

Abstract
The mechanisms that couple cell cycle progression with the organization of the peripheral cytoskeleton are poorly understood. In Saccharomyces cerevisiae, the Swe1 protein has been shown previously to phosphorylate and inactivate the cyclin-dependent kinase, Cdc28, thereby delaying the onset of mitosis. The nim1-related protein kinase, Hsl1, induces entry into mitosis by negatively regulating Swe1. We have found that Hsl1 physically associates with the septin cytoskeleton in vivo and that Hsl1 kinase activity depends on proper septin function. Genetic analysis indicates that two additional Hsl1-related kinases, Kcc4 and Gin4, act redundantly with Hsl1 to regulate Swe1. Kcc4, like Hsl1 and Gin4, was found to localize to the bud neck in a septin-dependent fashion. Interestingly, hsl1 kcc4 gin4 triple mutants develop a cellular morphology extremely similar to that of septin mutants. Consistent with the idea that Hsl1, Kcc4, and Gin4 link entry into mitosis to proper septin organization, we find that septin mutants incubated at the restrictive temperature trigger a Swe1-dependent mitotic delay that is necessary to maintain cell viability. These results reveal for the first time how cells monitor the organization of their cytoskeleton and demonstrate the existence of a cell cycle checkpoint that responds to defects in the peripheral cytoskeleton. Moreover, Hsl1, Kcc4, and Gin4 have homologs in higher eukaryotes, suggesting that the regulation of Swe1/Wee1 by this class of kinases is highly conserved.
Keywords: Cell cycle, checkpoint, cytoskeleton, Nim1, septin
The peripheral cytoskeleton is a dynamic network essential for a variety of cellular functions, such as motility, establishment of cell–cell contacts, polarized cell growth, and cell morphogenesis (Drubin et al. 1996; Botstein et al. 1997). During cell division, the peripheral cytoskeleton mediates morphogenetic events and participates in the orientation of the mitotic axis. It also plays a central role by forming the cleavage apparatus that ultimately separates the two daughter cells. These cytoskeletal events occur in a highly ordered fashion during cell division, suggesting that mechanisms operate to coordinate them with other events of the cell cycle. For example, positioning and assembly of the cleavage apparatus must be both spatially and temporally controlled to ensure proper segregation of sister chromosomes into daughter cells. Although much is known about both the cell cycle machinery and the peripheral cytoskeleton, the mechanisms that synchronize cortical and nuclear events remain obscure.
Nuclear events of the cell cycle are monitored through a series of well-defined checkpoints, in which one series of events must be completed before the next set of events can be initiated (Hartwell and Weinert 1989). Substantial progress has been made in elucidating the checkpoints that monitor DNA replication, DNA damage, and spindle integrity (Paulovich et al. 1997). Recently, a cell cycle checkpoint monitoring cell morphology has been discovered (Lew and Reed 1995), indicating that cytoplasmic events are also monitored during the cell cycle. However, the mechanisms by which cells detect morphological defects remain undetermined. It is tempting to speculate that the peripheral cytoskeleton is involved in such sensing processes (McMillan et al. 1998), but how cells monitor the organization of their cytoskeleton is unclear.
Cytoskeletal processes and cell cycle progression have been well studied in the budding yeast, Saccharomyces cerevisiae. Bud formation establishes both the axis of growth and division, and the site of cytokinesis in this organism (Pringle et al. 1995; Chant 1996). Bud growth occurs in two successive phases (Lew and Reed 1993). The first phase involves bud emergence and apical bud growth, during which cell growth is restricted to the bud tip. In the second stage of bud formation, growth becomes isotropic (i.e., occurs over the entire surface of the bud, which becomes more rounded). The final shape of the bud thus depends on the relative length of the apical and isotropic growth phases; elongated buds result from delays in the switch to isotropic growth, whereas unusually spherical buds are formed when this switch occurs early (Lew and Reed 1993). Both formation and postmitotic separation of the bud require the septin- and actin-based peripheral cytoskeleton (Botstein et al. 1997). The septins represent a class of highly conserved proteins that have been shown to assemble into filaments in Drosophilla and are essential for cytokinesis in yeast, fruit flies and mammals (Hartwell 1971; Neufeld and Rubin 1994; Cooper et al. 1996; Longtine et al. 1996; Kinoshita et al. 1997). In S. cerevisiae, the septins Cdc3, Cdc10, Cdc11, and Cdc12 form a ring at the presumptive bud site that remains at the bud neck until cytokinesis (Kim et al. 1991; Longtine et al. 1996).
Bud growth is controlled by the cell cycle machinery. The G1 cyclins, Cln1 and Cln2, trigger bud emergence and apical bud growth through activation of Cdc28, the major cyclin-dependent kinase (CDK) of yeast (Lew and Reed 1993). The switch from apical to isotropic growth requires the activation of Cdc28 by the G2 cyclins, Clb1 and Clb2. Mutations that prevent activation of Cdc28 in G2 impair entry into mitosis. Cells with such mutations (e.g., cdc34, Schwob et al. 1994) have a prolonged apical growth phase and display an elongated bud morphology.
In eukaryotes, activation of the mitotic CDK, and thus the timing of mitosis, is controlled by inhibitory kinases. The highly conserved Wee1 kinase negatively regulates entry into mitosis through an inhibitory phosphorylation of the p34cdc2 CDK (Russell and Nurse 1987a; Coleman and Dunphy 1994). This regulatory mechanism is thought to help synchronize entry into mitosis with the completion of previous cell cycle events. In Schizosaccharomyces pombe, mutations that inactivate Wee1 lead to premature and catastrophic mitosis (Enoch and Nurse 1990). Entry in mitosis is promoted through inactivation of Wee1 by the Nim1 kinase (Russell and Nurse 1987b; Coleman et al. 1993; Parker et al. 1993; Wu and Russell 1993). How Nim1 kinase activity is regulated is not known.
In S. cerevisiae, the Wee1 kinase, Swe1, specifically inhibits the G2 form of Cdc28 (Booher et al. 1993) and delays entry into mitosis in response to bud emergence defects (Sia et al. 1996). Analogous to Wee1, Swe1 activity is negatively regulated by Hsl1, a kinase with strong homology to Nim1 in its catalytic domain (Ma et al. 1996; Tanaka and Nojima 1996). Despite our present understanding of the biochemical activity of Swe1, the molecular signals that it transduces to control entry into mitosis are not known.
Here, we provide evidence of a Nim1-related kinase that signals the organizational state of a cytoskeletal structure and thereby coordinates cell cycle progression with cortical events. We found that the Hsl1 kinase colocalizes and coprecipitates with the septins, and its activity is dependent on proper septin function. We propose that Hsl1, in association with two related kinases Gin4 and Kcc4, couples entry into mitosis with the organization of septin structures.
Results
Hsl1 colocalizes and coimmunoprecipitates with the septins in vivo
To learn how Nim1-related kinases function in yeast, we examined the subcellular distribution of a fully active Hsl1 kinase tagged with a triple HA epitope (3xHA) at its amino terminus by indirect immunofluorescence using antibodies directed against the HA epitope. 3xHA–Hsl1 localized as a ring at the bud neck of small-, medium-, and large-budded cells (Fig. 1A). Staining was not detected in unbudded cells, nor in cells lacking tagged Hsl1 (data not shown). This localization pattern does not depend on the nature or position of the epitope tag, because an identical staining pattern is observed when Hsl1 was tagged either at the amino terminus with a triple myc epitope (3xmyc) or at the carboxyl terminus with the 3xHA epitope (Fig. 1B; data not shown).
Figure 1.

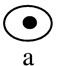
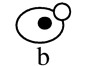
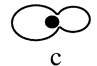
Hsl1 and Kcc4 localize to the bud neck. (A) A 3xHA–HSL1 3xmyc–CDC3 strain (Y1536) stained with anti-Myc (Myc–Cdc3) and anti-HA (HA–Hsl1) antibodies and DAPI (DNA staining). Cells at different stages of the cell cycle are presented as follows: Unbudded (G1; a,b), small budded (G1/S; c), mitosis (d,e), and cytokinesis (f). Cdc3 forms a ring in unbudded cells (b), a broad ring spanning the neck in budded cells (c), and a double ring in cells that undergo mitosis and cytokinesis (d–f). Hsl1 is not detected in unbudded cells, localizes as a ring at the neck on the bud side in budded cells (c,d,e), and is not evident after cells complete mitosis (f). (Note the ring staining in c). Localization of Hsl1 and Cdc3 is not detected in untagged strains. (B) Localization of Hsl1–3xHA in cdc12-1 cells incubated at either the permissive (left) or restrictive temperature (37°C, right). (C) Indirect immunofluorescence of KCC4–3xHA cells (Y1533) with anti-HA antibodies. Four representative cells from different fields are shown. Kcc4 localizes as a ring in unbudded cells (top left cell) and at the bud side of the neck in budded cells. All pictures are at the same scale. Bar, 5 μm.
