

Abstract
Mutations in the SMAD4/DPC4 tumor suppressor gene, a key signal transducer in most TGFβ-related pathways, are involved in 50% of pancreatic cancers. Homozygous Smad4 mutant mice die before day 7.5 of embryogenesis. Mutant embryos have reduced size, fail to gastrulate or express a mesodermal marker, and show abnormal visceral endoderm development. Growth retardation of the Smad4-deficient embryos results from reduced cell proliferation rather than increased apoptosis. Aggregation of mutant Smad4 ES cells with wild-type tetraploid morulae rescues the gastrulation defect. These results indicate that Smad4 is initially required for the differentiation of the visceral endoderm and that the gastrulation defect in the epiblast is secondary and non-cell autonomous. Rescued embryos show severe anterior truncations, indicating a second important role for Smad4 in anterior patterning during embryogenesis.
Keywords: Smad4/Dpc4 mutant mice, gastrulation, visceral endoderm, anterior development
Tumor suppressor genes are negative growth regulators that, when inactivated, release cells from proliferative constraints. DPC4 (for Deleted in Pancreatic Cancer, locus 4) has been isolated by virtue of its frequent homozygous deletion in pancreatic cancer (Hahn et al. 1996). Loss of heterozygosity (LOH) at the DPC4 locus occurred in >50% of the pancreatic carcinomas and has been found to a lesser extent in carcinomas of the colon, breast, ovary, lung, and head and neck (Nagatake et al. 1996; Schutte et al. 1996). DPC4 is a member of the vertebrate SMAD gene family and was assigned the nomenclature SMAD4 (Derynck et al. 1996). SMADs were first identified by their homology to the Drosophila Mad (Mothers Against Decapentaplegic) gene (Raftery et al. 1995; Sekelsky et al. 1995). Genetic epistasis analysis implicated Mad downstream of dpp, which encodes the fruit fly homolog of the TGFβ family member BMP2/4 (Hoodless et al. 1996; Wiersdorff et al. 1996). Three Mad homologs, sma-2, sma-3, and sma-4, were identified in Caenorhabditis elegans and their inactivation recapitulated the phenotype caused by mutation in the TGFβ family receptor daf4 (Savage et al. 1996). Subsequently, several vertebrate Mad homologs, referred to as SMADs (Derynck et al. 1996), have been isolated and shown to be transducers of TGFβ-related signals (Massagué et al. 1997).
TGFβ family of secreted factors includes TGFβs, activins, bone morphogenetic proteins (BMPs), and nodal. These factors elicit a broad spectrum of cellular responses, from cell proliferation and differentiation to specification of developmental processes (Kingsley 1994; Wall and Hogan 1994). TGFβ family members signal through heteromeric complexes of type I and type II transmembrane Ser/Thr kinase receptors (Attisano and Wrana 1996). In response to signals, specific SMAD proteins associate with, and are phosphorylated by, distinct activated type I receptors. For instance, SMAD2 and SMAD3 physically associate with TGFβ receptor type I (TβR-I), whereas SMAD1, and possibly SMAD5, interact with BMPR-I (Macias-Silva et al. 1996; Zhang et al. 1996; Kretzschmar et al. 1997). In contrast to the pathway-restricted SMADs, SMAD4 rapidly associates with both SMAD1 in response to BMPR-I signaling and SMAD2 in response to TβR-I and ActR-IB signaling (Lagna et al. 1996). Overexpression of SMAD1 induces a ventral mesoderm phenotype in Xenopus oocytes similar to the effect of BMP2/4, whereas overexpression of SMAD2 induces dorsal mesoderm typically observed in Vg/activin/nodal treated embryos (Baker and Harland 1996; Graff et al. 1996; Thomsen 1996). Overexpression of SMAD4 in Xenopus embryos activates both ventral and dorsal mesoderm, confirming its role as a common mediator of both BMP2/4 and Vg1/activin/nodal signaling pathways (Lagna et al. 1996).
SMAD proteins share a high degree of homology in their amino-terminal MH1 (Mad homology) and carboxy-terminal MH2 domains (Wrana and Attisano 1996). The MH2 domain is considered the effector domain, whose activity is opposed by its physical interaction with the MH1 domain (Baker and Harland 1996; Liu et al. 1996; Hata et al. 1997). Unlike SMAD4, SMAD1 and SMAD2 contain consensus phosphorylation sites for receptor type I Ser/Thr kinases within their MH2 domains (Macias-Silva et al. 1996; Kretzschmar et al. 1997). The model emerging from recent biochemical and crystallographic studies implies that phosphorylation of the receptor-regulated SMADs relieves them of the MH1 inhibitory effect, allowing their interaction with SMAD4 and subsequent translocation to the nucleus (Hata et al. 1997; Massagué et al. 1997; Shi et al. 1997). In activin-treated cells of the frog embryo, the heteromeric complex binds to Fast1, a member of the winged-helix transcription factor, and regulates transcription of an early-immediate gene (Chen et al. 1996, 1997).
