpG2T and CxpA2d contain one allele that is present in all Cx. pipiens complex members, whereas the other allele is specific to pure Cx. p. quinquefasciatus, allowing for the detection of a south to north gradient penetration of Cx. p. quinquefasciatus ancestry. This polarity in CxpG2T and CxpA2d suggests that Cx. p. pipiens f. pipiens and Cx. p. quinquefasciatus may have been derived from a single ancestral population.
In conclusion, we found that IGS-based SNP markers CxpG2T and CxpA2d are reliable and cost-effective (minimal cost incurred by the additional restriction digestion step) for the identification of Cx. pipiens complex mosquitoes bearing Cx. p. quinquefasciatus genetic signature and reexamination of the hybridization zone. Our finding also indicates that the IGS region can be successfully used for the development of convenient markers such as SNPs, and in conjunction with other molecular markers (e.g., microsatellite markers), it can accommodate characterization of genetic variation, identification of morphologically identical incipient species, and understanding of the evolution of Cx. pipiens complex mosquitoes. Finally, with the release of the Cx. p. quinquefasciatus genome sequence,41,42 it is anticipated that additional studies will take advantage of the genome sequence to develop higher-resolution markers for examining genetic variation pertinent to population differentiation as well as associations with heterogeneity in disease transmission.
ACKNOWLEDGMENTS
The authors thank Ary Farajollahi, Mercer County Mosquito Control for providing mosquitoes from Trenton, NJ; Dr. Jorge Arias, Fairfax County Disease Carrying Insects Program for sampling mosquitoes from Fairfax, VA; Dr. Charles Apperson, North Carolina State University for providing mosquitoes from Raleigh, NC; Dr. Rosemarie Kelly, Georgia Department of Health for providing mosquitoes from Atlanta, GA; and Dr. Daniel Kline, US Department of Agriculture, Agricultural Research Service Mosquito and Fly Research Unit for sampling mosquitoes from Gainesville, FL. We are also grateful for the technical assistance of the Connecticut Agricultural Experiment Station support staff, John Sheppard, and Michael Thomas.
Footnotes
Financial support: Funding for this research was provided in part by Laboratory Capacity for Infectious Diseases Cooperative Agreement number U50/CCU6806-01-1 from the Centers for Disease Control and Prevention, US Department of Agriculture Specific Cooperative Agreement number 58-6615-1-218, and US Department of Agriculture-administered Hatch funds CONH00768 (to T.G.A.).
Authors' addresses: Shaoming Huang, Goudarz Molaei, and Theodore G. Andreadis, Center for Vector Biology and Zoonotic Diseases, Connecticut Agricultural Experiment Station, New Haven, Connecticut, E-mails: Shuang@sjmosquito.org, Goudarz.Molaei@ct.gov, and Theodore.Andreadis@ct.gov.
References
- 1.Barr AR. The distribution of Culex p. pipiens and Cx. p. quinquefasciatus in North America. Am J Trop Med Hyg. 1957;6:153–165. doi: 10.4269/ajtmh.1957.6.153. [DOI] [PubMed] [Google Scholar]
- 2.Vinogradova EB. Taxonomy, Distribution, Ecology, Physiology, Genetics, Applied Importance and Control. Sofia, Bulgaria: Pensoft Publishers; 2000. (Culex Pipiens Pipiens Mosquitoes). [Google Scholar]
- 3.Hayes EB, Komar N, Nasci RS, Montgomery SP, O'Leary DR, Campbell GL. Epidemiology and transmission dynamics of West Nile virus disease. Emerg Infect Dis. 2005;11:1167–1173. doi: 10.3201/eid1108.050289a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang S, Hamer GL, Molaei G, Walker ED, Andreadis TG. Genetic variation associated with mammalian feeding in Culex pipiens from a West Nile virus epidemic region in Chicago, Illinois. Vector Borne Zoonotic Dis. 2009;9:637–642. doi: 10.1089/vbz.2008.0146. [DOI] [PubMed] [Google Scholar]