The cellular localization of Hsl1 is similar to septin distribution. To determine whether these proteins colocalize, double staining experiments were performed in a strain containing 3xmyc–CDC3 and 3xHA–HSL1 alleles. As shown in Figure 1A, Hsl1 and Cdc3 colocalize at the neck during most of the budding cycle. However, differences were detected. First, whereas 72% of unbudded cells harbored a septin ring at the cortex, <10% of these cells showed Hsl1 staining (n=170). Thus, the septin ring forms in unbudded cells (Fig. 1A,b), whereas Hsl1 is mainly detected in budded cells (Fig. 1A,c). Second, whereas Cdc3 is located at the cortex throughout the neck region, Hsl1 localizes specifically on the bud side of the neck (Fig. 1A,d,e; Fig. 1B). This asymmetry is particularly evident in cells undergoing mitosis, in which Cdc3 is present as a double ring at the neck and Hsl1 is only present as a single ring in the bud (Fig. 1A,e). Third, late in mitosis septin structures on both sides of the neck region lack Hsl1 and the protein is no longer detectable; Cdc3 rings are still present on both side of the neck.
The effect of disrupting septin structures on Hsl1 localization was also analyzed. A strain containing the 3xmyc–CDC3 and 3xHA–HSL1 alleles and a temperature-sensitive cdc12-1 mutation was incubated at either the permissive or restrictive temperature; cells were fixed and stained with anti-Myc and anti-HA antibodies. The localization of all septins is disrupted by cdc12-1 (Kim et al. 1991). In cdc12-1 cells grown at the permissive temperature, Hsl1 and Cdc3 localize to the bud neck, as in wild-type cells (Fig. 1B). When these cells are shifted to the restrictive temperature, Cdc3 is absent from the neck (data not shown), and the buds become elongated (Fig. 1B; see below). Localization of Hsl1 to the neck is also no longer detected; instead diffuse staining is observed throughout the cell (Fig. 1B). We considered the possibility that the delocalization of Hsl1 resulted from the formation of elongated buds. Strains with a deletion of HSL7 (Ma et al. 1996) also form elongated buds but are viable, indicating that septin function is not strongly affected. In an hsl7Δ mutant, Hsl1 still localizes to the bud neck (data not shown). Thus, the mislocalization of Hsl1 in the cdc12-1 strain is likely to directly result from the absence of septin structures at the neck.
The subcellular localization of the Hsl1 kinase suggested that this protein is associated with the septin complex in vivo. To address this possibility, we tested whether Hsl1 coprecipitates with the Cdc3 septin. Extracts from cells containing 3xmyc–CDC3, 3xHA–HSL1 or both alleles were subjected to immunoprecipitation with polyclonal antibodies directed against either the myc or HA epitope, and immunoblot analysis was used to visualize the precipitated proteins. As shown in Figure 2A, anti-HA antibodies precipitate 3xmyc–Cdc3 only if 3xHA–Hsl1 is present in the extract. Similarly, anti-myc antibodies precipitate 3xHA–Hsl1 only in the presence of 3xmyc–Cdc3. Thus, Hsl1 and Cdc3 are part of the same complex in vivo.
Figure 2.

Septins and Hsl1 coimmunoprecipitate and Hsl1 autophosphorylation and kinase activity depend on septins. (A) Protein extracts from either 3xmyc–CDC3 3xHA–HSL1 (Y1536), 3xmyc–CDC3 (Y1535), or 3xHA–HSL1 (Y1530) strains were immunoprecipiated with either anti-Myc or anti-HA antibodies. The pellets were separated by SDS-PAGE, and immunoblots were probed with both anti-HA and anti-Myc antibodies. (B) Immunoblot analysis of extracts of 3xHA–HSL1 (Y1530) and 3xHA–HSL1 cdc12-1 (Y1534) cells grown at 24°C and 37°C. High molecular weight isoforms are detected; these are absent in cdc12-1 cells incubated at 37°C and in samples (immunoprecipitates) treated with calf alkaline phosphatase. (C) Plasmids encoding either wild-type HSL1 or a putative kinase-inactive HSL1, hsl1–K110A, were introduced into hsl1Δ (Y1527) and wild-type (Y1521) strains. The phosphorylated isoforms are absent in hsl1–K110A strains and reduced in hsl1–K110A–HSL1 strains. (D) Hsl1 shows little kinase activity when isolated from septin mutants. Anti-HA immunoprecipitates were incubated in the presence of [32P]ATP under kinase reaction conditions. Protein samples were subjected to electrophoresis and exposed for autoradiography. Numbers under each lane correspond to 32P incorporation standardized to the amount of protein as quantified by immunoblotting.
Hsl1 kinases activity depends on septin function
Hsl1 abundance in wild-type and septin mutants was examined by immunoblot analysis. 3xHA–HSL1 and cdc12-1 3xHA–HSL1 strains were grown at the permissive temperature and then an aliquot of each culture was shifted to 37°C for 3 hr. Protein extracts were prepared and analyzed by immunoblotting with antibodies directed against the HA epitope. As shown in Figure 2B, the level of 3xHA–Hsl1 does not vary substantially between cdc12-1 mutant and wild-type strains. However, its migration during SDS-PAGE is dramatically affected by the septin mutation. 3xHA–Hsl1 from cdc12-1 cells grown at the permissive temperature or from wild-type cells grown at either temperature migrates as a very broad band in low percentage polyacrylamide gels. This migration property is due to phosphorylation, as phosphatase treatment reduces the 3xHA–Hsl1 band to a single fast-migrating isoform (Fig. 2B). 3xHA–Hsl1 from cdc12-1 cells shifted to 37°C is resolved as a fast-migrating form, suggesting that Hsl1 phosphorylation requires septin function.
Hsl1 modification might be due to autophosphorylation. To determine whether the presence of Hsl1 phosphoisoforms depends on Hsl1 activity, we constructed a mutant allele of HSL1, hsl1K110A, expected to encode a catalytically inactive kinase (Hanks and Quinn 1991). This hsl1–K110A mutation abolishes the ability of the protein to complement the phenotypes associated with an hsl1 deletion (see Materials and Methods), suggesting that Hsl1 kinase activity is required for in vivo function. When the 3xHA–Hsl1–K110A protein is the sole source of Hsl1, the protein migrates at the size of the unphosphorylated protein (Fig. 2C). Interestingly, in wild-type HSL1 cells, the 3xHA–Hsl1–K110A protein exhibits modest levels of phosphorylation, suggesting that it can be phosphorylated in trans by Hsl1. These results suggest that most phosphorylated forms of Hsl1 depend on Hsl1 activity and, therefore, are the result of autophosphorylation events.
Because Hsl1 autophosphorylates, its phosphorylation state provides a useful indicator of Hsl1 kinase activity. The hypophosphorylated form of Hsl1 in the cdc12-1 strain thus suggests that Hsl1 is inactive in septin mutants.
To determine whether Hsl1 activity depends on the septins, we immunoprecipitated 3xHA–Hsl1 from different strains and assayed its activity in vitro. Hsl1 autophosphorylation in vitro is extremely efficient. It is formally possible that a second kinase coprecipitates with Hsl1 and is responsible for Hsl1 phosphorylation. However, because Hsl1 kinase inactivation via the K110A mutation abolishes the observed phosphorylation (data not shown), Hsl1 phosphorylation most likely reflects Hsl1 activity. Using this assay, we found that Hsl1 is active when isolated from wild-type cells and cdc12-1 cells incubated at the permissive temperature (Fig. 2D; data not shown). In contrast, Hsl1 is inactive when precipitated from the cdc12-1 septin mutant incubated at the restrictive temperature (Fig. 2D). Thus, Hsl1 activity depends on septin organization.