Gene inactivation of several TGFβ family members or their receptors has shown the role of these factors in mouse embryogenesis. Homozygous deletion of either Bmp4 or its receptor Bmpr-I causes defective mesoderm formation, possibly the result of reduced cell proliferation of the ectoderm from which the mesoderm originates (Mishina et al. 1995; Winnier et al. 1995). In nodal-deficient mice, mesoderm induction occurs but no organized streak is formed (Conlon et al. 1994). Furthermore, during later stages of embryonic development, Bmp4 was shown to be required for proper posterior development (Winnier et al. 1995), whereas nodal was required for normal anterior patterning (Varlet et al. 1997). Homozygous deletion of one of the activin receptors (ActRIIB) also results in abnormal anterior/posterior patterning, as well as lateral asymmetry (Oh and Li 1997).
If Smad4 is a common component of multiple TGFβ family signaling pathways, one would predict that disruption of its function would have severe consequences for early embryonic patterning. To investigate this, we disrupted the Smad4 gene by homologous recombination in embryonic stem (ES) cells. We show that Smad4-deficient mouse embryos failed to gastrulate and died in utero by embryonic day 7.5 (E7.5). Although the inner cell masses (ICMs) of E3.5 mutant blastocysts proliferated similarly to wild-type ICMs in vitro, the ectoderm of E6.5 mutant embryos showed reduced proliferation in vivo, possibly explaining the gastrulation defect. The differentiation of the visceral endoderm in mutant embryos was impaired in vivo and in vitro. The functional requirement for the visceral endoderm was confirmed by complementation studies using tetraploid aggregation experiments, in which wild-type visceral endoderm rescued the gastrulation defect of Smad4 mutant embryos. These results indicate that the gastrulation defect in Smad4-deficient embryos was non-cell autonomous and suggest an essential role for the visceral endoderm in mesoderm formation. The rescued embryos exhibited additional phenotypes, typically affecting anterior development of the embryo, revealing a later requirement for Smad4 within the epiblast-derived lineage for normal anterior specification.
Results
The Smad4tm1Ari allele is a null mutation
The inactivation of the SMAD4 gene in human as well as the null alleles in the Drosophila Mad or in the C. elegans Sma2 and Sma3 are most frequently caused by mutations within the MH2 domain (Sekelsky et al. 1995; Savage et al. 1996; Wrana and Attisano 1996). This domain is encoded by exons 8–11 in human and mouse and was the target of the vector designed to disrupt the Smad4 locus in ES cells. Homologous recombination of the targeting vector resulted in the replacement of a 5-kb genomic fragment containing exon 8 and part of exon 9 with the neomycin resistance (neo) gene (Fig. 1A). Homologous recombination of the targeting vector at the Smad4 locus was confirmed by Southern blot analysis on three ES cell clones (Fig. 1B), two of which gave rise to germ-line transmission. Two Smad4 heterozygous ES clones (C8 and F9) were also subjected to higher G418 concentration to generate Smad4 homozygous mutant ES cell clones (Mortensen et al. 1992). Southern blot analysis performed on the DNA of these clones confirmed the loss of the wild-type locus, and three were chosen (F9-2, F9-5, and C8-24) for further analysis (Fig. 1B). A control clone (C8-3) subjected to the same selection conditions but remaining heterozygous for the Smad4 locus was also used for subsequent analysis (Fig. 1B).
Figure 1.

Targeted disruption of the Smad4 locus results in a null mutation. (A) Partial genomic organization of the wild-type Smad4 locus in mouse (top). The bar represents the 0.5-kb flanking probe used in Southern blot analysis generating a 12-kb EcoRI fragment for the wild-type allele. Arrows depict the location of the primers used in PCR analysis to identify the wild-type allele. The targeting vector (middle) was generated by cloning the 4.5-kb SpeI genomic fragment as the long-arm and the 385-bp BglII genomic fragment as the short-arm. The targeted locus after homologous recombination (bottom) would generate an 8.5-kb EcoRI fragment by use of the 0.5-kb flanking probe. Arrows depict the primers used for PCR screening of the homologous recombination in ES cells and for subsequent genotyping. An asterisk (*) indicates that the restriction site has been lost during the cloning process. (RI) EcoRI, (S) SpeI, (B) BglII. (B) Southern blot analysis of ES cell clones generated by homologous recombination at the Smad4 locus. Genomic DNA was isolated from wild type (E14K), heterozygous (C8, F9, and C8-3), and homozygous (C8-24, F9-2, and F9-5) mutant cell lines and digested with EcoRI. (C) Northern blot analysis of the ES cell clones. The blotted membrane containing total RNA was probed with the same 0.5-kb genomic fragment described above (Smad4) and stripped and reprobed with a neomycin specific probe (Neo). A strip and reprobe of this same membrane with the ribosomal L32 probe showed equal loading of RNA in all lanes (data not shown). (D) Western blot analysis of the ES cell clones. Total cell lysates were immunoblotted with a polyclonal α-Smad4 antibody. A band of an apparent molecular weight of 62 kd was detected in the wild-type (E14K) and the heterozygous mutants, but not in the Smad4 homozygous mutants.