- 5.Gomes B, Sousa C, Novo M, Freitas F, Alves R, Corte-Real A, Salgueiro P, Donnelly M, Almeida A, Pinto J. Asymmetric introgression between sympatric molestus and pipiens forms of Culex pipiens (Diptera: Culicidae) in the Comporta region, Portugal. BMC Evol Biol. 2009;9:262. doi: 10.1186/1471-2148-9-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang S, Molaei G, Andreadis TG. Genetic insights into the population structure of Culex pipiens (Diptera: Culicidae) in the northeastern United States by using microsatellite analysis. Am J Trop Med Hyg. 2008;79:518–527. [PubMed] [Google Scholar]
- 7.Kothera L, Godsey M, Mutebi J-P, Savage HM. A comparison of aboveground and belowground populations of Culex pipiens (Diptera: Culicidae) mosquitoes in Chicago, Illinois, and New York City, New York, using microsatellites. J Med Entomol. 2010;47:805–813. doi: 10.1603/me10031. [DOI] [PubMed] [Google Scholar]
- 8.Sanogo YO, Kim CH, Lampman R, Halvorsen JG, Gad AM, Novak RJ. Identification of male specimens of the Culex pipiens complex (Diptera: Culicidae) in the hybrid zone using morphology and molecular techniques. J Med Entomol. 2008;45:203–209. doi: 10.1603/0022-2585(2008)45[203:iomsot]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 9.Kothera L, Zimmerman EM, Richards CM, Savage HM. Microsatellite characterization of subspecies and their hybrids in Culex pipiens complex (Diptera: Culicidae) mosquitoes along a north-south transect in the central united states. J Med Entomol. 2009;46:236–248. doi: 10.1603/033.046.0208. [DOI] [PubMed] [Google Scholar]
- 10.Smith JL, Fonseca DM. Rapid assays for identification of members of the Culex (Culex) pipiens complex, their hybrids, and other sibling species (Diptera: Culicidae) Am J Trop Med Hyg. 2004;70:339–345. [PubMed] [Google Scholar]
- 11.Aspen S, Savage HM. Polymerase chain reaction assay identifies North American members of the Culex pipiens complex based on nucleotide sequence differences in the acetylcholinesterase gene Ace.2. J Am Mosq Control Assoc. 2003;19:323–328. [PubMed] [Google Scholar]
- 12.Savage HM, Aggarwal D, Apperson CS, Katholi CR, Gordon E, Hassan HK, Anderson M, Charnetzky D, McMillen L, Unnasch EA, Unnasch TR. Host choice and West Nile virus infection rates in blood-fed mosquitoes, including members of the Culex pipiens complex, from Memphis and Shelby County, Tennessee, 2002–2003. Vector Borne Zoonotic Dis. 2007;7:365–386. doi: 10.1089/vbz.2006.0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahnck CM, Fonseca DM. Rapid assay to identify the two genetic forms of Culex (Culex) pipiens L. (Diptera: Culicidae) and hybrid populations. Am J Trop Med Hyg. 2006;75:251–255. [PubMed] [Google Scholar]
- 14.Keyghobadi N, Matrone MA, Ebel GD, Kramer LD, Fonseca DM. Microsatellite loci from the northern house mosquito (Culex pipiens), a principal vector of West Nile virus in North America. Mol Ecol Notes. 2004;4:20–22. [Google Scholar]
- 15.Smith JL, Keyghobadi N, Matrone MA, Escher RL, Fonseca DM. Cross-species comparison of microsatellite loci in the Culex pipiens complex and beyond. Mol Ecol Notes. 2005;5:697–700. [Google Scholar]
- 16.Hillis DM, Dixon MT. Ribosomal DNA: molecular evolution and phylogenetic inference. Q Rev Biol. 1991;66:411–453. doi: 10.1086/417338. [DOI] [PubMed] [Google Scholar]
- 17.Collins FH, Paskewitz SM. A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Mol Biol. 1996;5:1–9. doi: 10.1111/j.1365-2583.1996.tb00034.x. [DOI] [PubMed] [Google Scholar]
