

Abstract
The retinoblastoma tumor suppressor protein (pRB) can inhibit cell cycle progression and promote differentiation. pRB interacts with a variety of transcription factors, including members of the E2F and C-EBP protein families and MyoD, and can either repress or activate transcription depending on the promoter under study. These biological and biochemical activities of pRB have been mapped previously to a core domain, referred to as the pRB pocket. Using a panel of synthetic pRB pocket mutants, we found that the acute induction of a G1/S block by pRB is linked to its ability to both bind to E2F and to repress transcription. In contrast, these functions were not required for pRB to promote differentiation, which correlated with its ability to activate transcription in concert with fate-determining proteins such as MyoD. All tumor-derived pRB mutants tested to date failed to bind to E2F and did not repress transcription. Despite an inability to bind to E2F, pRB mutants associated with a low risk of retinoblastoma, unlike high-risk mutants, retained the ability to activate transcription and promote differentiation. Thus, the pRB pocket participates in dual tumor suppressor functions, one linked to cell cycle progression and the other to differentiation control, and these functions can be genetically and mechanistically dissociated.
Keywords: Retinoblastoma, transcription, E2F, tumorigenesis
The retinoblastoma gene (RB-1) is frequently mutated in human cancer. Furthermore, many tumors that retain a wild-type RB-1 allele contain genetic alterations, such as amplification of the cyclin D1 gene, homozygous deletion of the p16/MTS1 tumor suppressor gene, or activating mutations of cdk4, which can lead to the inappropriate phosphorylation and, hence, functional inactivation, of the RB-1 gene product (pRB). Thus, inactivation of pRB may be a necessary step in human carcinogenesis (for review, see Strauss et al. 1995; Weinberg 1995).
The five cloned members of the E2F transcription factor family (E2F1–E2F5) bind to DNA as heterodimers with either DP1 or DP2. For simplicity, these heterodimers are collectively referred to as E2F (for review, see Adams and Kaelin 1995; La Thangue 1994). E2F DNA-binding sites are present in the promoters of a number of genes involved in DNA synthesis, such as DNA polymerase α and dihydrofolate reductase (DHFR) or in cell cycle control, such as cyclins E and cdc2 (for review, see Adams and Kaelin 1995; Ohtani et al. 1995; Botz et al. 1996; Geng et al. 1996). pRB binds to E2F and converts E2F from a transcriptional activator to a transcriptional repressor (for review, see Weintraub et al. 1992; Sellers and Kaelin 1996). All tumor-derived pRB mutants examined to date are unable to bind to E2F and are unable to repress transcription when bound to DNA (Sellers and Kaelin 1996). Furthermore, transcriptional activation of E2F responsive promoters can bypass a pRB-induced G1/S cell cycle block in vitro (Zhu et al. 1993; Qin 1995). Thus, formation of pRB/E2F complexes, and consequent repression of E2F dependent promoters, likely contributes to pRB-mediated tumor suppression in vivo. Such a view may account for the unexpected finding that E2F1 −/− mice are prone to tumors (Yamasaki et al. 1996). Specifically, loss of E2F1 may affect the ability of pRB to bind to certain E2F-responsive promoters.
p107 and p130, two members of the pRB family, can likewise bind to E2F and repress E2F-dependent transcription and yet, to date, have not been identified as targets of mutations in human tumors (Cao et al. 1992; Schwartz et al. 1993; Ginsberg et al. 1994; Vairo et al. 1995; Zhu et al. 1995). Furthermore, p107 −/− mice and p130 −/− mice are viable and do not develop tumors (Cobrinik et al. 1996; Lee et al. 1996). In contrast, RB −/− mice are not viable and RB +/− mice develop tumors (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992). In addition, the intracellular concentration of pRB appears to vastly exceed that of E2F and a number of non-E2F proteins have been identified that can bind to pRB, at least in vitro, including ATF2, BRG-1, hBrm, c-Abl, C/EBP, D-type cyclins, Elf-1, Id-2, Mdm2, MyoD, NF-IL6, PU.1, and UBF (for review, see Weinberg 1994; Chen et al. 1996; Sellers and Kaelin 1996; Taya 1997). These observations, taken together, suggest that pRB may perform additional function(s), unrelated to its role as a repressor of E2F-dependent transcription, which contribute to its ability to suppress tumor growth in vivo.
pRB can cooperate with certain transcription factors, such as C-EBP family members and MyoD, to transcriptionally activate genes involved in differentiation (Gu et al. 1993; Chen et al. 1996a,b; Novitch et al. 1996). Furthermore, pRB is required for expression of late markers of skeletal muscle and adipocyte differentiation in vitro (Gu et al. 1993; Chen et al. 1996a; Novitch et al. 1996). Finally, analyses of pRB-deficient mice suggest a role for pRB in myogenic and neuronal differentiation in vivo (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992; Nikitin and Lee 1996; Zacksenhaus et al. 1996). Whether transcriptional activation and differentiation promotion by pRB are direct consequences of its ability to repress E2F-responsive promoters and inhibit cell cycle progression is currently unknown.
The majority of tumor-derived pRB mutants are defective for multiple biochemical activities, making it difficult to discern the relative contributions of these activities to pRB-mediated tumor suppression. Therefore, we created a panel of pRB mutants that was then scored in a series of biochemical and biological assays. By use of this approach, the ability of pRB to induce an acute G1/S block was linked to its ability to bind to E2F and to repress transcription when bound to DNA. In contrast, neither stable binding to E2F nor an intact transcriptional-repression domain were required for pRB to activate transcription and promote differentiation.
Results
To determine which pRB properties might cosegregate with pRB-mediated growth suppression, a set of substitution mutants was generated by site-directed mutagenesis. Residues in the pRB that are highly conserved in p107 and p130 were replaced with the sequence NAAIRS (Wilson et al. 1985). NAAIRS is thought to be a flexible linker based on its appearance in both α-helical and β-sheet structures. Each mutant was named according to the first substituted residue. For example, residues 651–656 were replaced with NAAIRS in pRBΔ651. In the experiments described below, only data obtained with representative informative pRB mutants are shown. Over 20 pRB substitution mutants were generated in total. No conclusion drawn in this manuscript was based on the analysis of a single pRB mutant. A comprehensive biochemical and biological characterization of these mutants will be described elsewhere (W.R. Sellers and W.G. Kaelin, in prep.).
The ability of the pRB mutants to bind to E2F was measured in two assays. RB (−/−) tumor cells were transiently transfected with plasmids encoding hemagglutinin (HA)-epitope tagged versions of the pRB mutants, lysed, and immunoprecipitated with an anti-HA antibody. Wild-type pRB, and a tumor-derived pRB mutant (Δex22) were tested in parallel. All of the RB proteins were produced at comparable levels as determined by anti-HA western blot analysis of the corresponding whole cell extracts (Fig. 1C). pRB-associated proteins were released from the immunoprecipitates with a mild detergent, deoxycholate (DOC), and scored for E2F DNA-binding activity in gel-shift assays with a 32P-labeled E2F DNA-binding site (Fig. 1A; Shirodkar et al. 1992; Neuman et al. 1994). In parallel, cell extracts prepared from these transfectants were tested directly for total E2F DNA-binding activity by use of gel-shift assays (Fig. 1B). In this latter assay, interaction of pRB with E2F results in a loss of free E2F complexes (solid arrows) and the formation of a more slowly migrating complex shown previously to contain pRB and E2F (open arrow). All of the pRB mutants that retained the ability to form such stable pRB/E2F/DNA complexes were likewise able to bind to E2F in solution as measured in the immunoprecipitation (IP)–DOC release experiments (e.g., pRBΔ685). On the other hand, some pRB mutants that scored positively in the IP–DOC assays (Fig. 1A) were unable to form stable pRB/E2F/DNA complexes (e.g., pRBΔ651; Fig. 1B). These results suggest that either the IP–DOC assay is more sensitive than conventional gel-shift assays or that there are additional structural requirements for stable binding of pRB to E2F when the latter is bound to DNA as opposed to free in solution.
Figure 1.