Kcc4, a homolog of Hsl1, also localizes to the bud neck
Database searches revealed that the yeast genome encodes two other Nim1-like proteins closely related to Hsl1, namely Gin4 and Kcc4 (YCL024w, Fig. 3). Gin4 was identified as a binding partner of Nap1, a protein that specifically interacts with Cdc28–Clb1 and Cdc28–Cbl2 complexes (Altman and Kellogg 1997), and also through a genetic screen for mutations that are lethal in combination with G1 cyclin mutations (Benton et al. 1997). Kcc4 has not been described previously. Resequencing of the KCC4 locus (see Materials and Methods) indicated that the KCC4 ORF encodes a protein of 1038 amino acids that is similar to Gin4 throughout its entire length (53% identity, 68% similarities over 1024 amino acids). These proteins each contain a conserved 42 amino acid sequence at their carboxyl terminus that is found in many other protein kinases of the same family (Fig. 3B).
Figure 3.

Analysis of Hsl1-related kinases. (A) A dendrogram depicting members of the Nim1/Hsl1 family shows that nim1, Hsl1, Kcc4, and Gin4 are closely related. These alignments were obtained by comparison of the catalytic domains of these enzymes. (B) Sequence analysis reveals that the MARK (microtubule affinity regulatory kinase) kinases, PAR-1, Hsl1, Kcc4, Gin4, and several other kinases of the same family share a 42-amino-acid domain at their noncatalytic, carboxyl terminus. This domain is absent in other members of the family such as Nim1 and Snf1.
The Kcc4 protein was tagged at its carboxyl terminus with the 3xHA epitope. A strain carrying this tag exhibits wild-type growth rate and cell morphology. Like Hsl1, Kcc4–3xHA localizes on the bud side of the neck, and disappears from that region toward the end of mitosis (Fig. 1C). This localization also depends on the septins; Kcc4 is absent from the bud neck in a cdc12-1 mutant incubated at the restrictive temperature (data not shown). However, unlike Hsl1, Kcc4 accumulates at the presumptive bud site before bud emergence: 44% (n = 128) of unbudded cells show a Kcc4 ring at the cortex (e.g., see Fig. 1C, top left cell).
Hsl1, Gin4, and Kcc4 are functionally redundant and act upstream of Swe1
Because Hsl1 and Kcc4 colocalize, and Hsl1, Kcc4, and Gin4 are extremely similar, we investigated the possibility that the three kinases are involved in the same process. Deletion of either HSL1, KCC4, or GIN4 alone has no striking effect on strain viability or growth. During exponential growth, the cellular morphology of each of these single mutants is similar to wild type (Fig. 4a–d). However, careful cytological analysis of asynchronous populations reveals that gin4Δ and hsl1Δ mutants exhibit a delay in the onset of mitosis (Table 1). Large-budded cells that have not yet entered mitosis (category c) account for only 15% of an asynchronous wild-type population. However, this category is tripled in a hsl1Δ population and doubled in a gin4Δ population, suggesting that these strains are delayed prior to mitosis. For the hsl1Δ population, the increase in the proportion of large-budded cells is likely to be due to an accumulation of G2 cells, because FACS analysis of DNA content indicates that most of the cells in a haploid hsl1Δ population have a 2N content of DNA (Ma et al. 1996; Tanaka 1996). These data indicate that hsl1Δ and gin4Δ mutations affect cell cycle progression prior to nuclear division. gin4Δ and hsl1Δ mutants also have a mild cytokinesis defect, as indicated by the slightly increased proportion of large budded cells with divided nuclei (Table 1,e).
Figure 4.

hsl1Δ kcc4Δ gin4Δ cells have an elongated cell morphology. (a) Wild-type (Y1521), (b) kcc4Δ (Y1528), (c) gin4Δ (Y1529), (d) hsl1Δ (Y1527), (e) hsl1Δ kcc4Δ gin4Δ (1538), and (f) hsl1Δ kcc4Δ gin4Δ swe1Δ (1537) cells were grown at 22°C and viewed by differential interference contrast microscopy. gin4Δ and hsl1Δ cells exhibit a slight morphological defect, whereas double-mutant (not shown) and triple-mutant cells (e) exhibit an elongated cell morphology; this defect is suppressed by swe1Δ (f). hsl1Δ exacerbates the cdc12-1 morphological defect. cdc12-1 cells grown at the permissive temperature (g) do not have a morphological defect; however, the cdc12–1 hsl1Δ double mutant has elongated buds (h).
Table 1.
Cytological analysis of single and multiple mutants
 |
 |
 |
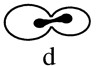 |
 |
N | |
|---|---|---|---|---|---|---|
| Wild type | 36.3 | 29.8 | 15.4 | 4.0 | 14.5 | 262 |
| kcc4Δ | 39.9 | 19.7 | 16.5 | 7.3 | 16.5 | 218 |
| gin4Δ | 21.9 | 14.0 | 31.8 | 4.8 | 27.4 | 248 |
| hsl1Δ | 10.1 | 20.6 | 44.0 | 4.4 | 21.0 | 292 |
| hsl1Δ gin4Δ kcc4Δ | 9.0 | 14.2 | 46.0 | 8.4 | 22.5 | 547 |
| hsl1Δ gin4Δ kcc4Δ swe1Δ | 32.6 | 20.6 | 17.9 | 7.7 | 21.2 | 541 |
| hsl1Δ gin4Δ kcc4Δ CDC28–Y19F | 33.2 | 20.3 | 15.4 | 7.0 | 24.1 | 443 |
Asynchronous cultures show the different stages of cell cycle progression: (a) Unbudded cells (G1); (b) small budded cells (G1/S); (c) large-budded cells prior to mitosis (G2); (d) mitosis; (e) postmitotic (cytokinesis/G1). The increase in c reveals a delay prior to mitosis; the increase in e reveals cytokinesis defects.
The hsl1Δ gin4Δ kcc4Δ triple mutant is viable, with only a mild growth defect as revealed by a slight difference in colony size compared with wild type. However, triple mutant cells exhibit extremely strong morphological defects; they form very long buds and are defective for cell separation, resulting in the formation of branched chains of elongated cells (Fig. 4e). Analysis of an asynchronous population of the triple mutant strain revealed a large increase in the proportion of premitotic cells (Table 1).
It has been shown that Hsl1 acts as a negative regulator of Swe1 (Ma et al. 1996), which is known to inhibit Cdc28 by phosphorylation of the conserved tyrosine at position 19 (Booher et al. 1993). Consistent with this, overexpression of Swe1 leads to the formation of elongated buds, by inhibiting G2/Cdc28 complexes and promoting accumulation of the G1 cyclins (Booher et al. 1993; Sia et al. 1996). To determine whether the morphological defects resulting from the simultaneous disruption of HSL1, KCC4, and GIN4 require the Swe1 kinase, we deleted SWE1 in a hsl1Δ gin4Δ kcc4Δ triple mutant. The resulting quadruple mutant grows slightly slower than the triple mutant, as judged by colony size, but this growth defect remains slight compared with wild type. Cytological analysis indicates that the elongated bud morphology (Fig. 4e,f) and cell cycle delay prior to mitosis (Table 1,c) of the hsl1Δ gin4Δ kcc4Δ triple mutant are eliminated by deletion of SWE1. These phenotypes are also suppressed by replacement of CDC28 by CDC28–Y19F, an allele encoding a nonphosphorylatable form of Cdc28 (Table 1). Thus, hsl1Δ gin4Δ kcc4Δ triple deletion trigger a Swe1-mediated delay of mitosis due to inhibition of Cdc28 by tyrosine phosphorylation. In contrast, introduction of the swe1Δ allele does not alleviate the cytokinesis defect of the hsl1Δ gin4Δ kcc4Δ triple mutant, as measured by the frequency of large-budded cells that have finished nuclear division but are not yet separated (Table 1,e).
Each of the double mutants, hsl1Δ kcc4Δ, hsl1Δ gin4Δ, and kcc4Δ gin4Δ, has a phenotype that is intermediate between that of the single and triple mutants. The strongest defect is observed in the gin4Δ hsl1Δ cells, closely followed by the gin4Δ kcc4Δ double mutant. The elongated bud morphology and cell cycle defects of each of these double mutants is suppressed by swe1Δ (data not shown). Together, these results indicate that Hsl1, Kcc4, and Gin4 act redundantly upstream of Swe1; and that Kcc4 and Gin4, like Hsl1, act as negative regulators of Swe1.
Hsl1 and Kcc4 colocalize with the septins, and the hsl1Δ gin4Δ kcc4Δ strain exhibits a morphological phenotype similar to that of septin mutants. Therefore, we tested whether any of these mutations interact genetically with septin mutants. Strains carrying a cdc12-1, hsl1Δ, gin4Δ, or kcc4Δ single mutation exhibit few or no morphological defects when grown at 22°C; however, each of the cdc12-1 hsl1Δ, cdc12-1 kcc4Δ, and cdc12-1 gin4Δ double mutants form elongated buds at this temperature (see hsl1Δ cdc12-1 cells, Fig. 4h). These genetic interactions are additive; the cdc12-1 hsl1Δ kcc4Δ gin4Δ quadruple mutant displays extreme growth defects at the permissive temperature. Thus, Hsl1, Kcc4, Gin4, and the septins interact genetically with one another.