To determine whether the homozygous deletion of the Smad4tm1Ari allele (named according to the mouse gene nomenclature committee) resulted in a null mutation, Northern blot analysis was performed on the various ES clones. A transcript of ∼3.8 kb was detected in E14K wild-type cells and with decreasing intensity in the heterozygous and homozygous mutant ES clones (Fig. 1C). RT–PCR analysis, however, indicated that the targeted exons 8 and 9 were absent from the transcript in the homozygous mutant clones (data not shown). Probing the same filter with neo specific sequences revealed a strong neo transcript of 1.3 kb in the homozygous mutant (Fig. 1C). A faint 3.8-kb transcript containing neo sequences was also present with the strongest signal detected in the homozygous mutants but absent in the wild-type ES cells. These results indicate that neo sequences fused with some of the Smad4 mutant transcript. Regardless, any alternative splicing involving exons 2–7 with neo sequences, or with exon 10 would result in frameshifts with multiple termination codons.
Because a Smad4 transcript was detected in the homozygous mutants, Western blot analysis was performed on the ES clones. A strong band of the expected size for Smad4 protein was detected in wild-type cells (Fig. 1D). The intensity of the Smad4 protein decreased by half in the heterozygous ES cells, showing a dosage effect, and was completely absent in the homozygous mutant cells. Putative truncated Smad4 protein, translated from the rearranged transcript, was not detected on Western blots even after prolonged exposure (Fig. 1D; data not shown). These results indicate that the homozygous deletion of the Smad4tm1Ari allele results in a null mutation.
Smad4 is expressed throughout embryogenesis and in adult tissues
To investigate the possible role of Smad4 in mouse development, the expression of Smad4 was examined by Northern blot analysis. During embryogenesis, a major Smad4 transcript of 3.8 kb was detected as early as the ES cell stage and throughout development (Fig. 2A). A transient decrease was noted at E10.5 of development, however, a period when organogenesis is well advanced. In situ hybridization analysis revealed a widespread tissue distribution in the embryo (Fig. 5; see below). In adult mice, a Smad4 transcript of 3.8 kb was also present in all tissues examined including thymus and hematopoietic cells (Fig. 2B). Interestingly, a higher transcript of ∼6.8 kb was relatively abundant in adult tissue(s) (specifically in the brain), compared with the developing embryo. Thus, the expression of Smad4 at all stages suggests its requirement during mouse development and in the adult.
Figure 2.

Expression of Smad4 during embryogensis and in adult tissues. (A) Northern blot analysis of Smad4 ES cells, embryos at different developmental stages (E7.5–E12.5), and in adult tissues (B). The membranes were hybridized with the murine full length Smad4 cDNA. A prominent transcript of ∼3.8 kb was ubiquitously expressed at all developmental stages and in all adult tissues examined. Filters were stripped and reprobed with the ribosomal gene, L32, to normalize for loading variations. (BM) bone marrow, (LN) lymph node.
Figure 5.
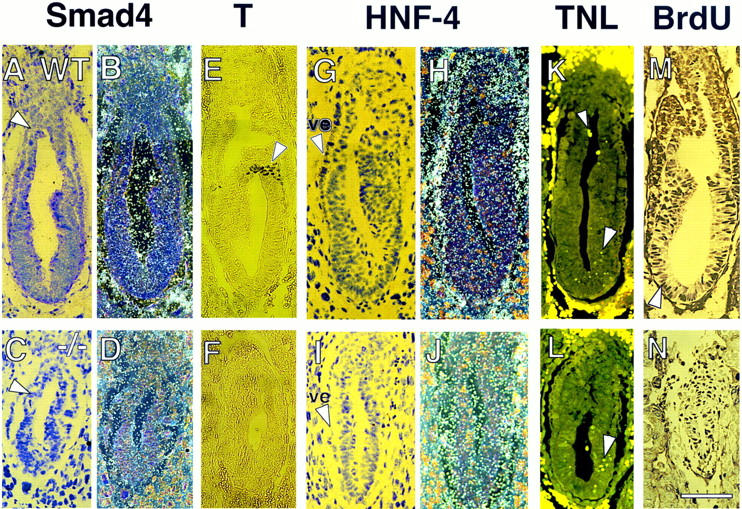
E6.5 Smad4 mutant embryos do not express Brachyury and show reduced Hnf4 expression and BrdU incorporation. In situ hybridization showing ubiquitous Smad4 expression in a wild-type embryo under bright-field (A) and dark-field (B) view. Smad4 expression in mutant embryo under bright-field (C) and dark-field (D) is drastically reduced. (A,C) Arrow points to the separation between the embryonic and extraembryonic regions. (E) Immunohistochemical analysis of T protein in wild-type embryo reveals T-positive cells in the nascent streak (arrowhead). (F) Absence of T expression in Smad4 mutant embryo. In situ hybridization showing Hnf4 expression in the visceral endoderm (ve, arrowhead in G) of a wild-type embryo under bright-field (G) and dark-field (H). Hnf4 expression in Smad4 mutant embryo under bright-field (I) and dark-field (J) is severely reduced in the visceral endoderm (arrowhead in I). (K) TUNEL staining of wild-type embryo reveals a few apoptotic nuclei in the ectoderm region (large arrowhead) and in the proamniotic cavity (small arrowhead). (L) TUNEL staining of Smad4 mutant embryo indicates a slight increase of apoptosis in the ectoderm (large arrowhead). (M) BrdU staining of wild-type embryo discloses strong BrdU-positive nuclei (arrowhead) throughout the embryo. (N) BrdU staining of Smad4 mutant embryo shows a significantly reduced number of BrdU positive nuclei. (Bar) 60 μm.