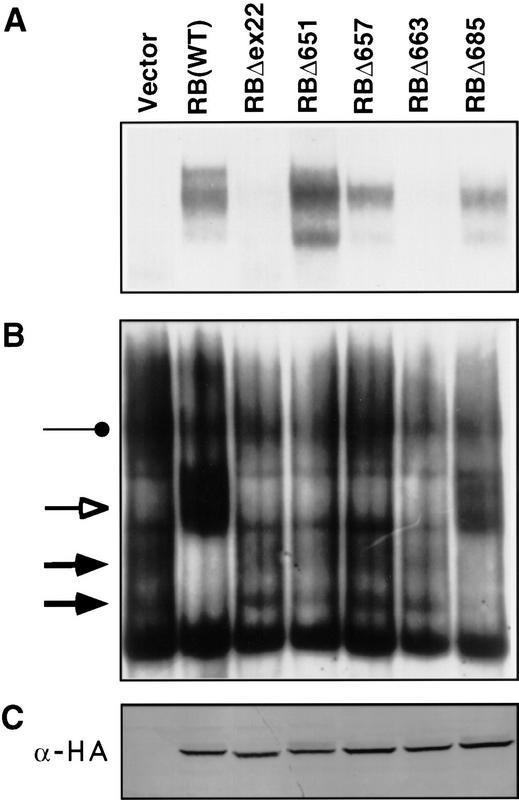
E2F-binding to pRB mutants. (A) IP–DOC release. SAOS2 cells were transfected with plasmids encoding epitope HA-tagged versions of the indicated pRB proteins. Anti-HA immunoprecipitates were prepared and bound proteins were released by treatment with deoxycholate. Released proteins were scored in gel-shift assays with a 32P-radiolabeled E2F DNA-binding site. (B) E2F DNA-binding activities in transiently transfected cells. Nuclear extracts were prepared from SAOS2 cells transiently transfected as in A and analyzed in gel-shift assays with a 32P-radiolabeled E2F DNA-binding site. (Solid arrows) Complexes containing free E2F; (open arrow and solid circles) pRB/E2F and p107/E2F complexes, respectively. (C) Production of pRB mutants. Whole cell extracts (150 μg) prepared from SAOS2 cells transiently transfected as in A were immunoblotted with an anti-HA antibody.
Acute induction of a G1/S block by pRB is linked to transcriptional repression
pRB represses transcription when bound to DNA. This activity maps to a region within pRB residues 379–928 (Adnane et al. 1995; Bremner et al. 1995; Sellers et al. 1995; Weintraub et al. 1995). The pRB mutants, as TETr–DNA-binding domain–pRB(379–928) chimeras, were next assayed for their ability to repress transcription. Plasmids encoding the TETr–RB(379–928) chimeras were transiently introduced into RB +/+ (data not shown) and RB −/− cells (Fig. 2B) along with an E2F1–promoter luciferase reporter plasmid in which the E2F sites were replaced with TETo sites (pGL2ANΔTETo) (Fig. 2C; Sellers et al. 1995). In parallel, the corresponding full-length pRB mutants were tested for their ability to induce a G1/S block. To this end, RB (−/−) SAOS2 osteosarcoma cells were transfected with a plasmid encoding wild-type HA–pRB, or the mutant derivatives thereof, and the cell cycle distribution of the transfected cells was determined by fluorescence-activated cell sorting (FACS) (Fig. 2A). Only those pRB mutants, such as pRBΔ685, that formed stable pRB/E2F/DNA complexes (Fig. 1B) and retained the ability to repress transcription (Fig. 2B) induced a G1/S block in these assays (Fig. 2A). In contrast, pRB mutants such as pRBΔ651, pRBΔ657, pRBΔ663, which were defective for these activities, were unable to induce a G1/S block. Notably, pRBΔ651 and pRBΔ657 were able to bind to E2F in solution as measured in the IP–DOC release assay (Fig. 1A). These results are consistent with the view that the acute induction of G1/S block following reintroduction of pRB into RB −/− tumor cells is linked to its ability to repress transcription once bound to E2F responsive promoters.
Figure 2.
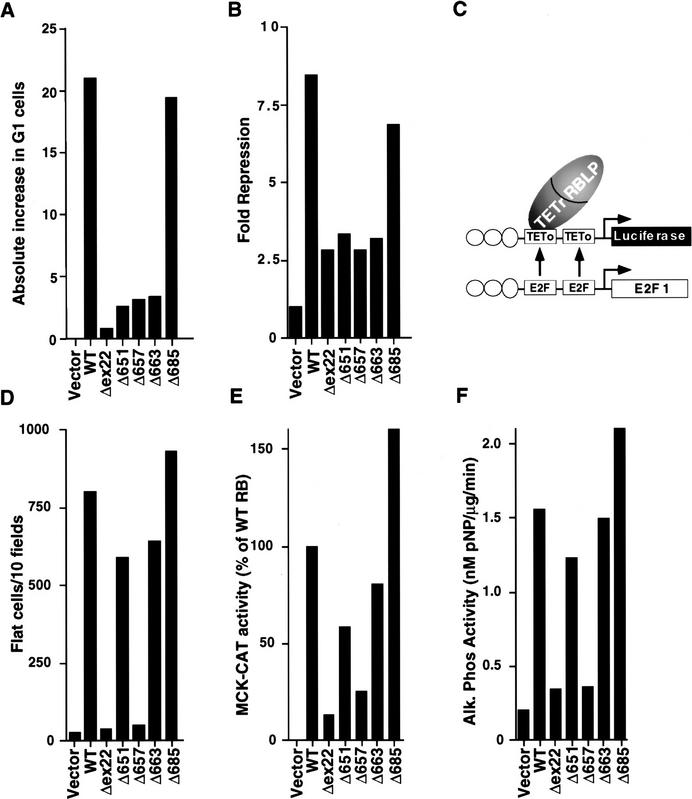
Transcriptional repression and activation functions predict the ability of pRB to induce a cell cycle block and promote differentiation. (A) Induction of a G1 arrest by pRB. SAOS2 cells were transiently transfected with plasmids encoding the indicated pRB proteins along with a plasmid encoding the cell-surface marker CD19. Seventy-two hours later, the DNA content of CD19-positive cells was determined by FACS. The y-axis indicates the absolute increase in the percentage of cells in G1 relative to cells transfected with the backbone expression plasmid. Each value represents the mean of 2–5 independent experiments. (B) Transcriptional repression mediated by TETr–RB(379-928) and mutant derivatives. SAOS2 cells were cotransfected with pGL2ANΔTETo, pCMV-β-Gal and plasmids encoding either wild-type TETr–RB(379-928) or the indicated mutant derivatives. Luciferase values were determined and normalized for β-galactosidase activity. Fold repression represents the corrected luciferase value obtained with TETr alone divided by the corrected luciferase obtained with the indicated TETr–RB chimeras. Data shown are means of replicate values and are representative of three independent experiments. (C) Experimental design for repression assays. Transcriptional repression was measured by use of a luciferase reporter plasmid (pGL2ANΔTETo) containing the region from −211 to +64 of the E2F1 promoter (Neuman et al. 1994). Both E2F DNA-binding sites were replaced by TETo DNA-binding sites. Wild-type pRB(379–928), and mutant derivatives thereof, were targeted to DNA as chimeras containing the DNAbinding domain of TETr (TETr–RBLP) (Sellers et al. 1995). (D) Flat cell induction. SAOS2 cells were transfected with plasmids encoding the indicated pRB proteins along with a neomycin resistance plasmid. The number of flat cells per ten 100× fields following 2 weeks of G418 selection was determined by manual counting. Values represent the mean of four independent experiments. (E) Transactivation of the MCK promoter. RB −/− MEFs were cotransfected with plasmids encoding the indicated pRB proteins along with a plasmid encoding MyoD and a plasmid containing the MCK promoter upstream of CAT. CAT activity was determined 36–48 hr following transfection. CAT activity relative to the activity obtained in the presence of wild-type RB is shown. Data are representative of two independent experiments. (F) Induction of alkaline phosphatase activity. SAOS2 cells were transfected and selected in G418 as in D. Alkaline phosphatase activity was measured using a colorimetric substrate and normalized for total protein content. Values represent the means of duplicate samples and are representative of three independent experiments.
Flat cell induction by pRB linked to transcriptional activation
Reintroduction of wild-type pRB into certain RB −/− cells such as SAOS2 osteosarcoma cells also causes profound morphological changes referred to as the flat cell phenotype (Fig. 3; Huang et al. 1988; Templeton et al. 1991; Hinds et al. 1992; Qin et al. 1992). To score for flat cell induction, the HA-pRB expression plasmids were transfected into SAOS2 cells along with a neomycin resistance marker. Following 2 weeks of drug selection, the number of flat cells per 10 100× fields was determined. As expected, wild-type pRB induced numerous flat cells, whereas pRBΔex22 did not (Figs. 2D and 3). A number of mutants, such as pRBΔ651 and pRBΔ663, efficiently generated flat cells (Figs. 2D and 3) despite an inability to form stable pRB/E2F/DNA complexes, an inability to repress transcription, and an inability to induce an acute G1/S block (Figs. 1B and 2, B and A, respectively). In contrast, mutants such as pRBΔ657 that, unlike pRBΔ663, were able to bind to E2F (Fig. 1A), at least in solution, were unable to induce flat cells (Fig. 2D). Finally, a chimeric protein containing the E2F1 DNA-binding domain and pRB transcriptional repression domain did not induce flat cells despite an ability to repress E2F-responsive promoters and induce a G1/S block in these cells (Fig. 4). Taken together, these results suggested that the induction of flat cells, unlike the acute induction of a G1/S block, was not linked to the ability of pRB to bind to E2F or to its ability to repress transcription.
Figure 3.