The septins are mislocalized in a hsl1Δ kcc4Δ gin4Δ triple mutant
To further characterize the cytokinesis defect of the hsl1Δ gin4Δ kcc4Δ triple mutant, a 3xmyc–CDC3 allele was introduced in this strain, and the septin rings were examined by immunolocalization with antibodies directed against the myc epitope. As shown in Figure 5A, the hsl1Δ gin4Δ kcc4Δ triple mutant shows normal septin staining in unbudded cells, but many budded cells contain abnormal septin structures. The majority (67% of the budded cells, n = 108) accumulate septins as elongated, irregular patches at the bud neck (Fig. 5b), whereas the rest of the population (33%) appear to have normal septin structures.
Figure 5.

Localization of 3xmyc–Cdc3 in wild-type (a) and hsl1Δ kcc4Δ gin4Δ cells (b). Cdc3 localization is aberrant in hsl1Δ kcc4Δ gin4Δ cells but similar to wild type in budded hsl1Δ kcc4Δ gin4Δ swe1Δ cells. Bar, 5 μm.
Septin defects induce a cell cycle delay dependent on tyrosine phosphorylation of Cdc28 by Swe1
Temperature-sensitive cdc3, cdc10, cdc11, and cdc12 septin mutants all form elongated buds at restrictive temperature (data not shown; see Fig. 6A for cdc12-1 results; see also Hartwell 1971; Kim et al. 1991). This elongated bud morphology indicates that the apical growth phase is prolonged in those cells. Because both the switch from apical to isotropic bud growth and the initiation of mitosis depend on the activation of Cdc28 by the G2 cyclins Clb1 and Clb2 (Lew and Reed 1993), we examined whether entry into mitosis was also delayed in a septin mutant. The timing of nuclear division was monitored in synchronized populations of wild-type and cdc12-1 strains. Pheromone-arrested cells were diluted into fresh medium at 37°C (the restrictive temperature for cdc12-1) and the timing of bud emergence and nuclear division were determined. The wild-type and the septin mutant strains initiate budding at the same time, but nuclear division was delayed ∼50 min in the mutant relative to wild type (Fig. 6B). Thus, both the switch to isotropic growth and nuclear division are delayed as a result of the disruption of septin structures.
Figure 6.
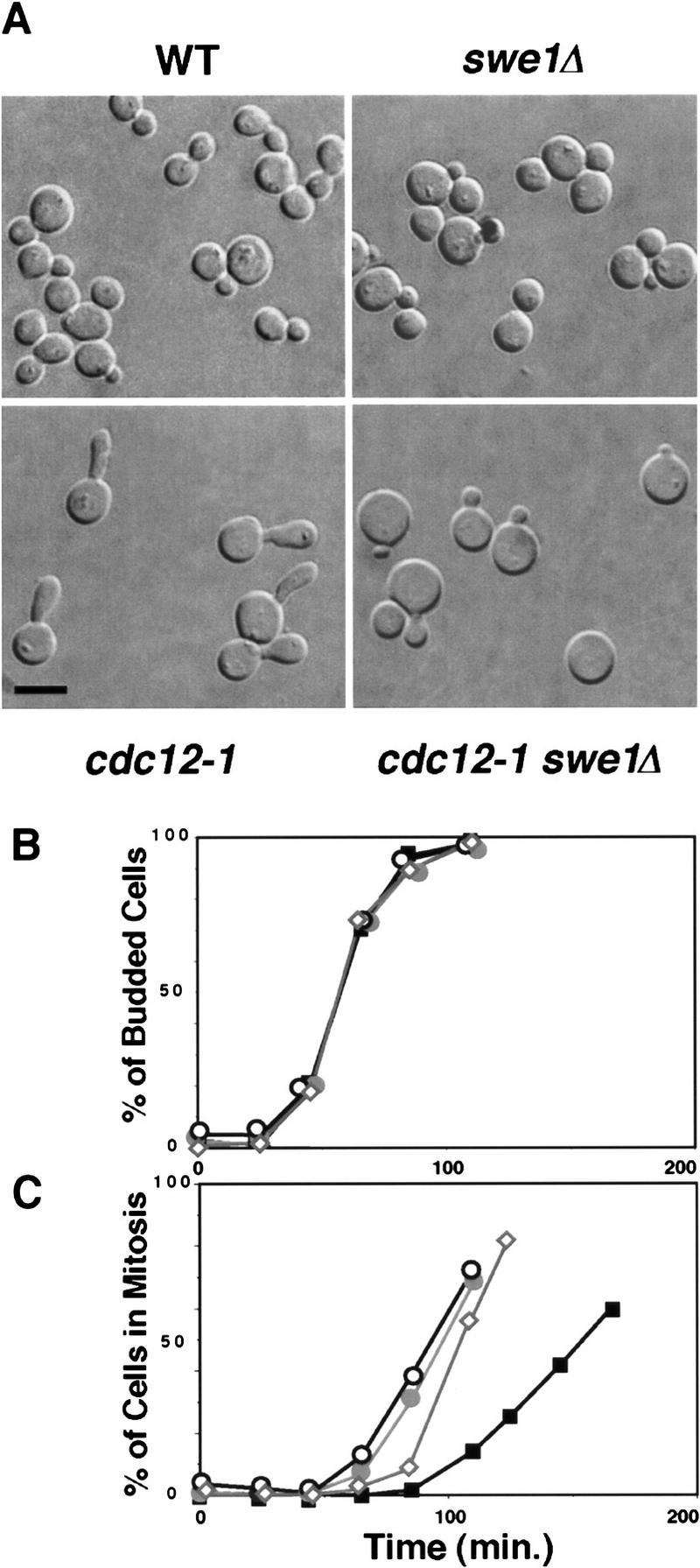
(A) Septin mutants have an elongated morphology that depends on Swe1. Wild-type (Y1521), cdc12-1 (Y1524), swe1Δ (Y1525), and swe1Δ cdc12-1 cells were grown at 24°C and shifted to 37°C for 3 hr. The temperature-sensitive cdc12-1 cells form elongated buds; this phenotype is suppressed by swe1Δ. Bar, 5 μm. (B,C) Septin mutants exhibit a SWE1-dependent delay in nuclear division. Wild-type (Y1521), cdc12-1 (1524), and swe1Δ cdc12-1 cells were grown at 24°C and arrested with mating pheromone. Cells were diluted into fresh medium lacking pheromone at 37°C, and the percentage of cells that had budded (B) or undergone nuclear division (C) was determined at the indicated times. cdc12-1 cells are delayed prior to nuclear division, which is partially dependent on SWE1. (⋄) wild type; (█) cdc12; (•) cdc12 swe1Δ; (○) cdc 12 CDC28Y–19F.
We determined whether Swe1 is necessary for the nuclear-division delay and the switch to isotropic growth seen in a septin mutant. Cell morphology and the timing of nuclear division were examined in cdc12-1 and cdc12-1 swe1Δ strains incubated at the restrictive temperature. As shown in Figure 6A, the formation of elongated buds by cells carrying a septin mutation depends on Swe1. The cdc12-1 strain forms very elongated buds when grown at 37°C, whereas the cdc12-1 swe1Δ double mutant develops smaller, rounder buds (Fig. 6A). The effect of the SWE1 deletion on bud growth in a septin mutant could result from a requirement for Swe1 during apical growth. However, a temperature-sensitive cdc34-2 mutant and a cdc34-2 swe1Δ double mutant both formed elongated buds at restrictive temperature (data not shown), indicating that Swe1 is not directly required for the formation of elongated buds. In addition to its effect on bud morphology, the deletion of SWE1 also abolishes the nuclear division delay due to the cdc12-1 mutation (Fig. 6B). Thus, the delays in both nuclear division and isotropic growth resulting from disruption of septin structures are mediated by Swe1.
To determine whether inhibition of Cdc28 is responsible for the delay in nuclear division observed in septin mutants, CDC28–Y19F was introduced in place of CDC28 in a cdc12-1 strain. In the cdc12-1 CDC28–Y19F mutant, the mitotic delay due to the septin mutation was also abolished. Thus, tyrosine phosphorylation of Cdc28 accounts for the Swe1-dependent nuclear division delay observed in a septin mutant.