Smad4 heterozygous mice display no increase in tumorigenicity
To address whether the loss of one Smad4 allele would lead to increased frequency of spontaneous tumors, 25 heterozygous mice, obtained from chimeras with germ-line transmission of the Smad4tm1Ari allele, were followed for a period of up to 11 months. As of this time, no increase in tumors has been observed compared with wild type on two different mouse backgrounds (C57/BL6 and CD1). Because the Smad4 protein level was reduced in the heterozygote ES cells (Fig. 1D), tumor formation in mice did not appear to be strictly dose-dependent. Inactivation of the other wild-type allele is presumably required for tumorigenesis. To increase the frequency of this rare event, induced mutagenesis might be required by use of genetic or chemical means. Even if both alleles are inactivated, however, this might be insufficient to be tumorigenic, and mutations in alternative pathways could be required. Possible candidate genes would include p16, ras, and p53 because in pancreatic tumors these genes are frequently mutated in conjunction with SMAD4 (Rozenblum et al. 1997). Further experiments along these lines are in progress.
Homozygosity for the Smad4 mutation leads to embryonic lethality by E7.5
Among 66 neonates examined, no homozygous mutant Smad4 mice were identified (Table 1), indicating that homozygosity for the Smad4tm1Ari allele resulted in embryonic lethality. To determine the lethality phase of Smad4 mutant embryos, the progeny from heterozygote intercrosses were analyzed at different days of gestation. At E6.5, 13% of the embryos were in resorption and 15% of the live embryos were morphologically abnormal and genotyped as mutants (Table 1; Fig. 3A,B). The mutant embryos were of reduced size compared with wild-type littermates and had a poorly defined boundary between the embryonic and the extraembryonic regions (Fig. 3B). At E7.5, mutant embryos had not significantly progressed in their development (Fig. 3C). By E8.5, most mutant embryos were in resorption or degenerating within the yolk sac (Fig. 3D,E; Table 1). These results show that homozygosity for the Smad4 mutation causes lethality at E7.5, and indicated that Smad4 is essential for postimplantation development. Mutant mice generated from the two independent targeted ES cell clones showed identical phenotypes.
Table 1.
Lethality phase of Smad4 mutant embryos
| Phenotype
|
Normal
|
Abnormal
|
Total
|
||
|---|---|---|---|---|---|
| genotype
|
+/+
|
+/−
|
−/−
|
resorbed
|
|
| E6.5 | 8 (31%) | 14 (54%) | 4 (15%) | 4 | 26 |
| E7.5 | 2 (7%) | 20 (66%) | 8 (27%) | 6 | 30 |
| E8.5 | 7 (25%) | 13 (46%) | 8a (29%) | 4 | 28 |
| Newborn | 27 (41%) | 39 (59%) | — (0%) | ||
Embryos from heterozygous breedings were dissected at different developmental stages and assessed morphologically as normal or abnormal (see Fig. 3). DNA was extracted from embryos or yolk sacs and typed by PCR using primers described in Fig. 1. Resorbed embryos were not included in the total count since they could not be typed. In parentheses appear the percentages corresponding to the different genotypic classes.
Five of eight yolk sacs did not contain any embryos.
Figure 3.
