

Abstract
Saccharomyces cerevisiae telomeric DNA replicates late in S phase, and telomeric genes are transcriptionally silent. Transcriptional repression of telomere-proximal genes results from silent chromatin initiating at the chromosome end, but the relationship between telomeric chromatin and DNA replication is unknown. Mutations in SIR3, a silent chromatin component, cause telomeric DNA on chromosome V to replicate much earlier because of earlier initiation of a nearby replication origin, the Y′ ARS. A second telomere-proximal ARS, from an X element, does not act as an origin in a wild-type strain, whereas in a sir3 cell it does. We conclude that telomeric chromatin has a Sir3-dependent inhibitory effect on DNA replication.
Keywords: Saccharomyces cerevisiae, heterochomatin, DNA replication, telomeres, silencing
Different regions of the eukaryotic genome are known to replicate at distinct, reproducible times within the period of S phase. This is observed in plants, insects, and vertebrates, as well as in the yeast Saccharomyces cerevisiae (Hand 1978; Fangman and Brewer 1992). In species with large genomes, extensive regions of the chromosomes replicate early in S phase, whereas other domains do not initiate replication until after the early domains have completed synthesis. Frequently, late replication of DNA is associated with its assembly into heterochromatin, which is highly condensed chromatin that often contains repeated DNA sequences such as those found at centromeres and telomeres (John 1988). This condensed chromosome replicates much later in S phase than its transcriptionally active homolog.
Telomeric chromatin in S. cerevisiae has several traits typical of heterochromatin (Grunstein 1998). In particular, telomeres confer epigenetic silencing of nearby genes (position effect variegation), and they replicate late in S phase (McCarroll and Fanagman 1988; Gottschling et al. 1990). The special chromatin found near telomeres is composed of hypoacetylated core histones as well as the SIR proteins, which are required for silencing telomeric genes (Grunstein 1998). Of the SIR proteins, Sir3p is probably the key component that defines a telomeric domain of transcriptional repression. It interacts with the tails of histones H3 and H4, spreading from the telomere inward along the chromosome, and its abundance in the cell determines how far a silent domain extends from the telomere (Renauld et al. 1993; Hecht et al. 1996).
In yeast, chromosome replication initiates at ARS (autonomous replicating sequences) elements (Fangman and Brewer 1991). ARS elements were originally identified as sequences that permit high-efficiency transformation of plasmids in yeast by serving as origins of DNA replication. However, in their normal chromosomal context, only a subset of the ARS elements initiate DNA replication within the ∼30 min duration of S phase (Fangman and Brewer 1992). Specific chromosomal origins of S. cerevisiae, like ARS1 on chromosome IV, initiate replication relatively early in S phase. Other ARS elements initiate later in S phase, such as ARS501 on chromosome V, which replicates ∼10 min after ARS1 (Ferguson et al. 1991).
It appears that telomeres can confer late replication on proximal origins. For instance, telomere-proximal middle repetitive sequences replicate relatively late in S phase (McCarroll and Fangman 1988; Louis 1995). In addition, whereas ARS elements on circular plasmids initiate replication early in S phase, they replicate late in S phase when the plasmid is linearized and telomeric sequences are added to its ends (Ferguson and Fangman 1992). In this study we investigated the role of silent chromatin in imposing late replication on origins near telomeres.
Results and Discussion
We asked whether silent telomeric chromatin was responsible for the relatively late replication time of telomere-proximal DNA by measuring the replication kinetics of sequences near the right telomere of chromosome V in a wild-type strain and in a strain in which the SIR3 gene was deleted. This region of the genome is represented schematically at the top of Figure 1A, and its late replication has been documented previously (McCarroll and Fangman 1988; Ferguson et al. 1991). Located adjacent to the telomeric repeats at the extreme end of the chromosome are members of two families of subtelomeric, middle repetitive sequences, the X and Y′ elements (Louis 1995). The X element is found at all native yeast telomeres, and the Y′ at most but not all. However, neither is essential for linear chromosome stability (Wellinger and Zakian 1989). Although each of these elements contains an ARS, it is not known whether either has origin activity at the right telomere of chromosome V (Ferguson et al. 1991). The regions examined for their replication kinetics are the restriction fragment R6, which contains the late-replicating chromosomal origin ARS501 and is ∼25 kb from the telomere, and R2, the penultimate unique DNA sequence adjacent to the telomeric X element.
Figure 1.