Swe1-mediated cell cycle regulation is necessary to maintain the viability of a septin mutant
Although deletion of SWE1 alleviates several aspects of the phenotype of the cdc12-1 mutant, the cdc12-1 swe1Δ double mutant is still defective for cytokinesis and growth at the restrictive temperature (data not shown). In fact, the cdc12-1 swe1Δ double mutant has additional defects compared with the original cdc12-1 strain. The SWE1 deletion greatly reduces the ability of the cdc12-1 strain to grow at a semipermissive temperature (30°C; Fig. 7A). Furthermore, deletion of SWE1 has a strong negative effect on the viability of the cdc12-1 strain maintained at the restrictive temperature (37°C; Fig. 7B). Incubation of cdc12-1 mutant cells for 14 hr at 37°C results in some decrease in viability, but 40% of the cells remain able to give rise to colonies when returned to 22°C. In contrast, after 14 hr at the restrictive temperature, only 0.7% of the cdc12-1 swe1Δ cells and 9% of the cdc12-1 CDC28–Y19F cells maintain the ability to form colonies at 22°C.
Figure 7.
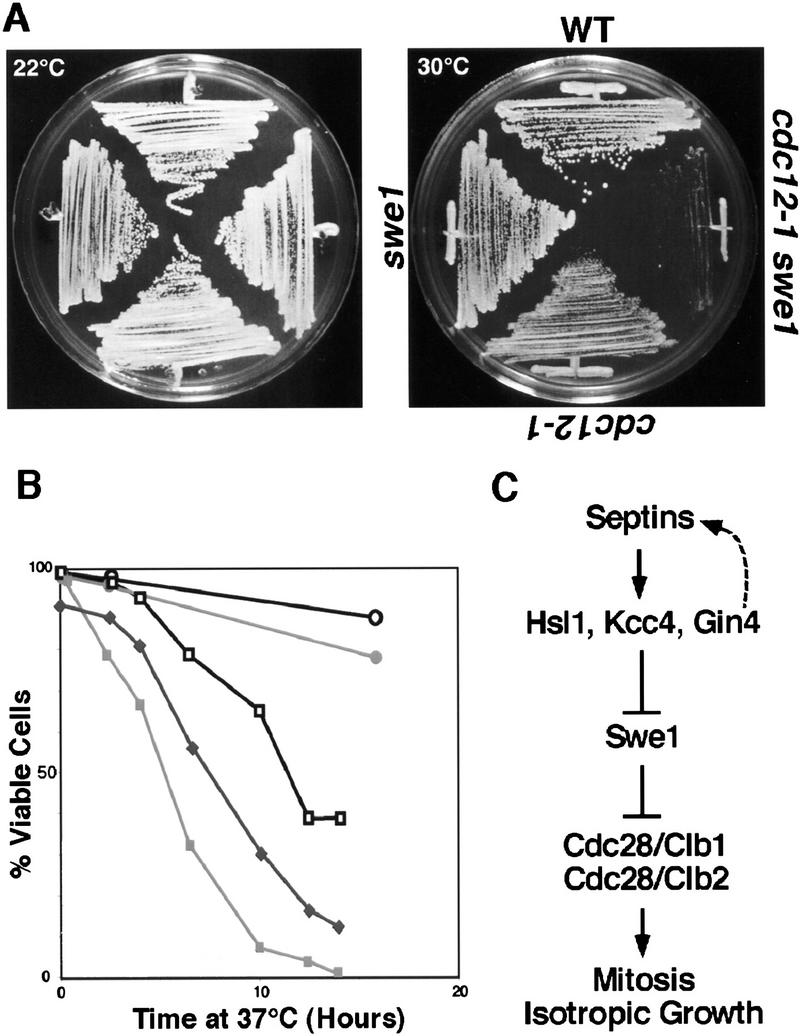
SWE1 is needed to maintain the viability of septin mutants exposed to the restrictive temperature. (A) The cdc12-1 swe1Δ double mutant has a reduced growth rate at 30°C compared with a cdc12-1 strain. Strains of the indicated phenotypes were incubated at either 22°C or 30°C. (B) Viability assay. Cells of the indicated genotype were incubated at 37°C, samples were taken at various time points, and the percentage of cells that maintained viability was determined by plating at 22°C. (○) wild type; (•) swe1Δ; (□) cdc12; (█) cdc12 swe1Δ; (♦) cdc12 CDC28–Y19F. (C) Model for the septin checkpoint of yeast. Proper assembly of the septin cytoskeleton results in localization and activation of Hsl1, Kcc4, and Gin4. This in turn inactivates Swe1, which thereby allows activation of the Cdc28 kinase. Thus, organization of the septin cytoskeleton is one mechanism by which cells detect that they have assembled both a bud and a proper cleavage apparatus for cytokinesis.
Discussion
Hsl1 is regulated by septin assembly
Hsl1, is a S. cerevisiae Nim1 homolog that regulates entry into mitosis (Tanaka and Nojima 1996) and has been shown to inhibit Swe1 (Ma et al. 1996). Here we show that Hsl1 interacts with the septins and depends on a functional septin cytoskeleton for activity. Hsl1 is a phosphoprotein, and Hsl1 phosphoisoforms disappear when Hsl1 kinase activity is abolished by a point mutation, indicating that Hsl1 phosphorylation directly or indirectly reflects Hsl1 activity. Interestingly, Hsl1 is hypophosphorylated in the cdc12-1 mutant, strongly suggesting that Hsl1 is inactive in those cells. Results from in vitro experiments are consistent with this idea. Hsl1 autophosphorylates in kinase assays in vitro; however, the kinase isolated from septin defective cells is inactive. The primary defect of mutated septins presumably is their inability to associate into filaments. Thus, our results suggest that Hsl1 requires interaction with the septin filaments or an associated structure to become active. Because Hsl1 is a negative regulator of Swe1, these results indicate that Hsl1 links Swe1 activity to septin organization.
HSL1, KCC4 and GIN4 act together to signal septin defects
The hypothesis that septin defects are detected through inactivation of the Hsl1 kinase predicts that deletion of HSL1 should lead to morphological and cell cycle defects mimicking the phenotype of septin-defective cells. However, a hsl1Δ mutant has much milder morphological defects and cell cycle delay than a septin mutant. We found that Kcc4 and Gin4, two kinases with similar architecture and extensive homology to Hsl1, function redundantly with Hsl1, as suggested by several lines of evidence. First, Kcc4 and Hsl1 colocalize with the same subcellular structure, the septin ring at the bud neck. Gin4 has also been reported to localize to the bud neck (Okuzaki et al. 1997; Longtine et al. 1998), suggesting that all three interact with the septin cytoskeleton. Like Hsl1 the activity of Gin4 has been found recently to also depend on septin function (Carroll et al. 1998). Second, gin4Δ and hsl1Δ mutants, and to a lesser extent, the kcc4Δ strain, have analogous cell cycle and morphological phenotypes, and each genetically interact with septin mutations. Third, the three disruptions have additive effects in wild type; unlike the single mutants, the hsl1Δ kcc4Δ gin4Δ strain forms extremely elongated buds, suggestive of a strong delay in the transition from apical to isotropic bud growth. This phenotype is extremely reminicent of the morphology of cells defective for septin function, supporting the idea that these kinases together signal septin defects. Further supporting the idea that a major role of these kinases is to signal septin organization status, the hsl1Δ kcc4Δ gin4Δ triple mutant remains viable, unlike septin-defective cells. Our data, in combination with other recently published results (Carroll et al. 1998) strongly indicate that the three kinases are most likely to be independent of each other for both localization and activity: (1) the activity of both Hsl1 and Gin4 requires their proper interaction with the septin cytoskeleton, but (2) in any of single mutant strain the two other kinases are still active, as demonstrated by the fact that the defects of the triple mutant are much stronger than those of any of the single mutants. Thus, the three kinases are unlikely to require each other to interact, colocalize with the septins, and be activated by them. Our results provide the first evidence of how Nim1-related kinases are regulated and thereby couple cell cycle progression with intracellular signals. We presume that because the three kinases, although closely related, are distinct, they might also integrate additional specific cellular signals for helping coordinate cell cycle events.
