

Abstract
Tahyna virus (TAHV) is widely distributed in Europe and Asia. A previous study reported a high level of conservation of the TAHV genome in isolates from Europe. During 2006 and 2007, three Tahyna virus isolates from mosquitoes were obtained from various locations in Xinjiang, People's Republic of China. We analyzed the complete coding sequence of full-length small, medium, and large segments of these isolates. Molecular and phylogenetic analyses of the three complete TAHV genomes showed that sequence identity between isolates from China and Europe was more divergent, and an unexpected level of medium segment diversity was found among isolates from China compared with high levels of sequence conservation for the small and large segments. This study indicated that effects of genotypic diversity on the ecology, transmission, and pathogenicity of TAHV in China should be studied.
Tahyna virus (TAHV) is a member of the California serogroup of the family Bunyaviridae and the genus Orthobunyavirus. Viruses of the genus Orthobunyavirus are arthropod-borne and possess a single-stranded, negative-sense RNA genome of three segments designated small (S), medium (M), and large (L), which encode the nucleocapsid (N), the surface polyprotein, and the polymerase protein, respectively. Tahyna virus was first isolated in Czechoslovakia in 1958 from a pool of Aedes caspius mosquitoes.1
Since that time, TAHV has been found and shown to be associated with human illness throughout parts of Europe and Asia.2–4 Typical clinical manifestations of TAHV infection include influenza-like symptoms of fever and respiratory congestion; the infection may also cause acute arthritis, pharyngitis, and complications of the central nervous system.5 Previous studies identified a high level of conservation of the TAHV genome, even though viruses were obtained from temporally distinct regions over a long period.6,7
In 2006, TAHV was isolated for the first time in the People's Republic of China, and human infection has been confirmed in this country.8,9 The first TAHV isolate (XJ0625) from China was obtained from Culex sp. mosquitoes in Kashi, Xinjiang.8 Subsequently, in 2007, two TAHV isolates (XJ0708 and XJ0710) were obtained from separate pools of Aedes vexans mosquitoes obtained in Bayinguoleng, Xinjiang, approximately 900 km east of Kashi. This report describes comprehensive genomic characterization of these TAHV isolates from China. Molecular and phylogenetic analyses of the N protein, polyprotein, and polymerase open reading frames (ORFs) indicate a relatively high level of M segment diversity compared with that of the S and L segments of these TAHVs from China.
The TAHV isolates were obtained from mosquito pools. These pools were obtained as part of arthropod-borne virus surveillance efforts in the People's Republic of China. All procedures used were performed according to manufacturer's instructions unless stated otherwise. Mosquitoes were collected in light traps, identified by morphologic characteristics, and separated by genus/species/collection site into pools of up to 50–100 specimens. The pools were triturated in minimal essential medium and clarified by centrifugation. One hundred microliters of resultant supernatants was then added to confluent layers of BHK-21 cells in six-well plates. Inoculated cells were then incubated at 37°C and inspected daily for cytopathic effects. Cells infected with supernatant from mosquito pools XJ0625 (containing 100 Culex mosquitoes that were not identified to species), XJ0708, and XJ0710 (containing 60 and 70 Ae. vexans mosquitoes in each pool, respectively) showed initial signs of cytopathic effects at day one post-inoculation. Supernatants from these cells were collected at day 2 post-inoculation and were inoculated into 25-cm2 flasks containing BHK-21 cells.
Viral RNA was extracted directly from 140 μL of infected, second passage BHK-21 cell culture supernatants of XJ0625, XJ0708 and XJ0710 by using the QiaAMP Viral RNA Extraction Kit (Qiagen, Valencia, CA). After extraction, cDNAs were randomly transcribed from purified viral RNAs by using Ready-To-Go™ You-Prime First-Strand Beads (GE Healthcare, Little Chalfont, United Kingdom) and random hexanucleotide primers. The XJ0625, XJ0708, and XJ0710 isolates were identified as TAHVs by polymerase chain reaction (PCR) amplification with Ex Taq DNA polymerase (Takara, Tokyo, Japan) and sequencing by using primers specific for the S segment of the TAHV genome.10
Whole genome sequencing was then performed by using PCR-based amplification of viral cDNAs with Ex Taq DNA polymerase (Takara, Japan) and primers specific for conserved regions of California serogroup virus genomes. Amplified products were examined by agarose gel electrophoresis (1% gel) and purified by using the QIA Quick Gel Extraction Kit (Qiagen). Purified amplification products were then sequenced directly by using primers designed as described above. Each nucleotide position was confirmed by generation of multiple sequences in the 5¢ and 3¢ directions. The origin of each nucleotide sequence was confirmed by repeated RNA extraction, cDNA generation, partial PCR reamplification, and nucleotide sequencing directly from BHK-21 cell isolates. Consensus sequences were assembled by using Contig Express Project software (Vector NTI Advance 1001 package; Invitrogen, Carlsbad, CA).