Sir3 delays replication of telomere-proximal DNA sequences. (A) Chromosome V-R telomeric constructs (centromere to the left). ARSs are indicated by black boxes; telomeric repeats by the checkered box at the end of the chromosome. X and Y′ elements are indicated by open boxes. ARS501 is ∼27 kb from the end of the chromosome. The locations of R6 and R2 EcoRI fragments, 7.9 and 4.4 kb, respectively, are depicted as shaded boxes beneath the chromosome. The distance between these two fragments is 9.5 kb. Beneath these fragments are shown their relative replication times (Δtrep, minutes after replication of ARS1) in both SIR+ and sir3. Beneath fragment R2 are arrows showing the predominant directions of replication fork movement through this fragment (percentages indicate the fraction of cells in the population with forks moving in the specified direction). Also indicated beneath fragment R6 is whether ARS501 is an active origin (YES/NO) in the strain as determined by 2D gel analysis (data not shown). (B) Density shift data for the R2 and R6 fragments. Δtrep for R2 and R6 are shown as in A. The density shift curves for ARS1 are given for each experiment. (Left) SIR+ strains; (right) sir3 mutants. AR120 and UCC4403 have a full-length chromosome V-R. UCC4431 and UCC4433 have the subtelomeric elements deleted. UCC4453 and UCC4457 have the Y′ ARS inserted back into the deleted chromosome. Percent replication is shown on the y-axis; the number of minutes after release into S phase is indicated on the x-axis. (C) 2D gels analyzing the direction of replication fork movement through the R2 region in SIR+ strains (AR120, UCC4431, and UCC4453, top row) and sir3 strains (UCC4403, UCC4433, and UCC4457, bottom row). (Inset) Idealized 2D fork direction gel with the arcs characteristic of forks approaching from the telomeric and centromeric sides of R2 indicated.
Replication times were measured by a density transfer protocol (McCarroll and Fangman 1988). The replication time of a chromosomal fragment (trep) is defined as the time when that fragment is replicated in 50% of the cells in a culture. Replication times are expressed as minutes after the replication time of the nontelomeric, early replicating fragment ARS1 (Δtrep).
SIR3 delays replication of a telomere-adjacent fragment
In a SIR+ strain (AR120), the R6 restriction fragment containing ARS501 replicated at approximately +10 min after ARS1 (Fig. 1A,B), consistent with earlier reports (Ferguson et al. 1991), and in the sir3 strain (UCC4403), this same fragment replicates at +8 min (Fig. 1A, B). In contrast, R2 replicated at +13 min in the SIR+ strain but at +4 min in sir3 (Fig. 1A,B). Because ARS501 (R6) replicated at essentially the same time in SIR+ and sir3 strains, the difference in the replication times at R2 was likely the result of changes in an origin firing distal to it. To verify whether this is true, the direction of replication forks passing through the R2 fragment in the two strains was determined by a two-dimensional gel method (Friedman and Brewer 1995). In both the SIR+ (AR120) and sir3 (UCC4403) strains, a fork originating from its telomeric side replicates the R2 fragment in >95% cells (Fig. 1A,C). This suggests that in the majority of cells, R2 is replicated by origins that initiated at or near the telomere and that the SIR3-dependent delay in replication timing is the result of changes in a telomere-proximal origin.
Although the replication timing of ARS501 was not affected by the absence of Sir3p, it was possible that deletion of the SIR3 gene might have global effects on late replication. To test this idea, the replication time of a fragment within a nontelomeric, late-replicating region was measured in both SIR+ and sir3 strains. This region on chromosome XIV near the KEX2 gene contains ARS1412, which lies ∼200 kb from the nearest telomere and is late replicating (Brewer et al. 1993; Friedman et al. 1996). The Δtrep of this fragment in a SIR+ strain was +11 min (s.d. = 1.7 min; data not shown). In a sir3 strain, the replication time of this fragment was +12 min (s.d. = 2.5 min; data not shown), essentially unchanged. These results, in combination with the fact that ARS501 (R6) did not respond to the Sir3-dependent change in replication timing, suggested that Sir3 was not responsible for late replication throughout the genome. This also suggested that the late firing of ARS501 (reported previously) is not under SIR3 control (Ferguson and Fangman 1992).
Which telomere-proximal origin was responsible for R2 replication and responded to SIR3 changes? It was likely to be the ARS in either the X or Y′ element, although in principle it could have been the end of the chromosome itself. To distinguish between these possibilities, the X and Y′ sequences at the right end of chromosome V were deleted, thus bringing the telomere adjacent to the R2 sequence (middle of Fig. 1A). This truncated chromosome was then analyzed by the same methods used to study the intact chromosome.
There were two notable differences in replication effects between this truncated chromosome and the normal chromosome. First, the R2 sequence in the truncated chromosome replicated even later in S phase (cf. +20 min of UCC4431 to +13 min of AR120; Fig. 1A,B) by a fork now originating primarily from the centromeric side of R2 (Fig. 1A,C). This reversal of fork directions suggested that either the X or the Y′ element contained the origin of replication that was normally responsible for most of R2’s replication.