- 18.Baldwin BG, Sanderson MJ, Porter JM, Wojciechowski MF, Campbell CS, Donoghue MJ. The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann Mo Bot Gard. 1995;82:247–277. [Google Scholar]
- 19.Crabtree MB, Savage HM, Miller BR. Development of a species-diagnostic polymerase chain reaction assay for the identification of Culex vectors of St. Louis encephalitis virus based on interspecies sequence variation in ribosomal DNA spacers. Am J Trop Med Hyg. 1995;53:105–109. [PubMed] [Google Scholar]
- 20.Moss T, Stefanovsky VY. Promotion and regulation of ribosomal transcription in eukaryotes by RNA polymerase I. Prog Nucleic Acid Res Mol Biol. 1995;50:25–66. doi: 10.1016/s0079-6603(08)60810-7. [DOI] [PubMed] [Google Scholar]
- 21.Mateos M, Markow TA. Ribosomal intergenic spacer (IGS) length variation across the Drosophilinae (Diptera: Drosophilidae) BMC Evol Biol. 2005;5:46. doi: 10.1186/1471-2148-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorokhova E, Dowling TE, Weider LJ, Crease TJ, Elser JJ. Functional and ecological significance of rDNA intergenic spacer variation in a clonal organism under divergent selection for production rate. Proc Biol Sci. 2002;269:2373–2379. doi: 10.1098/rspb.2002.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Favia G, della Torre A, Bagayoko M, Lanfrancotti A, Sagnon N, Toure YT, Coluzzi M. Molecular identification of sympatric chromosomal forms of Anopheles gambiae and further evidence of their reproductive isolation. Insect Mol Biol. 1997;6:377–383. doi: 10.1046/j.1365-2583.1997.00189.x. [DOI] [PubMed] [Google Scholar]
- 24.Wilkins EE, Howell PI, Benedict MQ. IMP PCR primers detect single nucleotide polymorphisms for Anopheles gambiae species identification, Mopti and Savanna rDNA types, and resistance to dieldrin in Anopheles arabiensis. Malar J. 2006;5:125. doi: 10.1186/1475-2875-5-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darsie RFJ, Ward RA. Identification and geographical distribution of the mosquitoes of North America, north of Mexico. Gainesville, FL: University Press of Florida; 2005. [Google Scholar]
- 26.Paule MR. In: Transcription of Ribosomal RNA Genes by Eukaryotic RNA Polymerase I. Paule MR, editor. Berlin, Germany: Springer-Verlag; 1998. pp. 1–8. (Class I genes). [Google Scholar]
- 27.Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW. Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA. 1984;81:8014–8018. doi: 10.1073/pnas.81.24.8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Black WC, McLain DK, Rai KS. Patterns of variation in the rDNA cistron within and among world populations of a mosquito, Aedes albopictus (Skuse) Genetics. 1989;121:539–550. doi: 10.1093/genetics/121.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Merida AM, De Mata MP, Molina E, Porter CH, Black WC., IV Variation in ribosomal DNA intergenic spacers among populations of Anopheles albimanus in South and Central America. Am J Trop Med Hyg. 1995;53:469–477. doi: 10.4269/ajtmh.1995.53.469. [DOI] [PubMed] [Google Scholar]
- 30.Scott JA, Brogdon WG, Collins FH. Identification of single specimens of the Anopheles gambiae complex by the polymerase chain reaction. Am J Trop Med Hyg. 1993;49:520–529. doi: 10.4269/ajtmh.1993.49.520. [DOI] [PubMed] [Google Scholar]
- 31.Whang IJ, Jung J, Park JK, Min GS, Kim W. Intragenomic length variation of the ribosomal DNA intergenic spacer in a malaria vector, Anopheles sinensis. Mol Cells. 2002;14:158–162. [PubMed] [Google Scholar]
- 32.Wu CC, Fallon AM. Analysis of a ribosomal DNA intergenic spacer region from the yellow fever mosquito, Aedes aegypti. Insect Mol Biol. 1998;7:19–29. doi: 10.1046/j.1365-2583.1998.71194.x. [DOI] [PubMed] [Google Scholar]