To compare TAHVs from China at the molecular level, we calculated nucleotide and amino acid identities for S, M, and L segment ORFs of selected TAHV isolates by using a CLUSTAL W alignment of sequences and MEGA version 4 software (Table 1). These analyses showed that the S segment nucleocapsid ORFs of TAHVs from China (XJ0625, XJ0708, and XJ0710) have greater than 99% nucleotide and amino acid sequence identities (Table 1). Similarly, the TAHV L segment polymerase ORFs had ≥ 96.7% nucleotide and amino acid sequence identities (Table 1). The S segment contains the N gene and the L segment contains the RNA-dependent RNA polymerase gene; both of these genes were relatively conserved within genus Orthobunyavirus. Although all virus isolates from Xinjiang or Europe were conserved within each group, the divergence between the two groups were still significant; isolates from Xinjiang showed no more than 92.5% or 83.1% nucleotide identities with Bardos 92 in S or L segments, respectively (Table 1).
Table 1.
Identity matrices for small, medium, and large segment open reading frames of Tahyna virus isolates from China compared with those of prototype Bardos 92 and variant Lumbo Tahyna virus strains*
| Genome segment and strain | % Nucleotide and amino acid sequence identities | ||||
|---|---|---|---|---|---|
| XJ0708 | XJ0625 | XJ0710 | Bardos 92 | Lumbo | |
| Small† | |||||
| XJ0708 | 99.2 | 99.3 | 92.5 | 90.5 | |
| XJ0625 | 100 | 99.6 | 91.8 | 90.5 | |
| XJ0710 | 100 | 100 | 91.9 | 90.5 | |
| Bardos 92 | 97.4 | 97.4 | 97.4 | 89 | |
| Lumbo | 96.6 | 96.6 | 96.6 | 95.7 | |
| Medium | |||||
| XJ0708 | 80.8 | 80.9 | 80.5 | 79.7 | |
| XJ0625 | 92.6 | 96.2 | 81.9 | 79.4 | |
| XJ0710 | 92.8 | 99 | 82.2 | 79.9 | |
| Bardos 92 | 92.6 | 94 | 94.2 | 80.8 | |
| Lumbo | 89.1 | 89.1 | 89.3 | 89.1 | |
| Large‡ | |||||
| XJ0708 | 97.4 | 96.8 | 82.6 | ||
| XJ0625 | 99.5 | 96.7 | 82.9 | ||
| XJ0710 | 99.7 | 99.7 | 83.1 | ||
| Bardos 92 | 97.1 | 97.1 | 97.3 | ||
% nucleotide sequence identities are shown above the diagonal, and % amino acid sequence identities are shown below the diagonal for each matrix. Values in bold represent comparisons between isolate XJ0708 from China and other Tahyna virus strains.
Compared regions include the nucleocapsid protein, the entire polyprotein open reading frame (Gn and Gc), and the polymerase open reading frame for the small, medium, and large segments, respectively.
Large segment data were not available for the Tahyna virus variant Lumbo strain.
In contrast to highly conservation of S and L segments in virus isolates from Xinjiang, analyses of glycoprotein ORFs of these viruses indicate a striking diversity of M segment genotypes within the Xinjiang group. Nucleotide identities of GN, NSm, and GC protein sequences in isolates from Xinjiang were 82.5–97% (GN), 82.4–96.2% (NSm), and 80.1–96.1% (GC). Identities for virus isolates from Xinjiang were much lower than those for virus isolates from Europe.6,7 Also, identities between virus isolates from Xinjiang and Europe were much lower than those within each group (Supplementary Table 1).
On the basis of host cell entry functions of envelope glycoproteins and diversity of hosts encountered in the arthropod-borne virus transmission cycle, relative diversity of TAHV M segment genotypes when compared with S and L segment genotypes is not unexpected. However, the degree of M segment diversity (< 20% nucleotide sequence diversity) among viruses isolated from the same geographic location of Xinjiang is surprising. The striking pattern of dissimilarity of M segment sequences in contrast to a relatively high degree of similarity in S and L segment sequences provides evidence of possible reassortment of genomic segments between variant strains of TAHV in China. Although significant divergence was observed in the M segment, no amino acid changes were observed for CT and TMD motifs in virus isolates from Xinjiang. These two domains were highly conserved. The CT domain plays an important role in targeting heterodimerized GN and GC proteins to the Golgi apparatus, and the TMD domain is important for virus assembly and morphogenesis.11,12
A previous study indicated that high degree of identity in the 5¢ and 3¢ regions of the M segment, although deletions may have occurred within 3¢ non-coding region.6 Nucleotide identities within virus isolates from Xinjiang were 95.1–100% in the 5¢ untranslated region (UTR) and 93.7–97.2% in the 3¢UTR. In addition, XJ0625 has two nucleotides deleted at positions 43 and 87 in the 3¢-non-coding region in the 3¢UTR (positions 4427 and 4471 for the whole M segment sequence). All virus isolates used in this study were passaged no more than three times after isolation from field mosquitoes. Therefore, it is likely that deletions to the genome did not occur during laboratory passage and that sequences reflect properties of viruses in nature. In addition, the variable region is not likely necessary for functions relevant to virus growth in such cell lines.