Second, there was no difference between the replication timing of R2 (or R6) when comparing SIR+ (UCC4431) and sir3 (UCC4433) versions of the truncated chromosome (Fig. 1A,B). This suggested that the Sir3-dependent change in timing of R2 seen in the normal chromosome acted through origins in the X or Y′ elements.
Telomeric Y′ ARS initiation is delayed by Sir3
Using the truncated chromosome as a starting point, we examined whether the ARS within the subtelomeric Y′ element was responsible for the Sir3-dependent delay in replication timing at R2. The Y′ ARS was a good candidate for being affected because it is located <1 kb from the junction of the terminal TG1–3 DNA tract, well within the normal domain of silent chromatin extending from the end of the chromosome known to be influenced by SIR3 (Renauld et al. 1993). The telomeric fragment of Y′ containing this ARS was reinserted into the truncated chromosome such that the sequences and spacing from the telomere to the Y′ ARS were maintained as in the normal chromosomal configuration. With this modified chromosome, the Sir3-dependent late replication of the R2 fragment was once again observed; R2 replicated at +9 min in SIR3 cells, whereas in sir3 cells it replicated at 0 min (Fig. 1A,B). This strongly suggests that silent chromatin delayed replication of the Y′ARS.
The R2 fragment with the reinserted Y′ ARS was replicated approximately 4 min earlier in this chromosome compared to the normal chromosome, for both the SIR3 and sir3 versions of the strains (+9 min vs. +13 minutes for SIR+ and 0 min vs. +4 min for sir3). This 4-min difference can be explained by considering that ∼9 kb of DNA is missing between R2 and the Y′ ARS in the altered chromosome and that the estimated rate of DNA replication fork movement in yeast is 1–3 kb/min (Rivin and Fangman 1980). In the final comparison of this altered construct, ARS501 does not act as an origin of replication in the sir3 version (Fig. 1A). Most likely, R6/ARS501 is passively replicated by a much earlier replicating fork from the Y′ ARS.
Next we asked whether the SIR3-dependent delay was restricted to the V–R telomere by measuring the replication kinetics of the total Y′ family of subtelomeric elements. The time of replication for the Y′ family shifted from +10 min in the SIR+ strain, to +2 min in the sir3 strain (data not shown). Although it was not possible to ascertain whether this shift was a result of all, or only a subset of the Y′ elements replicating earlier, as a group they replicated significantly earlier in the sir3 strain. Hence, the timing effect was not specific to the V–R telomere and suggested that replication timing of most telomeres was under SIR3 control.
Sir3 delays initiation of ARS1 near a telomere
The above results led us to ask whether other ARS elements, when placed near a telomere within the normal silencing domain of Sir3p, responded to the Sir3-dependent delay in firing. To answer this question, a fragment containing the early replicating ARS1 was inserted at the same location that the Y′ ARS fragment had been integrated, in both SIR+ and sir3 strains (Fig. 2). In this case, as with the integration of the Y′ ARS, R2 undergoes a shift in replication timing from late, in the SIR+ strain (UCC4451, +21 min), to 8 min earlier in the sir3 strain (UCC4455, +13 min). ARS1 was acting as an origin in both the SIR+ and sir3 strains as evidenced by forks coming from the telomeric side of R2 (Fig. 2C). Thus the Sir3-dependent delay in replication of R2 was not specific to the Y′ ARS, but can be extended to at least one other origin, ARS1 when it is located within 1 kb of the telomere.
Figure 2.



ARS1 integrated at the V-R telomere is delayed in replication by Sir3. (A) Diagram of the chromosome V-R telomeric ARS1 integration construct (centromere to the left). (Rightward arrow) SIR3 (UCC4451); (leftward arrow) sir3 (UCC4455). Details are as in Fig. 1A. (B) Density shift data for the R2 fragment. Details are as in Fig. 1B, except only replication times (Δtrep) for R2 and ARS1 (at its natural location on chromosome IV) are presented. (C) 2D gels analyzing the direction of replication fork movement through the R2 region in wild-type (UCC4451) and sir3 (UCC4455) strain. Details are as in Fig. 1C. (Left) SIR3; (right) sir3 (B,C).
There is also a Sir3-independent delay in the replication of the telomeric ARS1. In the sir3 strain, the R2 fragment adjacent to the telomeric ARS1 replicated significantly later (13 min) than a fragment next to ARS1 at its normal location on chromosome IV. Because the telomeric ARS1 does not act as an origin in all the sir3 cells (only 58%), we cannot determine whether the delay is the result of a later firing of the telomeric ARS1, a lack of its use, or a combination of both effects.