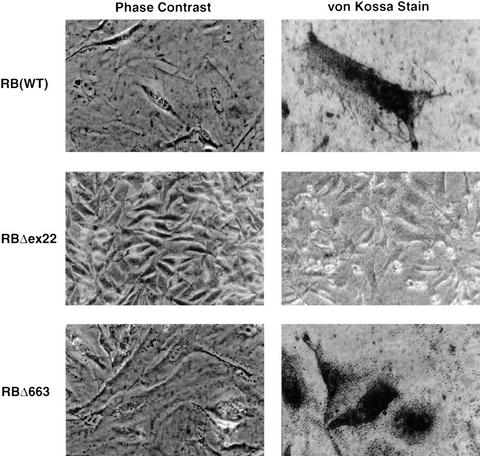
Morphological changes induced following restoration of pRB function in SAOS2 osteogenic sarcoma cells. SAOS2 osteogenic sarcoma cells were stably transfected with plasmids encoding the indicated pRB proteins. (Left) Phase-contrast micrographs of cells following 2 weeks of selection in G418. (Right) von Kossa Staining for mineral deposition after 2 weeks selection in G418 followed by 2 weeks incubation in media containing 50 μg/ml l-ascorbic acid and 10 mm β-glycerol-phosphate. Magnification, 300×.
Figure 4.
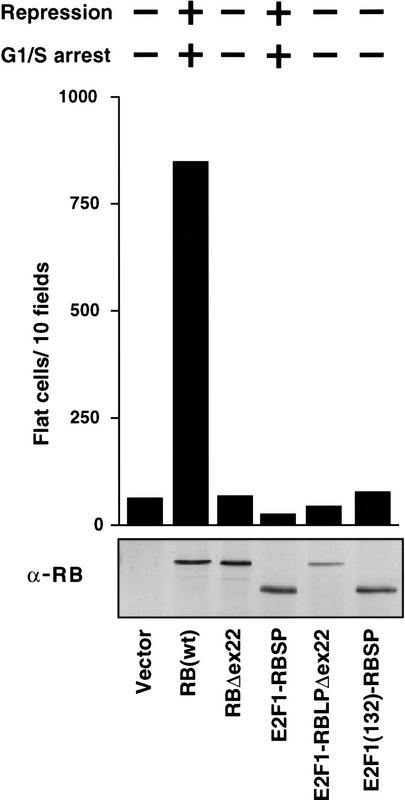
An E2F1–RB chimera does not induce flat cells. SAOS2 cells were transfected with plasmids encoding either wild-type pRB, pRBΔex22, or the indicated E2F1-RB chimeras, along with a neomycin resistance marker. (E2F1) E2F1(1–368); (RBSP) pRB(379–792); (RBLP) pRB(379–928). The number of flat cells per ten 100× fields following 2 weeks of G418 selection was determined by manual counting. Data shown are representative of three independent experiments. (Top) The ability of the various proteins to either repress transcription or to induce a G1/S arrest, as determined previously (Sellers et al. 1995). (Bottom) Production of each protein was confirmed by anti-RB immunoprecipitation followed by anti-RB immunoblot analysis.
MyoD, NF-IL6, C/EBP, and the glucocorticoid receptor (GRα), are examples of transcription factors that depend on the presence of wild-type pRB to activate certain promoters (Gu et al. 1993; Singh et al. 1995; Chen et al. 1996a,b; Novitch et al. 1996). Next, the pRB mutants were tested for their ability to cooperate with MyoD as one measure of this activity. To this end, a plasmid encoding MyoD was introduced into RB −/− murine embryo fibroblasts (MEFs) with a reporter plasmid containing the muscle creatine kinase (MCK) promoter upstream of the chloramphenicol acetyltransferase (CAT) gene. In the absence of pRB, there was little or no effect of MyoD on the MCK promoter (Fig. 2E). MyoD activation of the MCK promoter was, as expected, restored in the presence of wild-type pRB, but not pRBΔex22. Each pRB mutant that induced flat cells (such as pRBΔ651, pRBΔ663, pRBΔ685) was likewise capable of cooperating with MyoD to activate the MCK promoter (cf. Fig. 2D and E). In contrast, mutants that did not induce flat cells (such as pRBΔ657) also failed to rescue MyoD activation (Fig. 2D,E). These data suggested that the ability of pRB to cooperatively activate transcription might underlie its ability to induce flat cells.
Flat cells display markers indicative of differentiation
The ability of pRB to cooperate with MyoD to activate a late differentiation promoter (MCK) suggested that the flat cell phenotype observed in SAOS2 osteosarcoma cells might reflect the ability of pRB to cooperate with one or more endogenous transcription factors involved in bone differentiation. If so, flat cells might express markers indicative of osteoblast differentiation. In keeping with this view, flat cells induced by wild-type RB exhibited a 7- to 8-fold increase in bone alkaline phosphatase activity compared to cells transfected with the backbone expression plasmid (Fig. 2F). Similarly, all of the pRB mutants capable of inducing flat cells induced a 6- to 10-fold increase in alkaline phosphatase activity. Conversely, those mutants that failed to induce flat cells also failed to induce bone alkaline phosphatase activity. These results were corroborated by assaying SAOS2 cells for bone mineral deposition by use of a modified von Kossa stain. Flat cells induced by wild-type pRB or pRBΔ663 scored positively in this assay, whereas cells producing pRBΔEx22 did not (Fig. 3).
pRB, p107, and p130 differ in their ability to activate transcription and promote differentiation
p107 and p130, like pRB, interact with certain E2F family members and can induce a G1/S block (Schwarz et al. 1993; Zhu et al. 1993; Claudio et al. 1994; Vairo et al. 1995). To determine whether these proteins were likewise capable of inducing flat cells, SAOS2 cells were transiently transfected with plasmids encoding either HA–pRB, HA–p107, HA–p130, or mutant derivatives thereof (HA-pRBΔ22, p107–DE, and HA-p130Δ21, respectively), along with a neomycin resistance plasmid. All of these proteins were produced at comparable levels as determined by anti-HA western blot analysis of extracts prepared from transiently transfected cells (Fig. 5A). Two weeks after drug selection, the number of flat cells was determined. p107 and p130 induced flat cells far less effectively than pRB (Fig. 5C). The relative ability of p107 and p130 to induce flat cells mirrored their ability to cooperate with MyoD in transcriptional activation assays in the RB −/− MEFs (Novitch et al. 1996). p107 and p130, like pRB, were capable of arresting SAOS2 cells in G1 following transient transfection (Fig. 5B). In contrast, the mutant derivatives of these proteins failed to induce either flat cells or a G1/S block (Fig. 5B,C). Thus, pRB, p107, and p130 differ in their ability to activate transcription and promote differentiation. Notably, the ability of the various pRB family members to induce a G1/S block (Fig. 5B) did not fully predict their ability to promote flat cells (Fig. 5C). In particular, p130 was comparable with pRB in arresting cells in G1 and yet was barely capable of inducing flat cells.
Figure 5.
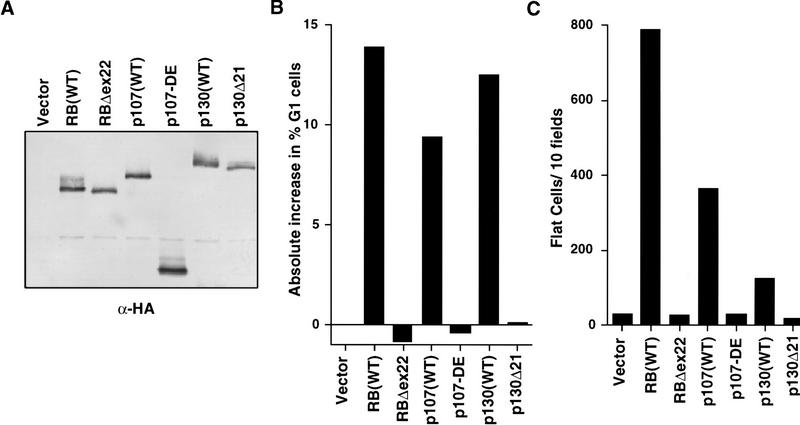
pRB family members differ in flat cell induction. (A) Production of pRB family members in SAOS2 cells. SAOS2 cells transiently transfected with plasmids encoding the indicated HA-tagged wild-type or mutant pRB family members were lysed and immunoprecipitated with an anti-HA antibody. The immunoprecipitates were resolved by electrophoresis in a 7.5% SDS–polyacrylamide gel and immunoblotted with an anti-HA antibody. (B) pRB family members induce a cell cycle block in SAOS2 cells. SAOS2 cells were transiently transfected with plasmids encoding the indicated wild-type or mutant pRB family members along with a plasmid encoding the cell surface marker CD19. Seventy-two hours later the DNA content of CD19-positive cells was determined by FACS. The y-axis indicates the absolute increase in the percentage of cells in G1 relative to cells transfected with the backbone expression plasmid. Data shown is from a single experiment and is representative of data obtained in three independent experiments. (C) Flat cell induction. SAOS2 cells were transfected with plasmids encoding the indicated wild-type or mutant pRB family members, along with a neomycin resistance plasmid. The number of flat cells per ten 100× fields following 2 weeks of G418 selection was determined by manual counting. Data shown are from a single experiment and is representative of data obtained in three independent experiments.
pRB mutants associated with a low-risk of retinoblastoma are defective for E2F-binding but retain the ability to activate transcription and promote differentiation
Certain germ-line RB-1 mutations give rise to retinoblastoma at a much lower frequency than expected for a null RB allele. Carriers of such alleles frequently develop benign retinal proliferations (retinomas) and the rare retinoblastomas that do develop are typically unifocal. Thus, these partially penetrant alleles preserve, to a significant degree, the ability to suppress tumor formation. We noted that one such allele, affecting pRB residue 661 (pRB;661W), mapped near pRBΔ663. Furthermore, pRB;661W suppresses colony formation in vitro when ectopically produced in RB −/− cells and yet, like pRBΔ663, does not bind to E2F (Kratzke et al. 1994) and is unable to repress transcription in assays similar to that depicted in Figure 2C (Sellers et al. 1995). This raised the possibility that growth suppression by partially penetrant mutants such as pRB661W might be linked to their ability to promote differentiation.