The observation that the hsl1Δ kcc4Δ gin4Δ triple mutant accumulates misshapen septin rings suggest that Hsl1, Kcc4, and Gin4 are also involved in the maintenance of septin organization. However, this function of Hsl1-related kinases in septin organization is not essential. We observe that the effect of the hsl1Δ kcc4Δ gin4Δ triple mutation on septin structures occurs mostly in medium- and large-budded cells. Thus, we propose that Hsl1, Kcc4, and Gin4 (or in some strains, only Gin4; Longtine et al. 1998) are necessary for the continued stability of septin structures in G2. We therefore suggest that these kinases are involved in both cell cycle control, through the regulation of Swe1, and the maintenance of cytoskeletal organization. Such a bifunctionality is not uncommon for proteins involved in cell cycle checkpoints. For example, DNA polymerase ε, a protein involved in DNA replication, also acts as a sensor of replication blocks in the DNA replication checkpoint (Navas et al. 1995). In yeast, the Mps1 kinase is required for both spindle pole body duplication and cell cycle arrest in response to spindle defects (Weiss and Winey 1996). In each case, such bifunctional proteins are found to act furthest upstream in the signaling pathway, suggesting that they are part of the sensor that detects defects in the monitored struture or process.
Septin defects activate a Swe1-dependent cell-cycle checkpoint
Here we show that yeast cells delay the entry into mitosis as a result of septin defects. Furthermore, this mitotic delay is dependent on Swe1. Deletion of SWE1 eliminates the mitotic delay and the elongated bud phenotype observed in the cdc12-1 mutant. However, the swe1Δ allele does not rescue the cytokinesis and viability defects of the cdc12-1 mutant, indicating that it does not bypass nor rescue septin function. Even at semipermissive temperatures, the deletion of SWE1 exacerbates cdc12-1 growth defects, excluding the hypothesis that it acts as a suppressor of septin defects. Moreover, Swe1 is required to prevent mortality of cells with septin defects. We therefore suggest that the Swe1 pathway triggers a delay in cell cycle progression in response to septin defects, a process that helps defective cells remain viable. This interpretation is in agreement with the known cellular function of Swe1, a kinase that delays mitosis by directly and specifically inhibiting the Cdc28 kinase associated with the mitotic cyclins by phosphorylation of Tyr 19 of Cdc28 (Booher et al. 1993). Consistent with this idea, we found that replacement of the Tyr 19 of Cdc28 by a similar but unphosphorylatable amino acid (Phe) bypasses the cell cycle delay due to septin defects, indicating that this delay is the result of Cdc28 inhibition by tyrosine phosphorylation. Thus, we propose that Hsl1, Kcc4, Gin4, and Swe1 act in a cell cycle checkpoint that detects septin defects and responds by inhibiting the mitotic CDK. Our model for this septin checkpoint pathway is presented in Figure 7c. We suggest that activation of Hsl1, Kcc4, and Gin4, due to the formation of the septin ring, leads to the inhibition of Swe1 and therefore permits the switch to isotropic growth and the onset of mitosis. In the presence of septin defects, Hsl1, Kcc4, and Gin4 remain inactive and Swe1 inhibits Cdc28, and thereby delays both the switch to isotropic bud growth and mitosis. Recent results from other laboratories suggest that this checkpoint machinery is regulated by the cell cycle machinery: Both Gin4 and Hsl1 interact physically with the G2 forms of Cdc28 (Tanaka and Nojima 1996; Altman et al. 1997; Okuzaki et al. 1997), and full activation of Gin4 depends on Clb function (Tjandra et al. 1998). We suggest that this is part of a feedback loop necessary for the rapid inactivation of Swe1 at the G2/M transition, in response to correct septin organization.
The mechanism by which Hsl1p-related kinases monitor septin assembly and become activated by septins remains to be determined, but several nonmutually exclusive possibilities exist. First, similar to that described for two other members of the Hsl1p kinase family, Snf1 and Nim1 (Jiang and Carlson 1996; Wu and Russell 1997), the binding of Hsl1p to a cofactor might be necessary to relieve the kinase from auto-inhibition. This cofactor might be the septin filaments themselves, which may activate Hsl1p-related kinases by binding a putative inhibitory domain in their nonkinase region. A second possibility is that Hsl1p activation depends on transautophosphorylation and/or activation by other kinases. It is possible that the septin ring facilitates the rapid phosphorylation and activation of the Hsl1-related kinases by concentrating them at the bud neck. Other kinases, such as Cla4 and Elm1p (Blacketer 1995), two kinases that have been shown to act upstream of Gin4 and Hsl1 (Tjandra et al. 1998; A. Myers pers. comm.), respectively, might be necessary to initiate the reaction. The septins might activate the Hsl1 pathway by serving as a scaffold that brings these different kinases together.
Relevance of septin checkpoints
The Swe1 kinase is involved in a morphological checkpoint that prevents mitosis in cells that fail to form a bud (Lew and Reed 1995). However, the mechanism for detection of the presence or absence of a bud was not defined. We suggest that the survey of septin function might participate in the sensing of morphological defects.
Recently, other studies have shown that drugs and mutations that affect the actin cytoskeleton trigger a Swe1-dependent cell cycle delay, suggesting that the actin cytoskeleton might be directly monitored by the morphogenetic checkpoint (McMillan et al. 1998). The mechanism by which cells monitor actin organization remain unclear. Our results demonstrate that the septins are also monitored by the Swe1 checkpoint. Several hypotheses can account for these observations.
First, it is possible that the actin- and septin-dependent checkpoints are distinct, although they both use Swe1 to trigger a mitotic delay. This would suggest that mechanisms of Swe1 regulation other than those involving Nim1-related kinases are responding to actin defects. Such mechanisms of Swe1 regulation might consist of direct control of Swe1 turnover in response to actin defects (McMillan et al. 1998).
Second, it is also possible that actin defects do not affect Swe1 regulation directly. Instead, cells might respond to such defects by inhibiting the Mih1 phosphatase that counterbalances Swe1 function. A similar situation exists in the case of the DNA replication checkpoint in S. pombe, in which in vivo phosphorylation of Cdc25 (to which Mih1 is homologous) by either of the Cds1 or Chk1 kinases is necessary to trigger a cell cycle delay in response to replication defects (Zeng et al. 1998). The fact that different levels of the actin depolymerizing drug Latrunculin A affects the time that the cell needs to adapt to the checkpoint response and resume cell cycle progression strongly support this hypothesis (McMillan et al. 1998).
Finally, it is also possible that some actin defects affect septin structures, or that the ability of Hsl1-related kinases to colocalize with and become activated by the septins is actin dependent. This model suggests that the septins are not directly monitored but are part of sensing machinery that monitors actin organization. However, the fact that SWE1 is necessary to maintain viability of septin mutants contradicts the simple view that the septins serve solely in a mechanism dedicated to monitoring actin function. It suggests that the Hsl1/Swe1 pathway monitors the septins themselves to prevent accidents that could result from septin defects.
The septins are extremely conserved proteins involved in cytoskeletal functions (Neufeld and Rubin 1994; Longtine et al. 1996; Cooper and Kiehart 1996; Kinoshita et al. 1997). Their most obvious function is in formation of the cleavage apparatus at cytokinesis, but the septins are likely to have additional functions during interphase or in differentiating cells (Flescher et al. 1993; Fares et al. 1995; Longtine et al. 1996; Kinoshita et al. 1997; Y. Barral and M. Snyder, in prep.). Cell cycle checkpoints monitoring the organization of the septin cytoskeleton would ensure that the cell does not enter mitosis if the septin-based cleavage apparatus is not present. To date, there is no evidence, in yeast or other eukaryotes, that defects in the cleavage furrow affect cell cycle progression. However, we demonstrate that in S. cerevisiae, assembly of the septin ring, an event preparing the formation of the contractile ring and necessary for proper bud growth (Y. Barral and M. Snyder, in prep.), is monitored by a checkpoint. This process might be conserved in other organisms. Alternatively, septin structures might also be monitored during interphase. In mammalian cells, septins are found in focal adhesion complexes and stress fibers. Signals emanating from these complexes might allow the cell to integrate morphological information with cell cycle progression, as the septin checkpoint appears to do in yeast.
Database searches indicate that proteins encoded by uncharacterized ORFs in Caenorhabditis elegans, and the product of the cdr2+ gene in S. pombe show strong similarities to Hsl1 and Gin4 that extend into their respective noncatalytic carboxy-terminal regions (Breeding et al. 1998; Kanoh and Russell 1998; and data not shown). Thus, proteins exist in nonbudding organisms that are good candidates for monitoring the assembly of septin structures. The conservation of these proteins in budding and fission yeasts and worms suggest that monitoring the assembly of septin structures is a ubiquitous process that is not limited to S. cerevisiae.