Sir3 prevents initiation of a telomeric X ARS
Finally, the fate of the X ARS near the normal chromosome V-R telomere (ARS5RX) was examined. X elements are much more heterogeneous in sequence, and their ARSs are more variable in distance from the telomere than is the case for Y′ elements (Louis 1995). Nonetheless we wished to determine whether ARS5RX was affected by SIR3. Specifically we examined whether ARS5RX acted as an origin of replication in a native chromosome in the SIR+ and sir3 strains. In the SIR+ strain the ARS5RX does not appear to act as an origin, just as has been reported for ARS3LX (Fig. 3; data not shown; Newlon et al. 1993). Thus, ARS5RX is normally passively replicated by a fork originating at the Y′ ARS. In contrast, ARS5RX was able to act as an origin when SIR3 was deleted, as shown by the replication bubble arc in Figure 3. Thus, we conclude that Sir3p prevents X ARS from acting as an origin of replication.
Figure 3.
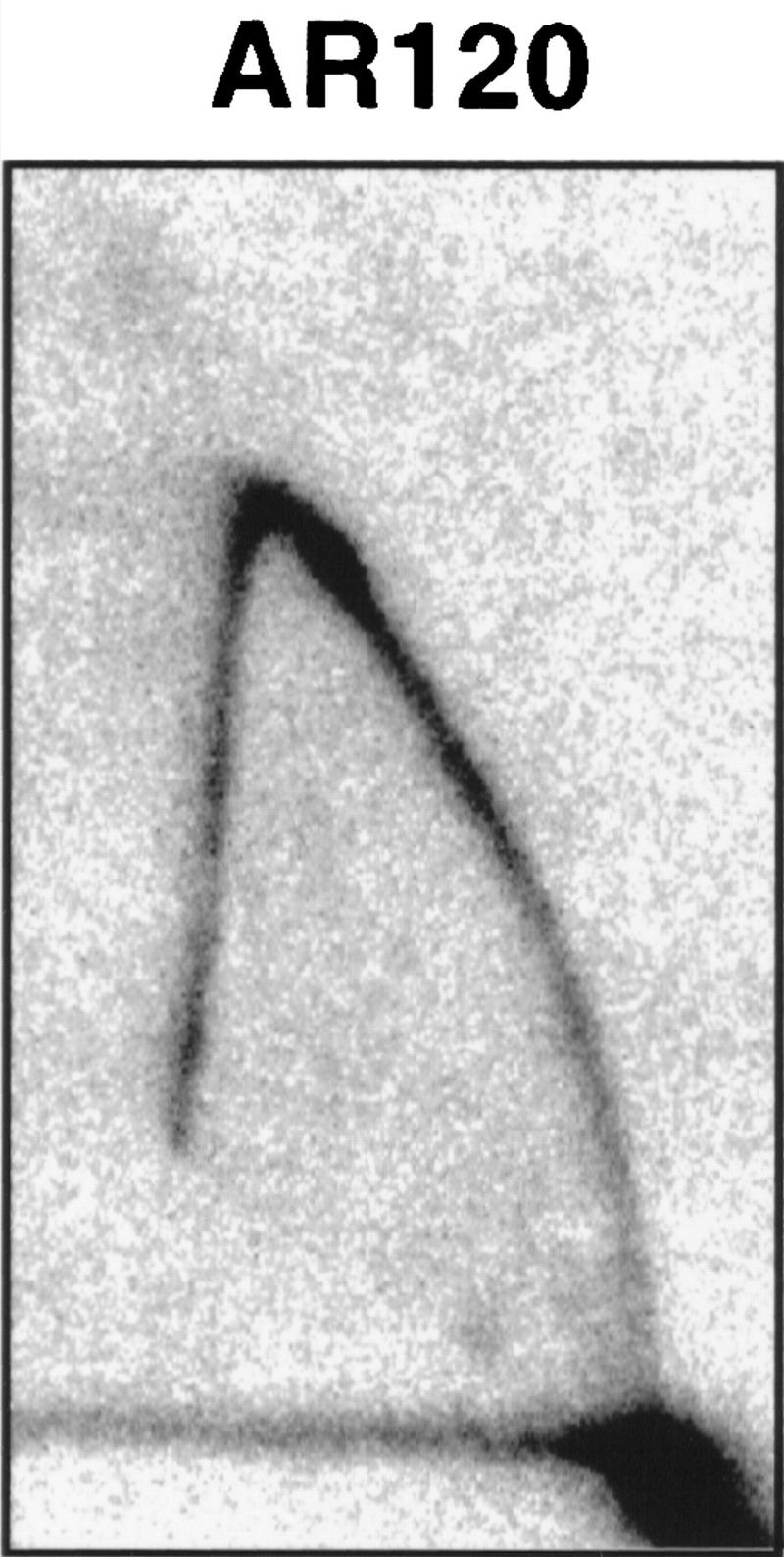