To address this, the protein products of two partially penetrant alleles, pRB;661W and pRBΔex4 (deletion of exon 4), were characterized and compared with the protein product of a presumed null allele isolated from the germ line of a child with bilateral retinoblastoma (pRB;567L; Yandell et al. 1989; Templeton et al. 1991; Onadim et al. 1992; Dryja et al. 1993; Kratzke et al. 1994; Lohmann et al. 1994). Nuclear extracts were prepared from SAOS2 cells transiently transfected with plasmids encoding HA-pRB(wt), or the aforementioned mutant derivatives, and tested in mobility-shift assays by use of an E2F-binding site. All of these mutants were produced at comparable levels in these cells as determined by anti-HA Western blot analysis (Fig. 6A). Extracts from backbone vector transfected cells contained free E2F complexes (solid arrows) but no E2F–pRB complex (open arrow; Fig. 6B). Wild-type pRB, as expected, led to a loss of free E2F and the formation of a pRB/E2F/DNA complex. In contrast, extracts from the pRBΔex22, pRB;661W, pRBΔex4, and pRB;567L transfectants did not contain this new complex (Fig. 6B). Note that the complete disappearance of free E2F in this transient transfection assay likely reflects the ability of pRB to bind to E2F derived from untransfected cells after cell lysis. If so, this would not undermine the conclusion that pRB;661W and pRBΔex4 are incapable of forming stable pRB/E2F/DNA complexes.
Figure 6.
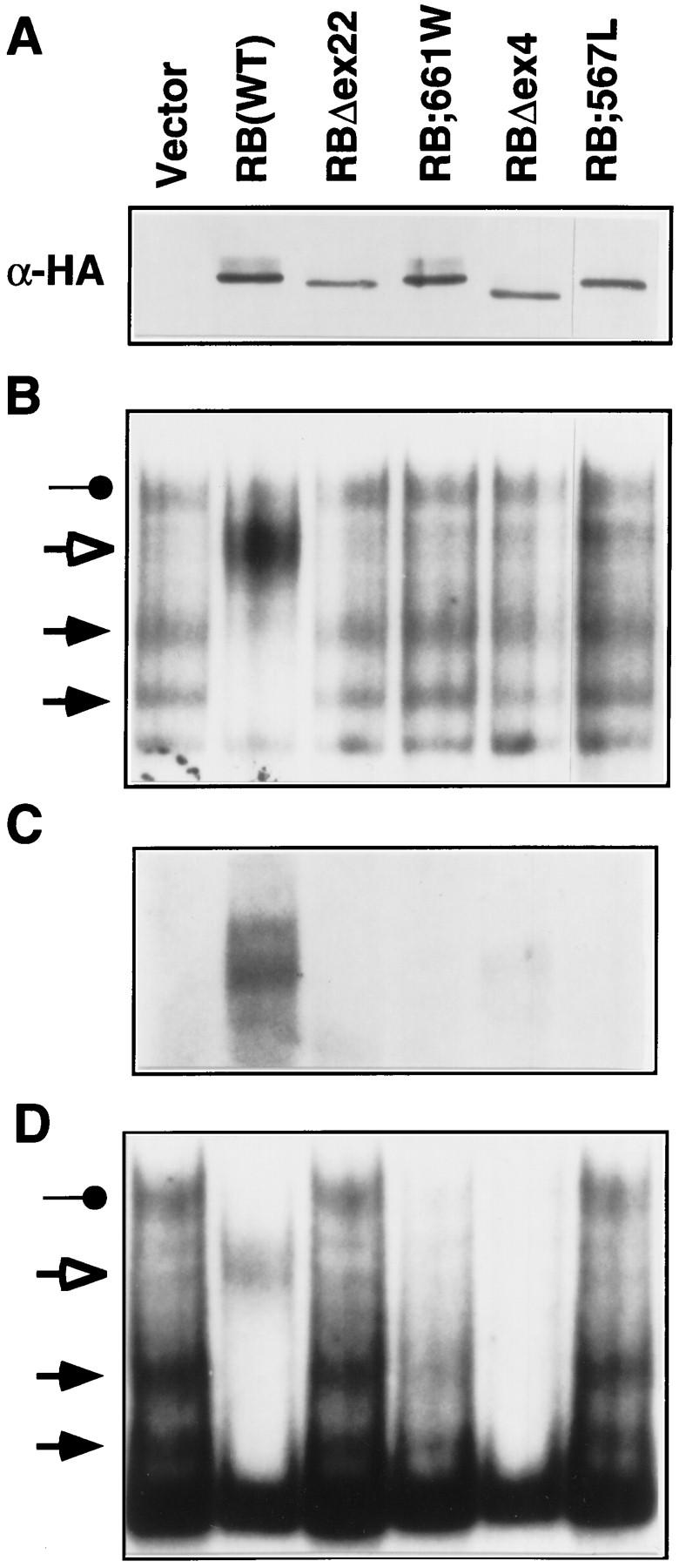
Proteins encoded by partially penetrant RB-1 alleles are defective for E2F binding. (A,C) SAOS2 cells transiently transfected with plasmids encoding the indicated HA-tagged pRB proteins were lysed and immunoprecipitated with an anti-HA antibody. The immunoprecipitates were immunoblotted with an anti-HA antibody (A) or were assayed for coimmunoprecipitated E2F by gel-shift analysis of deoxycholate released proteins by use of a 32P-radiolabeled E2F DNA-binding site (C). (B) E2F DNA-binding activity in transiently transfected cells. Nuclear extracts were prepared from SAOS2 cells transiently transfected with plasmids encoding the indicated HA-tagged pRB proteins and analyzed in gel-shift assays using an 32P-radiolabeled E2F DNA-binding site. (Solid arrows) Complexes containing free E2F; (open arrow and solid circle) pRB/E2F and p107/E2F complexes, respectively. (D) E2F complexes in stably transfected cells. SAOS2 cells were transfected with plasmids encoding the indicated HA-tagged pRB proteins along with a neomycin resistance plasmid. Following 2 weeks of selection in media containing G418, nuclear extracts were prepared and analyzed as in B.
In parallel, SAOS2 cells transiently transfected with plasmids encoding wild-type pRB, or the aforementioned mutant derivatives, were lysed and immunoprecipitated with an anti-HA antibody. Bound E2F was released by treatment with deoxycholate and scored in gel-shift assays. As expected, E2F bound to wild-type pRB but not to the null pRB mutants pRBΔex22 and pRB;567L. E2F binding to pRB;661W was likewise undetectable and binding to pRBΔex4 was grossly impaired (Fig. 6C). Finally, pRB;661W, like pRBΔex22, did not inhibit the ability of a GAL4–E2F1 chimera to activate transcription in vivo (data not shown). Thus, differences in E2F binding did not appear to account for the markedly different risks of developing retinoblastoma associated with germ line transmission of 567L compared with 661W or Δex4.
In contrast, the two partially penetrant mutants, pRB;661W and pRBΔex4, but not the two null mutants pRB;567L and pRBΔex22, induced flat cells and cooperated with MyoD and GRα with seemingly wild-type efficiency (Fig. 7A,B, and C, respectively). Nuclear extracts prepared from pRB;661W and pRBΔex4-induced flat cells did not contain an E2F–pRB complex (Fig. 6D), in keeping with the results obtained following transient transfection. Finally, pRB;661W and pRBΔex4, unlike pRBΔex22, cooperated with MyoD to induce muscle differentiation in vitro (Fig. 8; data not shown). Thus, pRB;661W and pRBΔex4 can induce both osteoblastic and myogenic differentiation in vitro.
Figure 7.