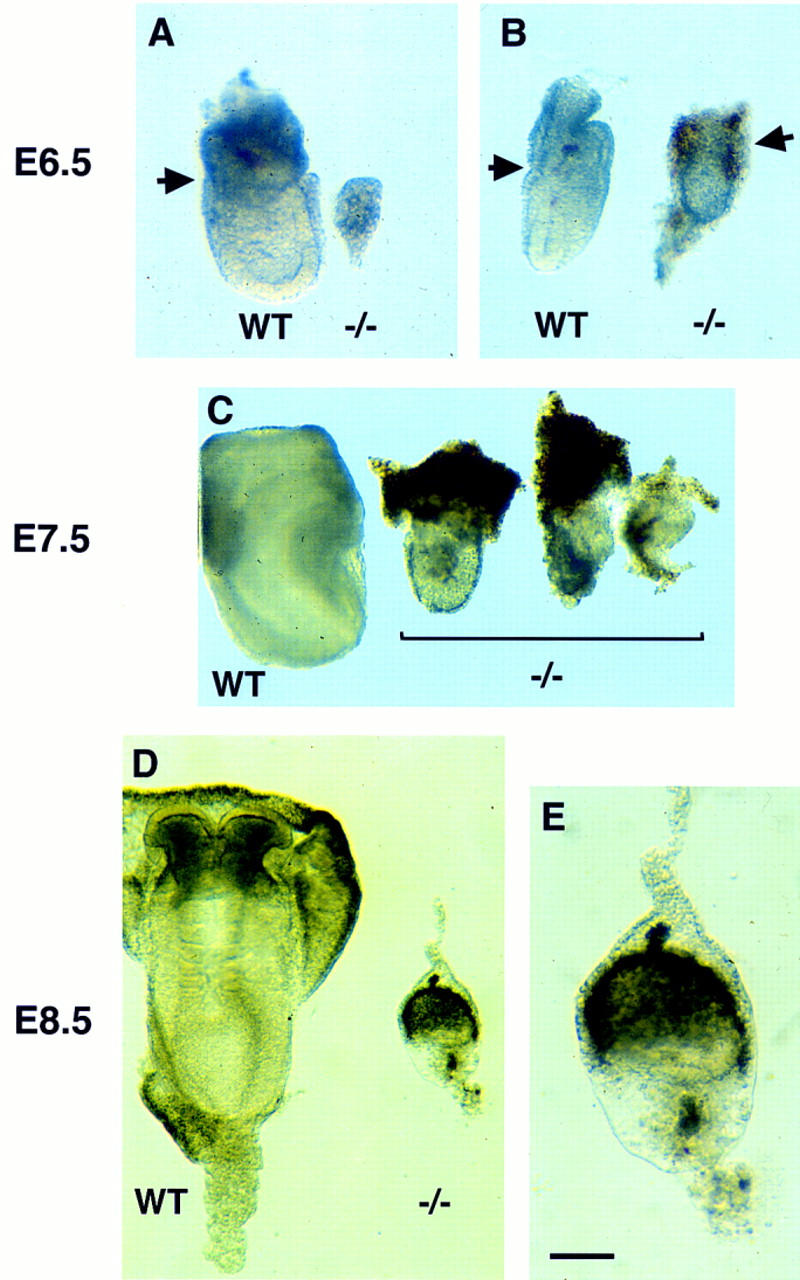
Growth retardation and poor differentiation in Smad4 mutant embryos. (A) E6.5 Smad4 mutant embryos severely growth-retarded and unorganized as compared with wild-type littermates (left); (B) mutant embryos with a poorly defined extraembryonic region. The arrows point to the separation between the embryonic and extraembryonic regions. (C) E7.5 Smad4 mutant embryos have not developed considerably and start to be resorbed. (D) E8.5 wild-type embryos start organogenesis (left). While most of the Smad4 mutant embryos are in resorption, very few remnant embryos remain with no distinguishable structures (right on D and detail in E). (Bar) 70 μm in A,B; 140 μm in C, 60 μm in D, and 20 μm in E.
Smad4 mutant embryos fail to form mesoderm and show abnormal visceral endoderm development
The structural organization of Smad4 mutant embryos was examined in detail by histological analysis of serially sectioned E5.5–E7.5 embryos obtained from heterozygous breedings (Fig. 4A–F). Embryos were classified morphologically as wild type or mutant. At E5.5, ∼30% (7/23) of the egg cylinder stage embryos were of mutant phenotype on the basis of their reduced size and a poorly organized visceral endoderm (Fig. 4A,B). At E6.5, phenotypic differences between wild-type and mutant embryos became more apparent. Wild-type embryos showed a well-organized ectoderm and endoderm, and a developing mesoderm (Fig. 4C). In contrast, 35% (11/32) of the sectioned embryos showed a reduced ectoderm region with no signs of mesoderm development and a disorganized visceral endoderm and extraembryonic region (Fig. 4D). At E7.5, wild-type embryos were advanced in gastrulation, showing a well-defined mesoderm layer (Fig. 4E). Approximately 40% (13/31) of the deciduas contained embryos in resorption or embryos that had not progressed significantly in their development (Fig. 4F) and were classified as Smad4 mutants. Because of the increasing severity of the Smad4 mutant phenotype at later stages of development, we focused our phenotypic analysis on E6.5.
Figure 4.