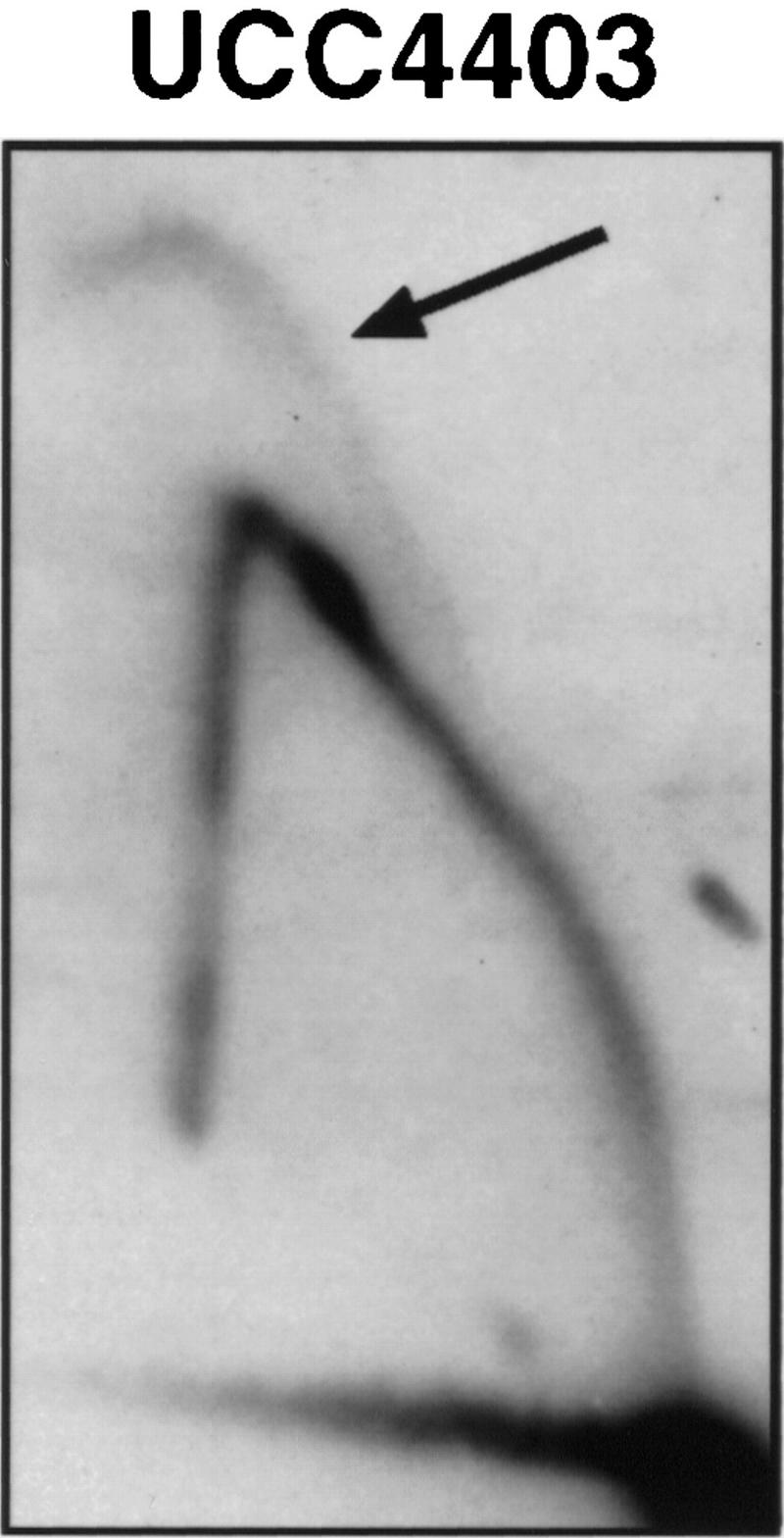
The ARS5RX becomes active in a sir3 strain. 2D gels analyzing activity of ARS5RX in SIR+ strain (AR120) and sir3 (UCC4403). The arrow points to the arc characteristic of an active replication origin in UCC4403. The shapes of the arcs are somewhat atypical because the fragment containing ARS5RX is relatively large (5.2 kb) and the origin is asymmetrically located on the fragment.
How does SIR3 cause late replication of telomeric DNA?
In this study we have shown that Sir3p causes a significant delay (8–9 min in an S phase that lasts ∼30 min) in the apparent initiation of replication at telomeric ARS elements, for both a Y′ ARS and ARS1. This is an ‘apparent’ delay, because given the limitation of the assays, we cannot distinguish between a true delay in initiation of DNA synthesis at the ARS and a localized initiation event at the ARS in which subsequent fork elongation is severely inhibited or slowed.