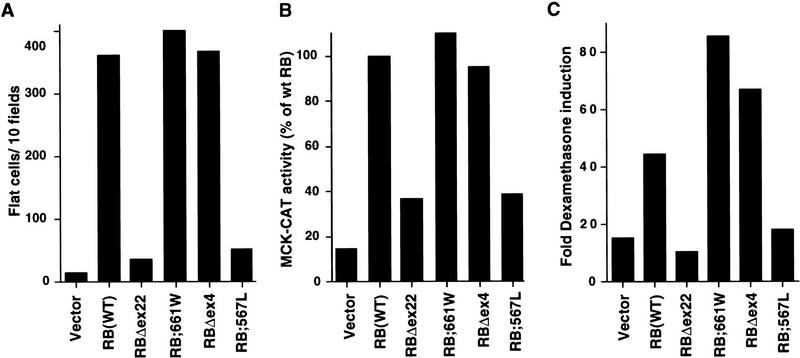
Proteins encoded by partially penetrant RB-1 alleles preserve the ability to induce flat cells and to cooperate with MyoD. (A) Flat cell induction. SAOS2 cells were transfected with plasmids encoding the indicated HA-tagged pRB proteins along with a neomycin resistance plasmid. The number of flat cells per ten 100× fields was determined after G418 selection for 2 weeks. (B) Transactivation of the muscle creatine kinase promoter. RB −/− MEFs were cotransfected with plasmids encoding the indicated pRB proteins along with a plasmid encoding MyoD and a plasmid containing the MCK promoter upstream of CAT. CAT activity relative to that obtained in the presence of wild-type RB is shown. Data are representative of two independent experiments. (C) Transactivation of a glucocorticoid responsive promoter. SAOS2 cells were transiently transfected in duplicate with plasmids encoding the indicated HA-tagged pRB proteins along with a plasmid encoding GRα, an MTV-promoter luciferase reporter plasmid and a plasmid encoding β-galactosidase. Dexamethasone was added 16 hr after transfection to one of each duplicate. Fold dexamethasone induced activity after correction for β-galactosidase activity is shown. Data shown are representative of two independent experiments.
Figure 8.
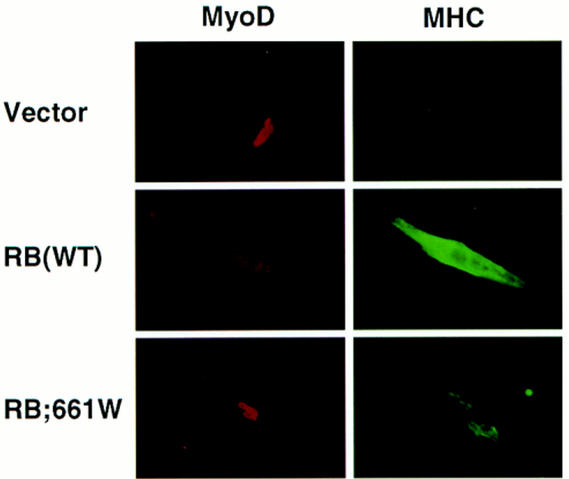
Myogenic conversion of embryonic fibroblasts. RB −/− MEFs were transiently transfected with plasmids encoding MyoD and the indicated pRB proteins. Differentiation media were added to the cells 24–48 hr after transfection. Two to four days later cells were immunostained for MyoD and myosin heavy chain (MHC) as indicated.
Discussion
E2F is thought to be a physiologically relevant target of pRB action. We found that the ability to bind to E2F in solution, at least as measured in IP-DOC release experiments, was not sufficient for pRB to induce an acute G1/S block. The latter required an intact transcriptional repression domain and the ability to form stable complexes with E2F bound to DNA. These observations argue against the notion that pRB simply sequesters E2F and are consistent with emerging data which suggest that pRB/E2F complexes actively repress the transcription of E2F-responsive promoters in G0/1 (Weintraub et al. 1992; Sellers and Kaelin 1996).
pRB-induced SAOS flat cells displayed two hallmarks of osteoblast differentiation relative to the parental cells, namely, increased expression of bone alkaline phosphatase and increased bone mineral deposition. Data presented here suggest that induction of osteoblast differentiation by pRB is not linked to its ability to repress E2F-dependent promoters and induce a G1/S block. An E2F1–RB chimera and p130, although capable of repressing E2F-dependent transcription and arresting cells in G1, were either unable to induce flat cells (E2F1–RB) or did so poorly (p130). Furthermore, pRB mutants such as pRBΔ651 were able to bind to E2F and yet were unable to induce flat cells. Conversely, pRB mutants such as pRBΔ663 were unable to bind stably to E2F, unable to repress transcription when bound to DNA, and unable to induce an acute G1/S block, and yet were able to induce flat cells.
In contrast, the ability of the various pRB mutants to induce flat cells was accurately predicted by their ability to activate transcription of the MCK promoter in cooperation with MyoD. This suggests that ability of pRB to induce SAOS-2 flat cells might reflect the ability of pRB to cooperate with one or more endogenous transcription factors involved in bone differentiation. Furthermore, pRB mutants that promoted osteoblastic differentiation were likewise capable of inducing myogenic conversion of fibroblasts in vitro.
Taken together, these data suggest that pRB performs two functions, namely, regulation of cell cycle progression through its ability to repress E2F-dependent promoters and promotion of differentiation through its ability to activate transcription in concert with certain non-E2F transcription factors (Fig. 9). pRB can cooperate with a variety of transcription factors including, ATF-2, MyoD, GRα, C/EBP, and NF-IL6 (for review, see Chen et al. 1996a; Sellers and Kaelin 1996). The biochemical basis of this cooperation, which may involve physical association with pRB, is not well understood.
Figure 9.
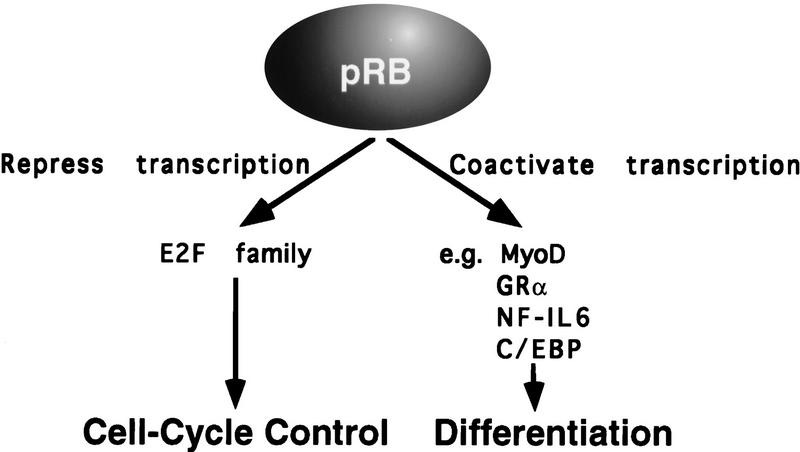
Model for pRB function.
The link between tumor suppression and various pRB biochemical functions has been inferred from studies of naturally occurring pRB mutants. Typical of such mutants, pRBΔex22, encoded by a tumor-derived, somatically altered, RB allele, and RB;567L encoded by a germ- line allele from a patient with bilateral retinoblastoma, lack any measurable ability to interact with E2F, to arrest cells in G1, to activate transcription, or promote differentiation. In contrast, two partially penetrant mutants, pRBΔex4 and pRB;661W, were grossly defective for E2F binding but were capable of activating transcription and promoting differentiation. Thus, in the simplest view, tumor suppression by pRB is linked both to its ability regulate cell cycle progression and to its ability to promote differentiation. According to this model, the marked reduction in the risk of retinoblastoma associated with pRBΔex4 and pRB;661W compared with null pRB mutants reflects the fact that they retain one of these pRB functions, namely, the ability to activate transcription and promote differentiation.
pRB;661W suppresses colony formation when reintroduced into RB −/− cells (Kratzke et al. 1994). Therefore, growth suppression by pRB;661W is measurable both in vitro and in vivo. Furthermore, SAOS2 cells induced to differentiate by pRB mutants such as pRB;661W and pRBΔex4 arrest with either 2N or 4N DNA content (data not shown). Similarly, murine cells induced to undergo myogenic differentation by pRB;661W ceased proliferating as determined by absent bromodeoxyuridine incorporation (data not shown). SAOS2 flat cells lack free E2F as measured in gel-shift assays (Fig. 6D). This loss of E2F DNA-binding activity need not reflect a biochemical interaction between E2F and pRB. For example, during adipocyte differentiation, phosphatase PP2A is down-regulated leading to phosphorylation of the E2F heterodimeric partner DP-1 and consequent loss of E2F DNA-binding activity (Altiok et al. 1997). Thus, one possibility, given the available biochemical data, is that loss of E2F DNA-binding activity, and subsequent growth arrest, is an indirect consequence of differentiation promotion by pRB;661W and pRBΔex4. If true, this would suggest that pRB has both direct and indirect effects on E2F.