Other putative regulators of wee1 in higher eukaryotes
The discovery that Hsl1, Kcc4, and Gin4 link Swe1 function to the assembly of a cytoskeletal structure might provide more general insight into the regulators of wee1-related kinases. Hsl1, Gin4, and Kcc4 share structural and sequence similarity both in their kinase domains, and for Kcc4 and Gin4, in their carboxyl termini with a number of other molecules, particularly the MARK kinases from mammals (Drewes et al. 1997), and PAR-1 in C. elegans (Fig. 3; Guo and Kemphues 1995). These kinases have been implicated in the control of cell polarity and the microtubule organization (Guo and Kemphues 1995; Drewes et al. 1997). On the basis of their homology to Hsl1, we suggest that they may also regulate cell cycle progression in conjunction with cytoskeletal organization. For example, the PAR-1 protein localizes specifically to the posterior cell of early C. elegans embryos and is involved in P-granule migration and the establishment of the division axis during early embryogenesis (Guo and Kemphues 1995). Interestingly, par-1 mutations not only affect P-granule migration and spindle orientation, but also the timing of mitosis. At the two-cell stage, mitosis is normally delayed in the PAR-1 containing blastomere; however, in par-1 embryos, the two blastomeres enter nuclear division at the same time (Guo and Kemphues 1995). We propose that another aspect of PAR-1 function is to couple the cell cycle machinery with proper alignment of the spindle apparatus. It will be interesting to determine whether PAR-1 and the MARKs directly affect cell cycle progression and form a general mechanism for coupling cytoskeletal events with cell cycle progression.
Materials and methods
Yeast strains and growth conditions
All yeast strains are congenic with S288c. Yeast media and standard techniques used are described (Rose et al. 1990). General cloning procedures can be found in Sambrook et al. (1989). For the cell cycle synchronization experiments, MATa cells were arrested with 5 μg/ml α-factor mating pheromone and subsequently released in fresh medium. Point and small deletion mutations (cdc12-1, trp1-Δ63) were transferred into wild-type strain Y1521 (MATa ura3-52 leu2 his3-Δ200 lys2-801 ade2-101) through a minimum of five backcrosses. The swe1Δ(swe1:: LEU2) and hsl1Δ(hsl1::URA3) deletions are described (Ma et al. 1996). Deletions of KCC4 (kcc4::TRP1) and GIN4 (gin4::HIS3) genes were produced by PCR (Baudin et al. 1993). Transformants with the correct disruption of HSL1 and KCC4 loci were identified by PCR analysis.
The 3xmyc–CDC3 allele is described (Roemer et al. 1996). Tagging of Hsl1 and Kcc4 was performed as described (Schneider et al. 1995). Appropriate oligonucleotides with homology to the chromosomal insertion site were used to PCR amplify the epitope-tagging cassette containing URA3, the PCR products were transformed into yeast. Hsl1 was tagged at its amino terminus. Analysis of growth and morphological characteristics of cdc12-1 3xHA–HSL1 double mutants at the permissive and restrictive temperatures indicates that the 3xHA–HSL1 allele is fully active. Two versions of epitope-tagged Kcc4 were constructed. Strain Y1533 expresses a full-length Kcc4 protein tagged at its carboxyl terminus. Correct tagging of the targeted protein was verified by immunoblot analysis.
Construction of the inactive kinase allele was carried out by PCR with a plasmid carrying the HSL1 gene and oligonucleotides that substitute the AAA (Lys) position 110 codon with a GCG (Ala) codon. This substitution introduces a PvuI site in the sequence of the HSL1 gene. This mutant fails to complement hsl1Δ strains, as determined by morphological analysis of stationary-phase cells.
For KCC4 sequencing, a 585-bp region encompassing the 3′ end of the KCC4 locus was PCR amplified from yeast genomic DNA. Purified PCR products from five independent reactions were sequenced on both strands. Data analysis indicated insertion of a C after base 2737 in the KCC4 ORF.
Immunoprecipitations and kinase assays
Cells from a 5–10 ml YPD culture (OD 0.1–0.25) were resuspended in 100 μl of protein gel sample buffer containing 2 mm PMSF and lysed by vortexing with glass beads. Lysates were boiled for 5 min and 15 μl were loaded onto an 8% polyacrylamide gel (32% acrylamide, 0.18% bis-acrylamide) for immunoblotting.
Immunoprecipitations were performed as described (Altman and Kellogg 1997). A total of 10 ml of YPD cultures (OD of 0.1–0.2) were pelleted, resuspended in 150 μl of ice-cold IP buffer (50 mm K+-HEPES at pH 7.6, 1 mm EGTA, 1 mm MgCl2, 0.1% Tween 20, 2 mm PMSF, 10% glycerol, Sigma Protease inhibitor cocktail 1/1000) and an equal volume of glass beads, vortexed for eight 30-sec pulses, and then diluted with 800 μl of IP buffer. Lysates were incubated on ice for 15 min, centrifuged for 10 min at 4°C, and 400 μl of the extract was incubated with 2 μl of either polyclonal anti-HA (BabCo) or polyclonal anti-cmyc (MBL) antibodies for 5 hr at 4°C (gentle rocking) followed by a 2 hr incubation with 30 μl of protein A–agarose beads (Pierce Scientific). The pellets were washed five times with IP buffer and then transferred to a fresh tube. The samples were resuspended in 15 μl of 5× Laemmli buffer, boiled for 5 min and separated by SDS-PAGE for immunoblot analysis.
To assay Hsl1 kinase activity, the tagged protein was immunoprecipitated, and whereas one-half of the sample was used for immunoblot analysis, the other half was resuspended in 20 μl of kinase buffer (50 mm K+-HEPES at pH 7.6, 1 mmegta, 2 mmMgCl2, 0.1% Tween 20, 1 mm DTT, 10 μm ATP, 5 μCi [32P]ATP, 0.1 mg/ml Histone H1, Sigma Protease Inhibitor cocktail) and incubated at 30°C for 30 min. Kinase assays were stopped by the addition of 5 μl of 5× sample buffer, boiled for 5 min and loaded on a 10% (or 12%) polyacrylamide gel. The gel was fixed, dried, and exposed for autoradiography.
For immunoblot analysis, proteins were transferred to Immobilon-P membranes (Millipore), blocked for 2 hr in PBS + 5% dry milk and 0.05% Tween 20. Membranes were incubated overnight at 4°C in the same solution containing a 1/1000 dilution of either mouse monoclonal anti-HA (16B12 BabCo) or anti-cmyc (9E10 BabCo) antibodies. Membranes were washed four times in PBS + 0.05% Tween 20 and reincubated for 2 hr with HRP-conjugated secondary antibodies (Amersham). Detection (ECL, Amersham) was performed according to the instructions of the manufacturer.
Indirect immunofluorescence of tagged proteins
Indirect immunolocalization experiments were performed as described (Pringle et al. 1991). Cells from log phase cultures grown at 24°C or shifted to the restrictive temperature were fixed with 3.7% formaldehyde (final concentration) for 15 min. Fixed cells were subsequently vortexed, washed twice with PBS, and spheroplasted for up to 20 min at 37°C with 5 μg/ml Zymolyase T-100 in PBS + 1.2 m Sorbitol. The cells were washed twice with PBS-Sorbitol and transferred to polylysine coated slides. After four additional washes with PBS + 0.1% BSA, PBS + 0.1% BSA, 0.1% NP-40 (twice), and PBS + 0.1% BSA, the cells were incubated with the primary antibodies overnight at 4°C. After additional washings with PBS+0.1% BSA, PBS+0.1% BSA, 0.1% NP-40 (twice), and PBS + 0.1% BSA, incubation with the secondary antibodies was carried out for 2 hr at room temperature in the dark. For detection, either monoclonal anti-HA antibodies (clone 16B12, BabCo), 9E10 monoclonal anti-Myc antibodies or polyclonal anti-Myc antibodies (MBL, Japan) were used as primary antibodies. Primary antibodies were subsequently detected with either CY3 conjugated or FITC-conjugated secondary antibodies. The slides were mounted with a solution containing DAPI, and viewed with a Leitz fluorescence microscope.
Acknowledgments
We thank Ruth Collins, Shelley des Etages, Ruth Kroschewski, Brendan Manning, Petra Ross-Macdonald, Shirleen Roeder, and Susana Vidan for comments on the mansucript. Dawn Symonaitis, Amy Sheehan, and Petra Ross-Macdonald prepared and localized the Hsl1 construct tagged at codon 1225. We thank Daniel Lew and Michael Grunstein for providing strains and constructs. This work was supported by GM36494 to M.S. Y.B. was supported by a Human Frontiers postdoctoral Fellowship. S.B. was supported by a National Institutes of Health training grant.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL michael.snyder@yale.edu; FAX (203) 432-6161.