Regardless of the precise mechanism, we propose that silent telomeric chromatin prevents key components of the replication machinery from readily gaining access to the underlying DNA. Most chromatin is comprised of nucleosomes, whereas silent chromatin has nucleosomes that are stabilized by molecules such as Sir3p. In essence, there is a reduced off-rate of chromatin components from the DNA for silent chromatin compared to ‘normal’ chromatin. A reduced off-rate means that the effective on-rate for limiting replication factors is reduced. Studies on the activation of a silenced telomeric gene support such an idea. Higher concentrations of a transcriptional activator are required to express a telomeric gene to a given level than when the same gene is located at an internal, nonsilenced locus (Aparicio and Gottschling 1994). The on-rate for any binding factor is a function of the factor’s concentration and the amount of time it has to bind to its substrate. Thus, in an event like S phase that has a defined starting point, the delay in replication seen in silent chromatin may be the result of a need to synthesize sufficient levels of a limiting component during S phase to achieve the necessary concentration for binding. Alternatively, the concentration of the factor may not change significantly during S phase; however, more time is required for it to have a chance to bind.
It is thought that all active origins of replication have a prereplication complex assembled on them (Santocanale and Diffley 1996). This consists of the Origin Recognition Complex (ORC), which binds the ARS DNA, Cdc6p, the Mcm proteins, Cdc45p, and possibly other proteins (Pasero and Gasser 1998). Assuming that active telomeric origins have fully assembled prereplication complexes, then components like the Mcm proteins and Cdc45p, which are known to be recruited to chromatin and act at or near the replication fork, are reasonable candidates for the limiting replication factor in the model we present (Aparicio et al. 1997). The competition may occur either in the factor’s initial binding or in its ability to perturb nucleosomes for fork progression.
Why do telomeres replicate late?
It is tempting to speculate that late replication of telomeric DNA serves a special function in the cell; it may be required to re-establish silent chromatin, help coordinate replication of the chromosome end by telomerase, or facilitate sister chromatid cohesion. However, it is also likely that the Sir3-dependent late replication is a secondary consequence of telomeric chromatin. Telomeric chromatin may have evolved to serve in canonical roles associated with telomeres: distinguishing telomeres from broken chromosome ends, and protecting telomeres from degradation and end-to-end fusion.
In considering this latter idea, it is worth noting that the Y′ element causes the telomeric region of chromosome V-R to replicate earlier than when the Y′ element was not present (Fig. 1). Although Y′ elements have been suggested to play a role in telomere maintenance in the absence of telomerase (Lundblad and Blackburn 1993), we propose that Y′ elements may reside at telomeres because they provide a selective advantage in duplicating the genome more rapidly. X elements, which do not have the ability to initiate replication at V-R or III-L (Fig. 3; Newlon et al. 1993), are much more heterogeneous in sequence and size and therefore appear to be much older in evolutionary history than Y′ elements. Thus, in laboratory strains of S. cerevisiae, we may be spectators to the evolutionary expansion of a recently acquired DNA element, under the selective pressure of completing S phase more quickly.
Materials and methods
DNA plasmids
pSC4120, pL110, and pR151 were gifts from W. Fangman (University of Washington, Seattle), and plasmid pYP1-L2 was a gift of E. Louis (Louis and Haber 1992). Plasmids pVRINTY′ARS, pY′H3/XbaI, pUCR2, pR2X-2, pUCR2X/P, pUCK/N, pUCR6, pVRΔXY′TEL, and pVRINTARS1 were created in this study. Detailed maps of these plasmids are available upon request.
Yeast strains and methods
Media used for the growth of S. cerevisiae were described previously (Gottschling et al. 1990). All yeast strains in this study were grown at 23°C in YEPD unless noted otherwise. Standard methods were used for genetic manipulation of S. cerevisiae (Renauld et al. 1993). Methods for DNA preparation and analysis have been described previously (Gottschling et al. 1990; Aparicio et al. 1991).
The strain AR120 (HMLa MATa HMRa cdc7-1 bar1 trp1-289 ura3-52 leu2-3,112 his6) was a gift from W. Fangman. The strain UCC4403 was created by transforming AR120 with BamHI–HindIII-digested pJH107.1 (Ivy et al. 1986). Leu+ transformants were selected and checked by Southern blot to confirm the loss of SIR3.
The strains UCC4431 and UCC4433 were created by transforming the strains AR120 (SIR+) and UCC4403 (sir3), respectively, with PvuII-digested pVRΔXY′TEL. Ura+ transformants were selected to give strains UCC4430 (SIR+) and UCC4432 (sir3). These strains were then patched to 5-FOA, and 5-FOA resistant colonies were selected to give the strains UCC4431 (SIR+) and UCC4433 (sir3). The resulting strains have all sequences distal to the R2 fragment replaced with telomeric TG1–3 repeats.
The strains UCC4453 and UCC4457 were created by transforming the strains AR120 and UCC4403, respectively, with PvuII-digested pVRINTY′ARS and processed as described above. These strains have all sequences distal to the R2 fragment replaced with the Y′ ARS and telomeric TG1–3 repeats.