SAOS2 osteogenic sarcoma cells contain wild-type p107 and p130 (H. Stubdal and J. DeCaprio, unpubl.). Thus, the endogenous levels of p107 and p130 in these cells do not support the formation of flat cells. Similarly, these two proteins were significantly impaired relative to pRB in their ability to activate transcription in concert with MyoD in RB −/− MEFs (Novitch et al. 1996). Thus, in at least certain cell lineages, the differentiation promotion activities of the pRB family members measurably differ. A similar conclusion has been reached by Harlow and coworkers by use of an adipocyte differentiation model (M. Classon and E. Harlow, pers. comm.).
p107 and p130 can, like pRB, repress E2F-dependent promoters and induce a cell cycle block. Unlike pRB, however, p107 and p130 have not been identified as recurrent targets of mutations in human tumors (Weinberg 1995). Furthermore, mice lacking either p107 or p130 are viable and do not develop tumors (Cobrinik et al. 1996; Lee et al. 1996). In contrast, RB knockout embryos are not viable, and RB heterozygous mice develop pituitary tumors (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992). RB −/− embryos do not die until gestational day 13 and neuronal and melanotrophic cells in RB +/− and RB −/− embryos exhibit increased DNA synthesis, selective loss of specific neuronal markers, failure of timely innervation, altered morphology, and increased apoptosis (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992, 1994; Nikitin and Lee 1996). Partial restoration of pRB function in such embryos allows one to observe a defect in muscle differentiation that becomes manifest later during development (Zacksenhaus et al. 1996). Thus, the selection pressure to mutate RB in human tumors, and the requirement for pRB during development, may reflect its ability to regulate both the cell cycle and, in certain tissues, differentiation. On the basis of the mouse data, and the tight association between RB germ-line mutations and the development of retinoblastoma and sarcomas, one might infer that p107 and p130 cannot fully compensate for the effect of pRB loss on differentiation in neuroectodermal and mesenchymal tissues.
Our data do not preclude, however, the possibility that p107 and p130 can, like pRB, regulate the cell cycle and differentiation in certain tissues. In this regard, p107 −/−, p130 −/− succumb to a specific defect in chondrocyte maturation suggesting that the proper differentiation of these cells depends on these two proteins (Cobrinik et al. 1996). It is likely that broad functional overlap exists amongst pRB, p107, and p130 as most developing tissues in both RB −/− and p107 −/−, p130 −/− embryos appear to be normal (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992; Cobrinik et al. 1996).
This dual function model for pRB-mediated tumor suppression may account for the fact that, to date, mutations have been identified in RB-1, and its upstream regulators, but not in downstream components such as E2F. Furthermore, this dual function model may account for the underepresentation of missense mutations among naturally occurring RB-1 mutations. Pathological examination of human tumors reveals that proliferation and differentiation are both commonly deregulated and are typically inversely correlated. The frequent loss of both pRB functions in human tumors, either as a direct result of mutation, or as an indirect consequence of alterations in upstream pRB regulators (p16, cdk4, Cyclin D1), may, at least in part, underlie these observations.
Materials and methods
Cell culture and transfection
SAOS2 cells were grown in Dulbecco’s modified Eagle media (DMEM), supplemented with 10% fetal bovine serum (FBS), and transfected by the calcium-phosphate method (Chen and Okayama 1987).
RB −/− MEFs isolated from day 13 embryos were grown in DME supplemented with 10% FBS and were used between passages 3 and 10. MEFs were plated at 1.5 × 105 to 1.75 × 105 cells per 60 mm plate the day before transfection with a total of 2 μg of DNA by use of lipofectamine (GIBCO BRL) (Skapek et al. 1995). Myogenic conversion of embryonic fibroblasts was induced 24–48 hr after transfection by changing the media to DMEM containing 2% horse serum plus 10 μg/ml of insulin (differentiation media; DM) for 2–4 days.
Plasmids
pSG5L is a derivative of pSG5 (Stratagene) with a modified polylinker (R. Scully and D.M. Livingston, unpubl.). To introduce an HA tag, the HindIII–EcoRI insert from pSP72–HA-E2F1 (Krek et al. 1993) was ligated into pSG5L restricted with these two enzymes to make pSG5L–HA–E2F1. pSG5L–HA–E2F1 was restricted with BamHI and EcoRI (removing the E2F1 cDNA, but leaving the HA-tag) and ligated to the BamHI–EcoRI RB cDNA insert from pSG5–TETr–RB(379–928) (Sellers et al. 1995) to make pSG5L–HA–RB(379–928). pSG5L–HA–RB(WT) was made by replacing the BamHI–NheI RB cDNA fragment in pSG5L–HA–RB(379–928) with the corresponding fragment in pSG5–RB (Shirodkar et al. 1992). A three-way ligation of the vector generated by BamHI–EcoRI digestion of pSG5L–HA–RB(379–928), the BamHI–MluI insert from pSG5L–HA–RB and the MluI–EcoRI insert from pSG5–TETr–RB(379–928;Δ22) (Sellers et al. 1995) was performed to make pSG5L–HA–RBΔ22.
pSG5L–HA–RB;567L, pSG5L–HA–RB;661W and the pRB NAAIRS substitution mutants were all generated by site-directed mutagenesis by use of single-stranded anti-sense DNA derived from pSG5L–HA–RB(wt) and the indicated sense oligonucleotides. The NAAIRS mutant RB cDNAs were then excised by restriction with Bst1107I and NheI and ligated into similarly restricted pSG5–TETr–RB(379–928) to make the corresponding TETr–RB(379–928) mutants. All mutations were confirmed by sequencing (Table 1).
Table 1.
Oligonucleotides used in site-directed mutagenesis
| Mutation
|
Oligonucleotide
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RBΔ651 (NAAIRS) | T | C | T | C | T | T | T | C | A | C | T | G | T | T | T | a | a | t | g | c | t | g | c | t | a | t | a | c | g | a | t | c | g | C | T | A | G | C | C | T | A | T | C | T | C | C | G | G | |
| RBΔ657 (NAAIRS) | A | A | A | A | A | A | G | T | G | T | A | T | C | G | G | a | a | t | g | c | t | g | c | t | a | t | a | c | g | a | t | c | g | A | A | T | A | C | A | C | T | T | T | G | T | G | A | A | |
| RBΔ663 (NAAIRS) | G | C | C | T | A | T | C | T | C | C | G | G | C | T | A | a | a | t | g | c | t | g | c | t | a | t | a | c | g | a | t | c | g | C | T | T | C | T | G | T | C | T | G | A | G | C | A | C | |
| RBΔ685 (NAAIRS) | A | T | C | T | G | G | A | C | C | C | T | T | T | T | C | a | a | t | g | c | t | g | c | t | a | t | a | c | g | a | t | c | g | G | A | G | T | A | T | G | A | A | C | T | C | A | T | G | |
| RB;567L | G | C | A | T | G | G | C | T | C | T | C | A | G | A | T | c | t | t | C | C | T | T | T | A | T | T | T | G | A | T | C | T | T | ||||||||||||||||
| RB;661W | G | T | G | T | A | T | C | G | G | C | T | a | G | C | C | T | A | T | C | T | C | c | G | G | C | T | A | A | A | T | A | C | A | ||||||||||||||||
The HpaI–MluI RB cDNA insert from pCI–neo–RBΔ4 (G.A. Otterson and F.J. Kaye, unpubl.) was ligated into similarly restricted pSG5L–HA–RB to make pSG5L–HA–RBΔex4.
The BamHI inserts from pcDNA–HAE2F1(1–368)RB(379–792), pcDNA–HAE2F1(1–368)RB(379–928;Δ22) and pcDNA–HAE2F1(1–368;132)RB(379–792) (Sellers et al. 1995) were ligated into the BamHI site of pCMV-neo–Bam (Baker et al. 1990) to make pCMV–E2F1–RBSP, pCMV–E2F1–RBLPΔ22 and pCMV–E2F1(132)–RBSP, respectively.
pCD19 (Tedder and Isaacs 1989), pCMVneo (Baker et al. 1990), pCMV–RB (Qin et al. 1992), pCMV–RBΔ22 (Qin et al. 1992), pGL2ANΔTETo (Sellers et al. 1995), pCMV–βGal, pCSA–MyoD (Skapek et al. 1995), p-3300–MCK–CAT (Jaynes et al. 1988), pCMVneoBam–HAp107 (Zhu et al. 1993), pRS–hGRα (Giguere et al. 1986), pCMVneoBam–HAp107DE (Zhu et al. 1995), and pCDNA1–HAp130 (Vairo 1995) have been described previously. pMTV–GRE–Luc was the gift of William Chin (Brigham and Women’s Hospital, Harvard Medical School, Boston, MA) and pCDNA1–HAp130Δ21 was the gift of Juan Zalvide and James DeCaprio (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA).