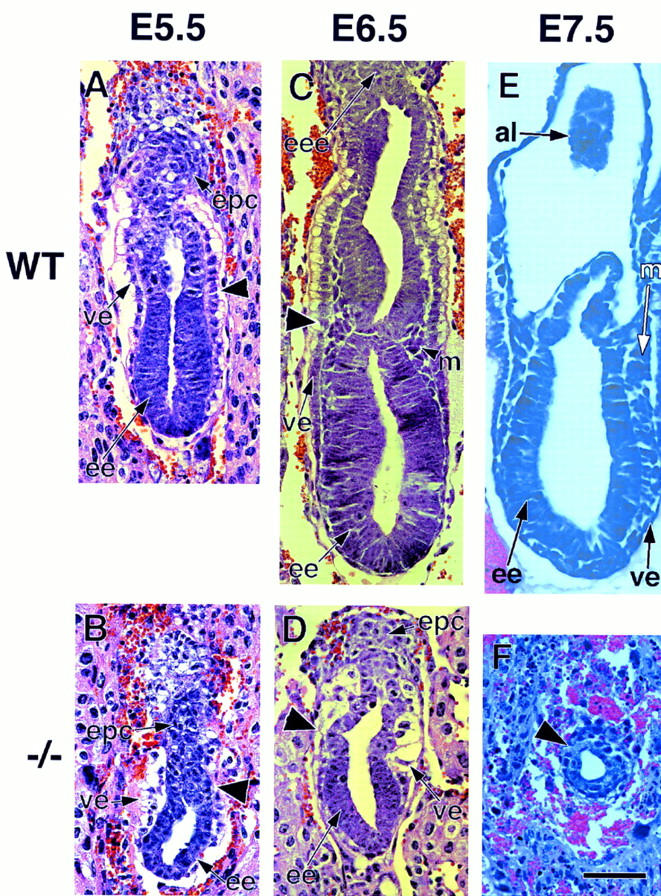
Defective mesoderm and visceral endoderm development in Smad4 mutant embryos. Embryos were sectioned and stained with hematoxylin–eosin. (A) E5.5 wild-type embryo is an early egg-cylinder stage. (B) E5.5 Smad4 mutant embryo, the embryonic region is reduced and the visceral endoderm is disorganized. (C) E6.5 wild-type late egg-cylinder stage embryo, both the extraembryonic and embryonic regions are well organized and mesoderm tissue can be distinguished. (D) E6.5 Smad4 mutant embryo, the extraembryonic region, including the visceral endoderm, is severely disorganized. The embryonic region is reduced in size and poorly developed but displays a proamniotic cavity. No sign of mesoderm can be observed. (E) E7.5 wild-type embryo, three germ layers are apparent. (F) E7.5 Smad4 mutant embryo. The arrow points to the extremely reduced embryonic region. (A–D) The large arrowhead points to the separation between the embryonic and extraembryonic region. (al) allantois; (ee) embryonic ectoderm; (eee) extraembryonic ectoderm; (epc) ectoplacental cone; (m) mesoderm; (pa) proamniotic cavity; (ve) visceral endoderm. (Bar) 60 μm in (A–D,F); 150 μm in E.
Absence of Brachyury and reduced Hnf4 expression in E6.5 Smad4 mutant embryos
To gain further insight into the Smad4 mutant phenotype, we analyzed the Smad4 spatial expression pattern in the E6.5 embryo. We used in situ hybridization over immunohistochemistry because our Smad4 antiserum was not appropriate for this technique. At E6.5, Smad4 was ubiquitously expressed in the ectoderm, visceral endoderm, and extraembryonic region of the E6.5 embryo (Fig. 5A,B). Smad4 expression in presumptive mutant embryos was reduced (five embryos analyzed), as shown in Figure 5, C and D.
Smad4 mutant embryos did not show any histological signs of mesoderm formation at E6.5 (Fig. 4D,E). To determine at the molecular level if any mesoderm was formed in mutant embryos, we examined the expression of the Brachyury (T) protein by immunohistochemistry in tissue sections (Fig. 5E,F). T is one of the earliest markers of mesoderm formation expressed at the onset of gastrulation at E6.5 (Kispert and Hermann, 1993). Intense T expression was detected in the nascent primitive streak in five of five wild-type embryos analyzed (Fig. 5E). No T expression was found in four mutant embryos examined (Fig. 5F). This result confirmed the histological analysis indicating that Smad4 mutant embryos did not make mesoderm or a primitive streak.
Although the extraembryonic region appeared to be disorganized in Smad4 mutant embryos (Fig. 4D,E), expression of the diploid trophoblast marker Mash2 (Guillemot et al. 1994) was normal (data not shown). To further investigate the defect in the extraembryonic tissue, we examined the expression pattern of Hnf4 in the visceral endoderm. Hnf4 is a transcription factor member of the steroid hormone receptor superfamily, whose expression is initially restricted to the visceral endoderm (Fig. 5G,H) (Duncan et al. 1994; Taraviras et al. 1994). In presumptive Smad4 mutant embryos, however, Hnf4 expression was severely reduced (Figure 5I,J).
Reduced in vivo proliferation in Smad4 mutant embryos
The reduced size of Smad4 mutant embryos could hypothetically be caused by an excess of apoptosis, or a decrease in cellular proliferation. We examined the first possibility by end-labeling fragmented nuclei with digoxygenin–UTP by use of the TUNEL assay. No significant difference in apoptotic figures was observed between wild-type and mutant embryos (Fig. 5K,L). To test the possibility of a cellular growth defect, we analyzed the proliferative capabilities of mutant embryos in vitro and in vivo. E3.5 wild-type blastocysts, when cultured in vitro, differentiate into adhering trophoblasts with a compact cellular mass resulting from extensive proliferation of the epiblast. Mutant Smad4 blastocysts grew similarly to wild-type littermates for a period of up to 12 days with a well developed endoderm surrounding the epiblast (data not shown), indicating no proliferative defect at this time. This result was consistent with the similar growth rate of the wild-type and homozygous mutant ES cells observed in the presence or the absence of serum (data not shown).