The strains UCC4451 and UCC4455 were created by transforming the strains AR120 and UCC4403, respectively, with PvuII-digested pVRINTARS1 and processing as described above. These strains have all sequences distal to the R2 fragment replaced with ARS1 and telomeric TG1–3 repeats.
Determination of replication times
Replication times were determined by a density transfer protocol as described previously (McCarroll and Fangman 1988). Isolated genomic DNA was digested with EcoRI for the replication timing protocols and probed with the following fragments to determine replication timing: a 2.8-kb EcoRI fragment from pSC4120, which hybridizes adjacent to ARS1; a 3.4-kb EcoRI fragment from pL110 was used for ARS1412; a 3.1-kb XbaI fragment from pR151 (Ferguson et al. 1991) was used for R6; a 3.9-kb EcoRI–NdeI fragment from pUCR2 for the R2 fragment and a 0.3-kb HindIII–XbaI fragment from pY′H3/XbaI was used as a probe for the Y′ family.
Slot blots were exposed for 2–7 days on a PhosphorImager cassette, and the images were quantified using ImageQuant software (Molecular Dynamics). For each time point, the unreplicated peak and the replicated peak were identified, and the relative hybridization for each peak was determined using NIH Image 1.61 software. Percent replication for each time point was calculated with the following equation: % replicated = (0.5 × % hybridization in the replicated peak)/[(0.5 × % hybridization in the replicated peak) + (% hybridization in the unreplicated peak)].
The percent replication was plotted on the y-axis versus the number of minutes after release from cdc7 arrest on the x-axis using Sigma Plot graphics software (Jandel Scientific). The equation for calculating a best fit curve for percent replication data was f(x) = [a/1 + eb(x–c)] + d, where a = 100, b = −0.1, c = 50; and d = 0.
Representative data sets were used for Figures 1–3. Each Δtrep value reported for ARS1412 were the average of three independent experiments, for SIR+ and sir3, from strains AR120, UCC4431, UCC4403, and UCC4433. The values for the total Y′ elements were an average of two independent experiments from strains UCC4403, UCC4431, and UCC4433.
2D gel methods
Cell collection and isolation of nuclei were performed as described (Marahrens and Stillman 1994). The collection of DNA by CsCl gradient and subsequent digestion, BND–cellulose chromatography, and DNA precipitation were done as described (Liang et al. 1993). 2D and fork direction gels were performed as described (Brewer and Fangman 1987; Friedman and Brewer 1995).
Firing of the ARS501 origin was determined on 2D gels with genomic DNA that was cleaved with XbaI to generate a 3.1-kb fragment containing ARS501. Blots were probed with this same 3.1-kb XbaI fragment isolated from plasmid pUCR6. Firing of ARS5RX was determined similarly, except the DNA was cleaved with HindIII and SalI to yield a 5.2-kb fragment containing ARS5RX. Blots were then probed with a 500-bp fragment from pRX2-2.
For all fork direction gels, except UCC4457, DNA was digested with EcoRV and XbaI before being run in the first dimension. In-gel digests were done with SpeI. Southern blots were probed with a 1.5-kb BamHI–HindIII fragment from pUCR2X/P and a 1.0-kb KpnI–NdeI fragment from pUCK/N. For UCC4457, DNA was digested with EcoRI and XhoI before being run in the first dimension. The in-gel digest was done with PstI. The blot was probed with a 1.5-kb BamHI–HindIII fragment from pUCR2X/P.
For quantification of fork direction gels, the intensity of hybridization was measured for each arc from approximately the point where the two curves, intersect to the apex of each curve, using ImageQuant software. The background signal was subtracted and the percent hybridization for each arc was calculated.
Acknowledgments
We are indebted to B. Brewer, W. Fangman, and M.K. Raghuraman for gifts of plasmids, strains, and their invaluable advice and patience during this work. E. Louis for plasmids, B. Stillman for technical advice, and members of the Gottschling laboratory and J. Roberts for comments on the manuscript. This work was supported by National Institutes of Health grant GM43893.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Dgottsch@fhcrc.org; FAX (206) 667-5894.