Phylogenetic analyses of the newly isolated TAHVs from China indicated that neighbor-joining and maximum-parsimony trees shared highly similar topologies and bootstrap values for major groupings (Figure 1). Phylogenies generated from TAHV N gene and polymerase gene ORFs infer that TAHV isolates from China are more closely related to one another than to the isolates from Europe, as indicated by the strong bootstrap support of these groupings (Figure 1). In contrast with geographically consistent groupings generated for S and L segments, although the M segment sequences of XJ0625 and XJ0710 group together with extreme support, the M segment ORF of isolate XJ0708 from China aligns independently within the context of a relatively large diversity of isolates from Europe, including the serologically related variant Lumbo TAHV strain. Interestingly, phylogenetic analysis based on GN, NSm, and GC generated different topology for XJ0708 (Supplementary Figures 1–3). These data further support diversity of M segment genotypes or virus reassortment in Xinjiang, China. In addition, preliminary phenotypic analyses showed markedly larger plaques for the divergent XJ0708 isolate (2.8 mm) than pinpoint plaques for XJ0625 (0.2 mm) and XJ0710 (0.2 mm) isolates in BHK-21 cells (Figure 2), which indicated diversity of phenotypes that correspond to different genotypes.
Figure 1.
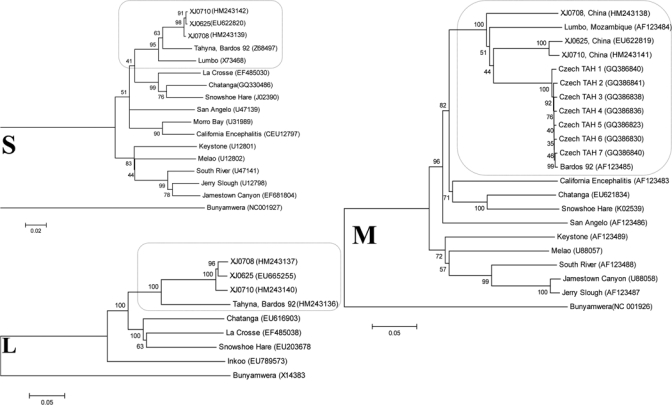
Phylogenies of California serogroup virus small (S), medium (M), and large (L) segment sequences. Full-length nucleocapsid, polyprotein (Gn and Gc) and polymerase open reading frames of S, M and L segments, respectively, of representative viruses of the California serogroup are compared. Highlighted comparisons of Tahyna virus isolates from China and closely related Lumbo virus isolates appear within selected regions of each tree. Distances and groupings were determined by using the p-distance algorithm and neighbor-joining method with MEGA version 4 software (www.megasoftware.net). Bootstrap values are indicated and correspond to 1,000 replications. Virus names or isolate designations are listed with GenBank accession numbers given in parentheses. The tree was rooted by using Bunyamwera virus as the outgroup virus. Scale bars indicate a genetic distance of 0.05 nucleotide substitutions per position.
Figure 2.

Plaque morphology of Tahyna viruses XJ0625, XJ0708 and XJ0710, China. A, XJ0625 (0.2 mm); B, XJ0708 (2.8 mm); C, XJ0710 (0.2 mm).
Although TAHVs and infections with TAHVs have been reported in Xinjiang and Qinghai Province, the association of TAHV with human illness is still not clearly defined.8,9 Our findings support the need for additional study of the diversity of TAHVs and the potential impact of that diversity on the ecology, epidemiology, and pathogenicity of TAHV in China.
Supplementary Material
ACKNOWLEDGMENTS
We thank Amy J. Lambert (Centers for Disease Control and Prevention) for help with the experiment design and assistance in the preparation and writing of this manuscript. We also thank staff members at the Xinjiang Center for Disease Control and Prevention, Xinjiang, China for the assistance in obtaining mosquito samples.