Flat cell assays
SAOS2 cells grown on p100 plates were transfected with 4 μg of pCMV–Neo and 20 μg of the indicated pSGL–HA–RB plasmids. Stable transfectants were selected and grown in DMEM containing 10% FCS and 500 μg/ml of G418 for 2 weeks. The number of flat cells per ten 100× microscopic fields was determined manually.
von Kossa staining
SAOS2 flat cells generated as above were grown in DMEM containing 10% FCS, 500 μg/ml of G418, 50 μg/ml of l-ascorbic acid, and 10 mm β-glycerol phosphate for 2 weeks. The cells were then fixed for 10 min in freshly prepared 4% paraformaldehyde in TBS, washed twice with TBS, once with ddH2O, and incubated in freshly prepared 3% AgNO3 for 30 min in the dark. The cells were then exposed to UV light on a UV transilluminator (Fotodyne) for 1 min to allow for color development (Asahina et al. 1996)
Immunoprecipitation and immunoblot analysis
Whole cell extracts were prepared as in Krek et al. (1993). Immunoprecipitation and immunoblotting were performed essentially as described previously (Harlow and Lane 1988). Immunoprecipation reactions contained 500 μl of cell extract (derived from the adherent cells in a nearly confluent p100 plate) and ∼1.5 μg of 12CA5 anti-HA antibody (Boehringer Mannheim) or 1 μl of N9 anti-RB sera (Sellers et al. 1995). Immunoprecipitates recovered on protein A–Sepharose were either eluted by boiling in SDS-containing protein sample buffer or with deoxycholate (see below).
For immunoblotting, filters were blocked and probed in TBS and 4% powdered milk for 1 hr. mAb12CA5 was used at a concentration of 1 μg/ml. N9 serum was used at a dilution of 1:1500. Bound protein was detected colorimetrically by use of an alkaline phosphatase conjugated goat anti-mouse antibody.
Immunofluorescence staining
Anti-MyoD and anti-MHC immunofluorescence staining was as described previously (Novitch et al. 1996).
FACS/Cell cycle analysis
SAOS2 cells grown on p100 plates were transfected with 4 μg of pCD19 and 20 μg of the indicated pSGL–HA–RB plasmid. Cells were harvested 72 hr later by treatment with trypsin–EDTA, and stained with anti-CD19 antibody (Tedder and Isaacs 1989), FITC-conjugated goat anti-mouse antibody and propidium iodide as described previously (Qin et al. 1995). Samples were analyzed by two-color FACS with a Beckton-Dickinson FACscan.
Gel shift assays
Nuclear protein extracts were prepared as described previously (Hurst et al. 1990). Two microliters of nuclear extract (10 μg of total protein) was added to 20 μl of Gel-shift buffer [20 mm HEPES (pH 7.5), 50 mm KCl, 10 mm MgCl2, 10% glycerol, 0.5 mm DTT, 0.1% NP-40, 0.1 mm EDTA at pH 8.0, 2 mg/ml of BSA, 200 μg/ml of sonicated salmon sperm DNA] containing 1 ng of 32P-end-labeled double-stranded E2F oligonucleotide (5′ E2F site from the E2F1 promoter or the E2F site from the adenoviral E2 promoter). The reactions were incubated at 20°C for 20 min before electrophoresis.
For the DOC release experiments, immune complexes bound to protein A–Sepharose were washed twice with buffer A(1) [20 mm HEPES (pH 7.5), 50 mm KCl, 1 mm MgCl2, 10% glycerol, 0.1% NP-40, 0.5 mm DTT). Bound E2F was released by incubating the Sepharose in 15 μl of buffer A(1) with 0.8% DOC for 10 min at 4°C. The resulting supernatant (10 μl) was then removed and incubated with 15 μl of buffer A(1) supplemented with 0.6% NP-40, 2 mg/ml of BSA, and 200 ng/ml of sonicated salmon sperm DNA for 15 min at 20°C. The end-labeled E2F oligonucleotide (1 ng) was then added and incubated for 15 min at 20°C. Complexes were resolved on a 0.25× TBE 4% acrylamide gel run at 300 V for 90 min.
Alkaline phosphatase activity
Alkaline phosphatase activity was determined as described previously (Asahina et al. 1996). SAOS2 cells grown in six well plates were washed twice with Tris-buffered Saline at pH 8.0 (TBS) and lysed in 100 μl of TBS+1% Triton X-100 for 20 min at 4°C. Lysate (5 μl) was added to 50 μl of assay buffer [50 mm Na2CO3 (pH 10.0), 10 mm MgCl2, 20 mm p-Nitrophenol Phosphate]. The reactions were stopped 15–30 min later with 50 μl of 0.5 n NaOH and the absorbance measured at 450 nm. A standard curve was generated by use of assay buffer containing from 1 to 10 nmoles of p-nitrophenol. Alkaline phosphatase activity was expressed as nmoles of nitrophenol generated per minute reaction time per microgram of total protein (as determined by Bradford assay).
MyoD and glucocorticoid receptor transactivation assays
For MyoD transactivation experiments, RB −/− MEFs were transfected with 0.5 μg pCSA–MyoD, 0.5 μg of p-3300–MCK–CAT, and 0.5–1.0 μg of the indicated pSG5L–HA–RB plasmids or the empty vector. Following 2 days under muscle differentiation culture conditions, cell were assayed for CAT activity. CAT assays were performed as described previously (Skapek et al. 1995).
For GRα transactivation experiments, SAOS2 cells were transiently transfected in six-well plates in duplicate with 3.0 μg of plasmids encoding the indicated HA-tagged pRB proteins along with 200 ng of pRS-GRα plasmid encoding GRα receptor, 1.0 μg of pMTV–GRE–luciferase, and 1.0 μg of pCMV–β–gal. The medium was changed 16 hr after transfection, and dexamethasone was added to a final concentration 10−6 m to one well of each duplicate set. Luciferase actvity and β-gal activity was determined for each sample as described previously (Qin et al. 1995).
Transcriptional repression assays
Transcriptional repression assays were performed essentially as described previously (Sellers et al. 1995). Briefly, SAOS2 cells grown on p100 plates were transfected with 8 μg of pGL2ANΔTETo, 4 μg of pCMV–βGal, and 10 μg of the various pSG5–TETr plasmids. Thirty-six hours later, luciferase and β-gal activities were determined for each sample as described previously (Qin et al. 1995).
Acknowledgments
We thank Peter Adams, Mark Ewen, Erik Flemington, Barrett Rollins and David Livingston for critical reading of this manuscript, Drs. Juan Zalvide and James DeCaprio for providing p130 plasmids, Drs. William W. Chin for providing the MTV–luciferase plasmid, and John Gribben for the pCD19 plasmid. W.R.S. and W.G.K. thank their colleagues in the Kaelin, DeCaprio, Ewen, and Livingston laboratories for the many hours of thoughtful discussion. This work was supported by the Sandoz Research Insitute, the McDonnell Foundation (W.G.K.), and a Physician Scientist Award from the National Cancer Institute, National Institutes of Health (W.R.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL William_Kaelin@dfci.harvard.edu; FAX (617) 632-4381.