To examine the effect of the Smad4 mutation on cellular proliferation in vivo, we analyzed the incorporation into DNA of 5-bromo-2′-deoxyuridine (BrdU). A decreased BrdU incorporation was observed in the epiblasts of the presumptive Smad4 mutant embryos compared with their littermates as judged by weaker staining of the BrdU-positive cells (Fig. 5M,N). Moreover, a quantitative difference was also obtained when a proliferative index based on the ratio of proliferating cells (BrdU-positive nuclei) to total cell number was calculated in wild-type and mutant embryos. Both the extraembryonic and embryonic regions of the wild-type and mutant Smad4 embryos were included in the analysis. A total of four wild-type embryos were analyzed and showed a proliferative index of 0.8 ± 0.05%, whereas three presumptive mutant embryos gave an index of 0.6 ± 0.03%. These values proved to be statistically significant (P < 0.05; Student’s t-test). This result indicates that the proliferation ability of Smad4 mutant embryos in vivo is impaired at E6.5, the time when morphological abnormalities become more apparent.
Impaired in vitro differentiation of the visceral endoderm and mesoderm in mutant ES cells
ES cells, when cultured in the absence of leukemia inhibitory factor (LIF), have the ability to differentiate into embryoid bodies containing the different germ layers (Doetschman et al. 1985; Robertson 1987). Morphological analysis revealed that by day 12, 50% and 85% of the heterozygous mutant and wild-type ES cells, respectively, formed cystic embryoid bodies with a prominent cyst cavity surrounded by visceral yolk sac endoderm (Fig. 6A). In contrast, in the homozygous mutant clones, no (clones F9-2 and F9-5) or few (clone C8-24) cystic embryoid bodies were apparent (Fig. 6B). Histological sections of the heterozygous deficient embryoid bodies revealed a well-developed layer of brush border cuboidal epithelium with a thick basement membrane, characteristic of the visceral endoderm (Fig. 6C). Although the homozygous mutant embryoid bodies formed localized brushed border of cuboidal epithelium, most of the surrounding tissue was poorly differentiated and resembled primitive endoderm (Fig. 6D). Interestingly, a thick layer of extracellular matrix was present under the endoderm layer. Its nature and origin, however, is unclear at this time.
Figure 6.
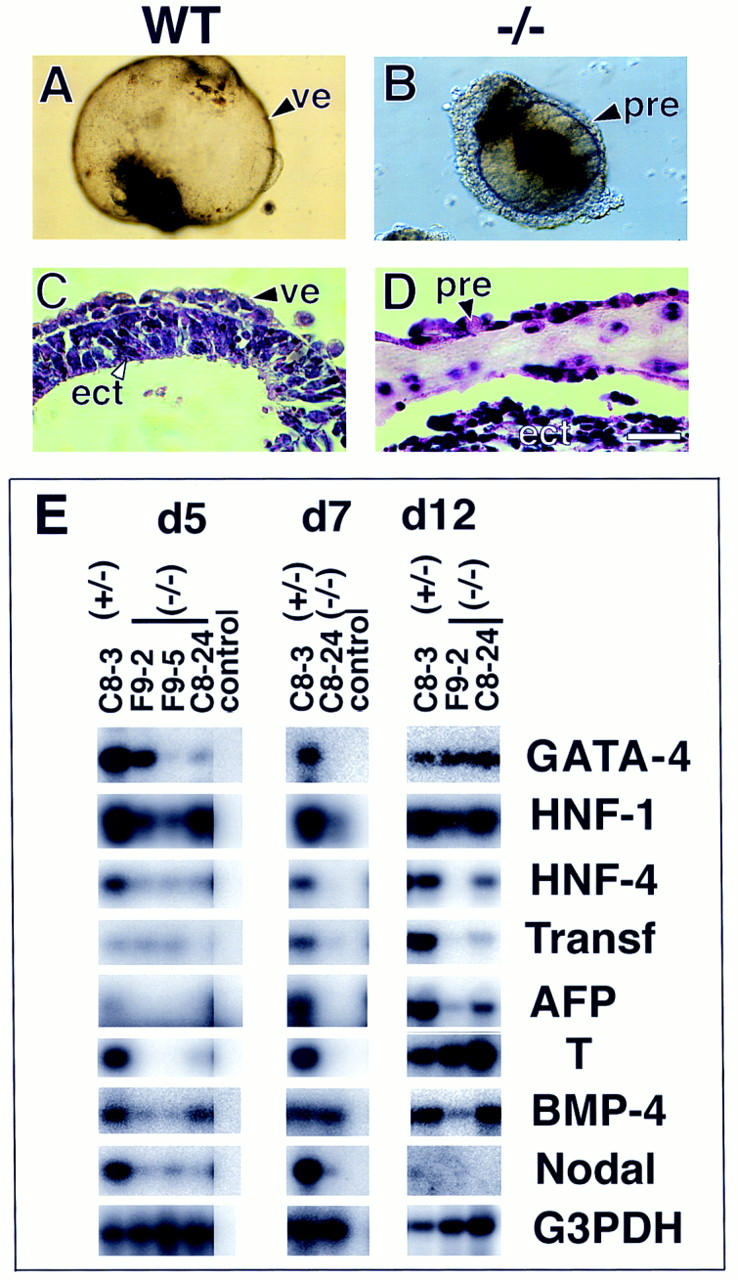
In vitro differentiation of the visceral endoderm and mesoderm is impaired in Smad4 mutant embryoid bodies. Morphological analysis of the EBs at day 10 of culture revealed that most (A) wild-type EBs formed a large cystic cavity. (B) Mutant embryoid bodies gave no sign of cavitation and were smaller in size. (C,D) H+E staining. (C) Wild-type embryoid bodies have a well defined visceral endoderm extending around the cystic cavity. (D) Embryoid bodies from Smad4-deficient cells have a discontinuous visceral endoderm and do not form any cystic cavity. (E) Semiquantitative RT–PCR analysis of markers for visceral endoderm and mesoderm development. RNA was extracted from embryoid bodies