Note: Supplemental table and figures are available at www.ajtmh.org.
Footnotes
Financial support: This study was supported by grants from the Ministry of Science and Technology of China (no. 2008ZX10004-001), The Young Scholar Scientific Research Foundation of China CDC (no.2010A105), a Development Grant from the State Key Laboratory for Infectious Disease Prevention and Control (2008SKLID105), and China CDC–US CDC Cooperative Agreement U19-GH000004.
Authors' addresses: Zhi Lu, Shi-Hong Fu, Feng-Tian Wang, Qing Tang, and Guo-Dong Liang, State Key Laboratory for Infectious Disease Prevention and Control, Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, People's Republic of China, E-mails: liflit@hotmail.com, shihongfu@hotmail.com, wangft82@163.com, qtang04@sina.com, and gdliang@hotmail.com. Roger S. Nasci, Division of Vector-Borne Infectious Diseases, National Center for Zoonotic, Vector-Borne, and Enteric Diseases, Centers for Disease Control and Prevention, Public Health Service, Fort Collins, CO, E-mail: rsn0@cdc.gov.
References
- 1.Bardos V, Danielova V. The Tahyna virus: a virus isolated from mosquitoes in Czechoslovakia. J Hyg Epidemiol Microbiol Immunol. 1959;3:264–276. [PubMed] [Google Scholar]
- 2.Bulychev VP, Alekseev AN, Kostiukov MA, Tukhtaev TM, Gordeeva ZE. Isolation of Tahyna virus from mosquitoes collected in Dushanbe [in Russian] Med Parazitol (Mosk) 1985;4:81–83. [PubMed] [Google Scholar]
- 3.Hubálek Z, Zeman P, Halouzka J, Juřicová Z, Št'ovíčková E, Bálková H, Šikutováet S, Rudolf I. Mosquitoborne viruses, Czech Republic, 2002. Emerg Infect Dis. 2005;11:116–118. doi: 10.3201/eid1101.040444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lvov DK, Kostiukov MA, Pak TP, Gordeeva ZE, Bun'etbekov AA. Isolation of Tahyna virus (California antigenic group, family Bunyaviridae) from the blood of febrile patients in the Tadzhik SSR [in Russian] Vopr Virusol. 1977;6:682–685. [PubMed] [Google Scholar]
- 5.Acha PN, Szyfres B. Chlamydioses, Rickettsioses, and Viruses. 3rd ed. Volume II. Washington, DC: Pan American Health Organization; 2003. pp. 110–131. (Zoonoses and communicable diseases common to man and animals). [Google Scholar]
- 6.Kilian P, Růzek D, Danielová V, Hypsa V, Grubhoffer L. Nucleotide variability of Tahyna virus (Bunyaviridae, Orthobunyavirus) small (S) and medium (M) genomic segments in field strains differing in biological properties. Virus Res. 2010;149:119–123. doi: 10.1016/j.virusres.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Bennett RS, Gresko AK, Murphy BR, Whitehead SS. Tahyna virus genetics, infectivity, and immunogenicity in mice and monkeys. Virol J. 2011;8:135. doi: 10.1186/1743-422X-8-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Z, Lu XJ, Fu SH, Zhang S, Li ZX, Yao XH, Feng YP, Lambert AJ, Ni DX, Wang FT, Tong SX, Nasci RS, Feng Y, Dong Q, Zhai YG, Gao XY, Wang HY, Tang Q, Liang GD. Tahyna virus and human infection, China. Emerg Infect Dis. 2009;15:306–309. doi: 10.3201/eid1502.080722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lv Z, Fu SH, Wang FT, Kosoy OL, Nasci RS, Liang GD. Investigation of Tahyna virus infection among unknown fever cases in Xinjiang, China. Bing Du Xue Bao. 2011;27:71–74. [PubMed] [Google Scholar]
- 10.Lambert AJ, Lanciotti RS. Consensus amplification and novel multiplex sequencing method for S segment species identification of 47 viruses of the Orthobunyavirus, Phlebovirus, and Nairovirus genera of the family Bunyaviridae. J Clin Microbiol. 2009;47:2398–2404. doi: 10.1128/JCM.00182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi X, Kohl A, Li P, Elliott RM. Role of the cytoplasmic tail domains of Bunyamwera orthobunyavirus glycoproteins Gn and Gc in virus assembly and morphogenesis. J Virol. 2007;81:10151–10160. doi: 10.1128/JVI.00573-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi X, Lappin DF, Elliott RM. Mapping the Golgi targeting and retention signal of Bunyamwera virus glycoproteins. J Virol. 2004;78:10793–10802. doi: 10.1128/JVI.78.19.10793-10802.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.