References
- Aparicio OM, Gottschling DE. Overcoming telomeric silencing: A trans-activator competes to establish gene expression in a cell cycle-dependent way. Genes & Dev. 1994;8:1133–1146. doi: 10.1101/gad.8.10.1133. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell. 1991;66:1279–1287. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Diller JD, Friedman KL, Kolor KM, Raghuraman MK, Fangman WL. The topography of chromosome replication in yeast. Cold Spring Harbor Symp Quant Biol. 1993;58:425–434. doi: 10.1101/sqb.1993.058.01.049. [DOI] [PubMed] [Google Scholar]
- Fangman WL, Brewer BJ. Activation of replication origins within yeast chromosomes. Annu Rev Cell Biol. 1991;7:375–402. doi: 10.1146/annurev.cb.07.110191.002111. [DOI] [PubMed] [Google Scholar]
- ————— A question of time—Replication origins of eukaryotic chromosomes. Cell. 1992;71:363–366. doi: 10.1016/0092-8674(92)90505-7. [DOI] [PubMed] [Google Scholar]
- Ferguson BM, Fangman WL. A position effect on the time of replication origin activation in yeast. Cell. 1992;68:333–339. doi: 10.1016/0092-8674(92)90474-q. [DOI] [PubMed] [Google Scholar]
- Ferguson BM, Brewer BJ, Reynolds AE, Fangman WL. A yeast origin of replication is activated late in S phase. Cell. 1991;65:507–515. doi: 10.1016/0092-8674(91)90468-e. [DOI] [PubMed] [Google Scholar]
- Friedman KL, Brewer BJ. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 1995;262:613–627. doi: 10.1016/0076-6879(95)62048-6. [DOI] [PubMed] [Google Scholar]
- Friedman KL, Diller JD, Ferguson BM, Nyland SV, Brewer BJ, Fangman WL. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes & Dev. 1996;10:1595–1607. doi: 10.1101/gad.10.13.1595. [DOI] [PubMed] [Google Scholar]
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Yeast heterochromatin: Regulation of its assembly and inheritance by histones. Cell. 1998;93:325–328. doi: 10.1016/s0092-8674(00)81160-5. [DOI] [PubMed] [Google Scholar]
- Hand R. Eucaryotic DNA: Organization of the genome for replication. Cell. 1978;15:317–325. doi: 10.1016/0092-8674(78)90001-6. [DOI] [PubMed] [Google Scholar]
- Hecht A, Strahl-Bolisinger S, Grunstein M. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature. 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- Ivy JM, Klar AJ, Hicks JB. Cloning and characterization of four SIR genes of Saccharomyces cerevisiae. Mol Cell Biol. 1986;6:688–702. doi: 10.1128/mcb.6.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B. The biology of heterochromatin. In: Verma RS, editor. Heterochromatin: Molecular and structural aspects. Cambridge, UK: Cambridge University Press; 1988. pp. 1–67. [Google Scholar]
- Liang C, Spitzer JD, Smith HS, Gerbi SA. Replication initiates at a confined region during DNA amplification in Sciara DNA puff II/9A. Genes & Dev. 1993;7:1072–1084. doi: 10.1101/gad.7.6.1072. [DOI] [PubMed] [Google Scholar]
- Louis EJ. The chromosome ends of Saccharomyces cerevisiae. Yeast. 1995;11:1553–1573. doi: 10.1002/yea.320111604. [DOI] [PubMed] [Google Scholar]
- Louis EJ, Haber JE. The structure and evolution of subtelomeric Y′ repeats in Saccharomyces cerevisiae. Genetics. 1992;131:559–574. doi: 10.1093/genetics/131.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1− senescence. Cell. 1993;73:347–360. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- Marahrens Y, Stillman B. Replicator dominance in a eukaryotic chromosome. EMBO J. 1994;13:3395–3400. doi: 10.1002/j.1460-2075.1994.tb06642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll RM, Fangman WL. Time of replication of yeast centromeres and telomeres. Cell. 1988;54:505–513. doi: 10.1016/0092-8674(88)90072-4. [DOI] [PubMed] [Google Scholar]
- Newlon CS, Collins I, Dershowitz A, Deshpande AM, Greenfeder SA, Ong LY, Theis JF. Analysis of replication origin function on chromosome III of Saccharomyces cerevisiae. Cold Spring Harbor Symp Quant Biol. 1993;58:415–423. doi: 10.1101/sqb.1993.058.01.048. [DOI] [PubMed] [Google Scholar]
- Pasero P, Gasser SM. New systems for replicating DNA in vitro. Curr Opin Cell Biol. 1998;10:304–310. doi: 10.1016/s0955-0674(98)80004-5. [DOI] [PubMed] [Google Scholar]
- Renauld H, Aparicio OM, Zierath PD, Billington BL, Chhablani SK, Gottschling DE. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes & Dev. 1993;7:1133–1145. doi: 10.1101/gad.7.7a.1133. [DOI] [PubMed] [Google Scholar]
- Rivin CJ, Fangman WL. Replication fork rate and origin activation during the S phase of Saccharomyces cerevisiae. J Cell Biol. 1980;85:108–115. doi: 10.1083/jcb.85.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF. ORC- and Cdc6-dependent complexes at active and inactive chromosomal replication origins in Saccharomyces cerevisiae. EMBO J. 1996;15:6671–6679. [PMC free article] [PubMed] [Google Scholar]
- Wellinger RJ, Zakian VA. Lack of positional requirements for autonomously replicating sequence elements on artificial yeast chromosomes. Proc Natl Acad Sci. 1989;86:973–977. doi: 10.1073/pnas.86.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]