References
- Altman R, Kellogg D. Control of mitotic events by Nap1 and the Gin4 kinase. J Cell Biol. 1997;13:119–130. doi: 10.1083/jcb.138.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in S. cerevisiae. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton B, Tinkelenberg A, Gonzalez I, Cross F. Cla4p, a S. cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol Cell Biol. 1997;17:5067–5076. doi: 10.1128/mcb.17.9.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacketer MJ, Madaule P, Myers AM. Mutational analysis of morphologic differentiation in S. cerevisiae. Genetics. 1995;140:1259–1275. doi: 10.1093/genetics/140.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of S. cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D, Amberg D, Mulholland J, Huffaker T, Adams A, Drubin D, Stearns T. The yeast cytoskeleton. In: Pringle JR, Broach JR, Jones EW, editors. The molecular and cellular biology of the yeast Saccharomyces. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1997. pp. 1–90. [Google Scholar]
- Breeding CS, Hudson J, Balasubramanian MK, Hemmingsen SM, Young PG, Gould KL. The Cdr2+ gene encodes a regulator of G2-M progression and cyotkinesis in Schizosaccharomyces pombe. Mol Biol Cell. 1998;9:3399–3415. doi: 10.1091/mbc.9.12.3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Altman R, Schieltz D, Yates JR, Kellogg D. The septins are required for the mitosis-specific activation of the Gin4 kinase. J Cell Biol. 1998;143:709–717. doi: 10.1083/jcb.143.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J. Septin scaffolds and cleavage planes in Saccharomyces. Cell. 1996;84:187–190. doi: 10.1016/s0092-8674(00)80972-1. [DOI] [PubMed] [Google Scholar]
- Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6:877–882. doi: 10.1016/0955-0674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Coleman TR, Tang Z, Dunphy WG. Negative regulation of the wee1 protein kinase by direct action of the nim1/cdr1 mitotic inducer. Cell. 1993;72:919–929. doi: 10.1016/0092-8674(93)90580-j. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Kiehart DP. Septins may form a ubiquitous family of cytoskeletal filaments. J Cell Biol. 1996;134:1345–1348. doi: 10.1083/jcb.134.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Enoch T, Nurse P. Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell. 1990;60:665–673. doi: 10.1016/0092-8674(90)90669-6. [DOI] [PubMed] [Google Scholar]
- Fares H, Peifer M, Pringle JR. Localization and possible functions of Drosophila septins. Mol Biol Cell. 1995;6:1843–1859. doi: 10.1091/mbc.6.12.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flescher EG, Madden K, Snyder M. Components required for cytokinesis are important for bud site selection in yeast. J Cell Biol. 1993;122:373–386. doi: 10.1083/jcb.122.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kemphues KJ. par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell. 1995;81:611–620. doi: 10.1016/0092-8674(95)90082-9. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM. Protein kinase catalytic domain sequence database: Identification of conserved features of primary structure and classification of family members. Methods Enzymol. 1991;200:38–81. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- Hartwell LH. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 1971;69:265–276. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Jiang R, Carlson M. Glucose regulates protein interactions within the yeast SNF1 protein kinase complex. Genes & Dev. 1996;10:3105–3115. doi: 10.1101/gad.10.24.3105. [DOI] [PubMed] [Google Scholar]
- Kanoh J, Russell P. The protein kinase Cds2, related to Nim1/Cdc1 mitotic inducer regulates the onset of mitosis in fission yeast. Mol Biol Cell. 1998;9:3321–3334. doi: 10.1091/mbc.9.12.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HB, Haarer BK, Pringle JR. Cellular morphogenesis in the S. cerevisiae cell cycle: Localization of the CDC3 gene product and the timing of events at the budding site. J Cell Biol. 1991;112:535–544. doi: 10.1083/jcb.112.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Kumar S, Mizoguchi A, Ide C, Kinoshita A, Haraguchi T, Hiraoka Y, Noda M. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes & Dev. 1997;11:1535–1547. doi: 10.1101/gad.11.12.1535. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: Regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol. 1995;5:739–749. doi: 10.1083/jcb.129.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, DeMarini DJ, Valencik ML, Al AO, Fares H, De VC, Pringle JR. The septins: roles in cytokinesis and other processes. Curr Opin Cell Biol. 1996;8:106–119. doi: 10.1016/s0955-0674(96)80054-8. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Lu Q, Grunstein M. A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in S. cerevisiae. Genes & Dev. 1996;10:1327–40. doi: 10.1101/gad.10.11.1327. [DOI] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol. 1998;142:1487–1499. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas TA, Zhou Z, Elledge SJ. DNA polymerase ε links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, Rubin GM. The Drosophila peanut gene is required for cytokinesis and encodes a protein similar to yeast putative bud neck filament proteins. Cell. 1994;77:371–379. doi: 10.1016/0092-8674(94)90152-x. [DOI] [PubMed] [Google Scholar]
- Okuzaki D, Tanaka S, Kanazawa H, Nojima H. Gin4 of S. cerevisiae is a bud neck protein that interacts with the Cdc28 complex. Genes Cells. 1997;12:753–770. doi: 10.1046/j.1365-2443.1997.1590358.x. [DOI] [PubMed] [Google Scholar]
- Parker LL, Walter SA, Young PG, Piwnica-Worms H. Phosphorylation and inactivation of the mitotic inhibitor Wee1 by the nim1/cdr1 kinase. Nature. 1993;363:736–738. doi: 10.1038/363736a0. [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell. 1997;88:315–321. doi: 10.1016/s0092-8674(00)81870-x. [DOI] [PubMed] [Google Scholar]
- Pringle J, Adams AEM, Drubin DG, Haarer BK. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–601. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Pringle J, Bi E, Harkins H, Zahner J, Devirgilio C, Chant J, Corado K, Fares H. Establishment of cell polarity in yeast. Cold Spring Harbor Symp Quant Biol. 1995;60:729–744. doi: 10.1101/sqb.1995.060.01.079. [DOI] [PubMed] [Google Scholar]
- Roemer T, Madden K, Chang J, Snyder M. Selection of axial growth sites in yeast requires Axl2p, a novel plasma membrane glycoprotein. Genes & Dev. 1996;10:777–793. doi: 10.1101/gad.10.7.777. [DOI] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in yeast genetics: A laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Russell P, Nurse P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell. 1987a;49:559–567. doi: 10.1016/0092-8674(87)90458-2. [DOI] [PubMed] [Google Scholar]
- ————— The mitotic inducer nim1 functions in a regulatory network of protein kinase homologs controlling the initiation of mitosis. Cell. 1987b;49:569–576. doi: 10.1016/0092-8674(87)90459-4. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory press; 1989. [Google Scholar]
- Schneider BL, Seufert W, Steiner B, Yang QH, Futcher AB. Use of PCR epitope tagging for protein tagging in S. cerevisiae. Nucleic Acids Res. 1995;11:1265–1274. doi: 10.1002/yea.320111306. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Sia RA, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996;7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Nojima H. Nik1: A Nim1-like protein kinase of S. cerevisiae interacts with the Cdc28 complex and regulates cell cycle progression. Genes Cell. 1996;1:905–921. doi: 10.1046/j.1365-2443.1996.d01-213.x. [DOI] [PubMed] [Google Scholar]
- Tjandra H, Compton J, Kellogg D. Control of mitotic events by the Cdc42 GTPase, the Clb2 cyclin and a member of the PAK kinase family. Curr Biol. 1998;8:991–1000. doi: 10.1016/s0960-9822(07)00419-8. [DOI] [PubMed] [Google Scholar]
- Weiss E, Winey M. The S. cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol. 1996;132:111–123. doi: 10.1083/jcb.132.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Russell P. Nim1 kinase promotes mitosis by inactivating Wee1 tyrosine kinase. Nature. 1993;363:738–741. doi: 10.1038/363738a0. [DOI] [PubMed] [Google Scholar]
- ————— Nif1, a novel mitotic inhibitor in Schizosaccharomyces pombe. EMBO J. 1997;16:1342–1350. doi: 10.1093/emboj/16.6.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Forbes KC, Wu Z, Moreno S, Piwnica-Worms H, Enoch T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature. 1998;395:507–510. doi: 10.1038/26766. [DOI] [PubMed] [Google Scholar]