References
- Adams PD, Kaelin WG. Transcriptional control by E2F. Cancer Biol. 1995;6:99–108. doi: 10.1006/scbi.1995.0013. [DOI] [PubMed] [Google Scholar]
- Adnane J, Shao Z, Robbins PD. The retinoblastoma susceptibility gene product represses transcription when directly bound to the promoter. J Biol Chem. 1995;270:8837–8843. doi: 10.1074/jbc.270.15.8837. [DOI] [PubMed] [Google Scholar]
- Altiok S, Xu M, Spiegelman BM. PPARγ induces cell cycle withdrawl: Inhibition of E2F/DP DNA-binding activity via a down-regulation of PP2A. Genes & Dev. 1997;11:1987–1998. doi: 10.1101/gad.11.15.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina I, Sampath TK, Hauschka PV. Human Osteogenic Protein-1 induces chondroblastic, osteoblastic, and/or adipocyte differentiation of clonal murine target cells. Exp Cell Res. 1996;222:38–47. doi: 10.1006/excr.1996.0005. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Markowitz S, Fearon E, Willson B, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- Botz J, Zerfass-Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigergiou A, Jansen-Dürr P. Cell cycle regulation of the murine Cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol. 1996;16:3401–3409. doi: 10.1128/mcb.16.7.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner R, Cohen BL, Sopta M, Hamel PA, Ingles CJ, Gallie BG, Phillips RA. Direct transcriptional repression by pRB and its reversal by specific cyclins. Mol Cell Biol. 1995;15:3256–3265. doi: 10.1128/mcb.15.6.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Faha B, Dembski M, Tsai L-H, Harlow E, Dyson N. Independent binding of the retinoblastoma protein and p107 to the transcription factor E2F. Nature. 1992;355:176–179. doi: 10.1038/355176a0. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P-L, Riley DJ, Chen Y, Lee W-H. Retinoblastoma protein postively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes & Dev. 1996a;10:2794–2804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- ————— Retinoblastoma protein directly interacts with and activates the transcription factor NF-IL6. Proc Natl Acad Sci. 1996b;93:465–469. doi: 10.1073/pnas.93.1.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NMT, van der Valk M, Hooper ML, Berns A, Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Claudio PP, Howard CM, Baldi A, De Luca A, Fu Y, Condorelli G, Sun Y, Colburn N, Calabretta B, Giordano A. p130/pRb2 has growth suppression properties similar to yet distinctive from those of retinoblastoma family members pRb and p107. Cancer Res. 1994;54:5556–5560. [PubMed] [Google Scholar]
- Cobrinik D, Lee M-H, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA, Jacks T. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes & Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- Dryja TP, Rapaport J, McGee TL, Nork TM, Schwartz TL. Molecular etiology of low-penetrance retinoblastoma in two pedigrees. Am J Hum Genet. 1993;52:1122–1128. [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Eaton EN, Picon M, Roberts JM, Lundberg AS, Gifford A, Sardet C, Weinberg RA. Regulation of cyclin E transcription by E2Fs and the retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- Giguere V, Hollenberg S, Rosenfeld M, Evans R. Functional domains of the human glucocorticoid receptor. Cell. 1986;46:645–652. doi: 10.1016/0092-8674(86)90339-9. [DOI] [PubMed] [Google Scholar]
- Ginsberg D, Vairo G, Chittenden T, Xiao Z-X, Xu G, Wydner KL, DeCaprio JA, Lawrence JB, Livingston DM. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes & Dev. 1994;8:2665–2679. doi: 10.1101/gad.8.22.2665. [DOI] [PubMed] [Google Scholar]
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies—A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- Huang H-JS, Yee J-K, Shew J-Y, Chen P-L, Bookstein R, Friedmann T, Lee EY-HP, Lee W-H. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science. 1988;242:1563–1566. doi: 10.1126/science.3201247. [DOI] [PubMed] [Google Scholar]
- Hurst HC, Masson N, Jones NC, Kee KAW. The cellular transcription factor CREB corresponds to activating factor 47 (ATF-47) and forms complexes with a group of polypeptides related to ATF-43. Mol Cell Biol. 1990;10:6192–6203. doi: 10.1128/mcb.10.12.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jaynes JJ, Johnson J, Buskin C, Gartside C, Hauschka S. The muscle creatine kinase gene is regulated by multiple upstream elements, including a muscle specific enhancer. Mol Cell Biol. 1988;8:62–70. doi: 10.1128/mcb.8.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzke RA, Otterson GA, Hogg A, Coxon AB, Geradts J, Cowell JK, Kaye FJ. Partial inactivation of the RB product in a family with incomplete penetrance of familial retinoblastoma and benign retinal tumors. Oncogene. 1994;9:1321–1326. [PubMed] [Google Scholar]
- Krek W, Livingston DM, Shirodkar S. Binding to DNA and retinoblastoma gene product promoted by complex formation of different E2F family members. Science. 1993;262:1557–1560. doi: 10.1126/science.8248803. [DOI] [PubMed] [Google Scholar]
- La Thangue NB. DRTF1/E2F: An expanding family of heterodimeric transcription factors implicated in cell-cycle control. Trends Biochem Sci. 1994;19:108–114. doi: 10.1016/0968-0004(94)90202-x. [DOI] [PubMed] [Google Scholar]
- Lee EY-HP, Chang C-Y, Hu N, Wang Y-CJ, Lai C-C, Herrup K, Lee W-H, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lee EY-HP, Hu N, Yuan S-SF, Cox LA, Bradley A, Lee W-H, Herrup K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes & Dev. 1994;8:2008–2021. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- Lee M-H, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E, Jacks T. Targeted disruption of p107: Functional overlap between p107 and Rb. Genes & Dev. 1996;10:1621–1632. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
- Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94:349–354. doi: 10.1007/BF00201591. [DOI] [PubMed] [Google Scholar]
- Neuman E, Flemington EK, Sellers WR, Kaelin WG. Transcription of the E2F1 gene is rendered cell-cycle dependent by E2F DNA-binding sites within its promoter. Mol Cell Biol. 1994;14:6607–6615. doi: 10.1128/mcb.14.10.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitin AY, Lee W-H. Early loss of the retinoblastoma gene is associated with impaired growth inhibitory innervation during melanotroph carcinogenesis in RB+/- mice. Genes & Dev. 1996;10:1870–1879. doi: 10.1101/gad.10.15.1870. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Mulligan GJ, Jacks T, Lassar AB. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol. 1996;135:441–456. doi: 10.1083/jcb.135.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onadim Z, Hogg A, Baird PN, Cowell JK. Oncogenic point mutations in exon 20 of the RB1 gene in families showing incomplete penetrance and mild expression of the retinoblastoma phenotype. Proc Natl Acad Sci. 1992;89:6177–6181. doi: 10.1073/pnas.89.13.6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X-Q, Chittenden T, Livingston DM, Kaelin WG. Identification of a growth suppression domain within the retinoblastoma gene product. Genes & Dev. 1992;6:953–964. doi: 10.1101/gad.6.6.953. [DOI] [PubMed] [Google Scholar]
- Qin X-Q, Livingston DM, Ewen M, Sellers WR, Arany Z, Kaelin WG., Jr The transcription factor E2F1 is a downstream target of Rb action. Mol Biol Cell. 1995;15:742–755. doi: 10.1128/mcb.15.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JK, Devoto SH, Smith EJ, Chellappan SP, Jakoi L, Nevins JR. Interactions of the p107 and Rb proteins with E2F during the cell proliferation response. EMBO J. 1993;12:1013–1020. doi: 10.1002/j.1460-2075.1993.tb05742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers WR, Kaelin WG., Jr RB as a modulator of transcription. Biochim Biophys Acta. 1996;1288:M1–M5. doi: 10.1016/0304-419x(96)00014-5. [DOI] [PubMed] [Google Scholar]
- Sellers WR, Rodgers JW, Kaelin Jr WG. A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proc Natl Acad Sci. 1995;92:11544–11548. doi: 10.1073/pnas.92.25.11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirodkar S, Ewen M, DeCaprio JA, Morgan J, Livingston DM, Chittenden T. The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell. 1992;68:157–166. doi: 10.1016/0092-8674(92)90214-w. [DOI] [PubMed] [Google Scholar]
- Singh P, Coe J, Hong W. A role for retinoblastoma protein in potentiating protein transcriptional activation by the glucocorticoid receptor. Nature. 1995;374:562–565. doi: 10.1038/374562a0. [DOI] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Spicer DB, Lassar AB. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- Strauss M, Lukas J, Bartek J. Unrestricted cell cycling and cancer. Nature Med. 1995;1:1245–1246. doi: 10.1038/nm1295-1245. [DOI] [PubMed] [Google Scholar]
- Taya Y. RB kinases and RB-binding proteins: New points of view. [Review] [50 refs] Trends Biochem Sci. 1997;22:14–17. doi: 10.1016/s0968-0004(96)10070-0. [DOI] [PubMed] [Google Scholar]
- Tedder TF, Isaacs CM. Isolation of cDNAs encoding the CD19 antigen of human and mouse B lymphocytes. J Immunol. 1989;143:712–717. [PubMed] [Google Scholar]
- Templeton DJ, Park SH, Lanier L, Weinberg R. Nonfunctional mutants of the retinoblastoma protein are characterized by defects in phosphorylation, viral oncoprotein association, and nuclear tethering. Proc Natl Acad Sci. 1991;88:3033–3037. doi: 10.1073/pnas.88.8.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairo G, Livingston DM, Ginsberg D. Functional Interaction between E2F4 and p130: Evidence for distinct mechanisms underlying growth suppression by different Rb family members. Genes & Dev. 1995;9:869–881. doi: 10.1101/gad.9.7.869. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Weintraub SJ, Prater CA, Dean DC. Retinoblastoma protein switches the E2F site from positive to negative element. Nature. 1992;358:259–261. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- Weintraub S, Chow K, Luo R, Zhang S, He S, Dean D. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Haft DH, Getzoff ED, Tainer JA, Lerner RA, Brenner S. Identical short peptide sequences in unrelated proteins can have different confirmations: A testing ground for theories of immune recognition. Proc Natl Acad Sci. 1985;82:5255–5259. doi: 10.1073/pnas.82.16.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- Yandell DW, Campbell TA, Dayton SH, Petersen R, Walton D, Little JB, McConkie-Rosell A, Buckley EG, Dryja TP. Oncogenic point mutations in the human retinoblastoma gene: Their application to genetic counseling. N Engl J Med. 1989;321:1689–1695. doi: 10.1056/NEJM198912213212501. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes & Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- Zhu L, van der Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes & Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]
- Zhu L, Enders G, Lees JA, Beijersbergen RL, Bernards R, Harlow E. The pRB-related protein p107 contains two growth suppression domains: Independent interactions with E2F and cyclin/cdk complexes. EMBO J. 1995;14:1904–1913. doi: 10.1002/j.1460-2075.1995.tb07182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]