- 33.Burton RS, Metz EC, Flowers JM, Willett CS. Unusual structure of ribosomal DNA in the copepod Tigriopus californicus: intergenic spacer sequences lack internal subrepeats. Gene. 2005;344:105–113. doi: 10.1016/j.gene.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Byrne K, Nichols RA. Culex pipiens in London underground tunnels: differentiation between surface and subterranean populations. Heredity. 1999;82:7–15. doi: 10.1038/sj.hdy.6884120. [DOI] [PubMed] [Google Scholar]
- 35.Kent RJ, Harrington LC, Norris DE. Genetic differences between Culex pipiens f. molestus and Culex pipiens pipiens (Diptera: Culicidae) in New York. J Med Entomol. 2007;44:50–59. doi: 10.1603/0022-2585(2007)44[50:gdbcpf]2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barr AR. Occurrence and distribution of the Culex pipiens complex. Bull World Health Organ. 1967;37:293–296. [PMC free article] [PubMed] [Google Scholar]
- 37.Fonseca DM, Keyghobadi N, Malcolm CA, Mehmet C, Schaffner F, Mogi M, Fleischer RC, Wilkerson RC. Emerging vectors in the Culex pipiens complex. Science. 2004;303:1535–1538. doi: 10.1126/science.1094247. [DOI] [PubMed] [Google Scholar]
- 38.Ross HH. The colonization of temperate North America by mosquitoes and man. Mosq News. 1964;24:103–118. [Google Scholar]
- 39.Fonseca DM, Smith JL, Wilkerson RC, Fleischer RC. Pathways of expansion and multiple introductions illustrated by large genetic differentiation among worldwide populations of the southern house mosquito. Am J Trop Med Hyg. 2006;74:284–289. [PubMed] [Google Scholar]
- 40.Fonseca DM, LaPointe DA, Fleischer RC. Bottlenecks and multiple introductions: population genetics of the vector of avian malaria in Hawaii. Mol Ecol. 2000;9:1803–1814. doi: 10.1046/j.1365-294x.2000.01070.x. [DOI] [PubMed] [Google Scholar]
- 41.Bartholomay LC, Waterhouse RM, Mayhew GF, Campbell CL, Michel K, Zou Z, Ramirez JL, Das S, Alvarez K, Arensburger P, Bryant B, Chapman SB, Dong Y, Erickson SM, Karunaratne SHPP, Kokoza V, Kodira CD, Pignatelli P, Shin SW, Vanlandingham DL, Atkinson PW, Birren B, Christophides GK, Clem RJ, Hemingway J, Higgs S, Megy K, Ranson H, Zdobnov EM, Raikhel AS, Christensen BM, Dimopoulos G, Muskavitch MAT. Pathogenomics of Culex quinquefasciatus and meta-analysis of infection responses to diverse pathogens. Science. 2010;330:88–90. doi: 10.1126/science.1193162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arensburger P, Megy K, Waterhouse RM, Abrudan J, Amedeo P, Antelo B, Bartholomay L, Bidwell S, Caler E, Camara F, Campbell CL, Campbell KS, Casola C, Castro MT, Chandramouliswaran I, Chapman SB, Christley S, Costas J, Eisenstadt E, Feschotte C, Fraser-Liggett C, Guigo R, Haas B, Hammond M, Hansson BS, Hemingway J, Hill SR, Howarth C, Ignell R, Kennedy RC, Kodira CD, Lobo NF, Mao C, Mayhew G, Michel K, Mori A, Liu N, Naveira H, Nene V, Nguyen N, Pearson MD, Pritham EJ, Puiu D, Qi Y, Ranson H, Ribeiro JMC, Roberston HM, Severson DW, Shumway M, Stanke M, Strausberg RL, Sun C, Sutton G, Tu Z, Tubio JMC, Unger MF, Vanlandingham DL, Vilella AJ, White O, White JR, Wondji CS, Wortman J, Zdobnov EM, Birren B, Christensen BM, Collins FH, Cornel A, Dimopoulos G, Hannick LI, Higgs S, Lanzaro GC, Lawson D, Lee NH, Muskavitch MAT, Raikhel AS, Atkinson PW. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science. 2010;330:86–88. doi: 10.1126/science.1191864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011 doi: 10.1093/molbev/msr121. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Junier T, Pagni M. Dotlet: diagonal plots in a web browser. Bioinformatics. 2000;16:178–179. doi: 10.1093/bioinformatics/16.2.178. [DOI] [PubMed] [Google Scholar]