at day 5, 7, and 12 of in vitro differentiation and analyzed for the expression of early (GATA-4, HNF-1) and late (HNF-4, transferrin, α-fetoprotein) markers for the visceral endoderm development. The expression of Brachyury (T) was examined for mesoderm differentiation. To normalize for the amount of mRNA used as starting material, the cDNA of G3PDH was amplified. Verification of the PCR products was assessed by Southern blot analysis by use of end-labeled primers as probe(s), internal to the sequence amplified. Control RT reactions, without reverse transcriptase, were performed on all RNA preparation for each set of primers used to assess the absence of DNA contamination (data not shown). Only one control RNA from the heterozygous mutant was included for each specific PCR reaction (control lane). Abbreviations. (pre) primitive endoderm, (ect) ectoderm. (Bar) 120 μm in A, 60 μm in B, and 40 μm in C,D).
To delineate the in vitro defect of the visceral endoderm, biochemical and molecular differentiation markers were examined by semi-quantitative RT–PCR at different time periods of the embryoid body cultures (Fig. 6E; Table 2). Gata4 expression is essential for the earliest stages of visceral endoderm differentiation in vitro, as well as Hnf1 that appear to be expressed early in the specification of the visceral endoderm lineage (Soudais et al. 1995; Duncan et al. 1997). Although Hnf4 is expressed early in the development of the visceral endoderm, it is required only later for complete differentiation (Chen et al. 1994; Duncan et al. 1994; Taraviras et al. 1994). Other late markers include secreted serum proteins such as α-fetoprotein and transferrin. In Smad4 heterozygous embryoid bodies (C8-3), early, as well as late, markers of the visceral endoderm are expressed at all time points examined (days 5, 7, and 12) (Fig. 6E). In the three homozygous mutant clones examined, the expression of Gata4 and Hnf1 was generally decreased or absent at early time points but was comparable with wild type by day 12 of culture (Fig. 6E), suggesting that early specification of the visceral endoderm lineage was delayed. Moreover, in all homozygous deficient clones, Hnf4, transferrin, and α-fetoprotein were absent or greatly reduced (Fig. 6E), indicating that Smad4 is also required for the maintenance or late differentiation events of the visceral endoderm.
Table 2.
Primers used in the expression analysis of the embryoid bodies
| Genes
|
Primers
|
|
|---|---|---|
| amplification (sense; antisense)
|
probe (internal)
|
|
| HNF-4 | TAGCAGAGATGAGCCGTGTGTCC; TAGCAGAGATGAGCCGTGT | |
| GTC | ATGGCTTTGAGGCAGGCGTATTC | |
| AFP | ATACTCAAGAACTCACCCCAACCT; CTCACACCAAAGCGTCAA | |
| CACATT | TGAGACAGGAAGGTTGGGGTGAG | |
| Transferrin | ACCTCCTACTACGCTGTGGCTGTG; GGGTTCTTTCCTTCGGTG | |
| TTATCC | ACTGTTCAGCTCTCCTCTTGG | |
| GATA-4 | CAGCCCCTACCCAGCCTACAT; GTGCCCCAGCCTTTTACTTTG | AAACCAGAAAACGGAAGCCCAAGAACC |
| HNF-1 | GAAAGCAACGGGAGATCCTCCGAC; CCTCCTCCACTAAGGCCT | |
| CCCTCTCTTCC | ACGACCGGCAAAAGAATCCCAGCAAGG | |
| T | AGAAAGAAACGACCACAAAGATG; ATTTATTTATTTTTCCCTT | |
| GTCC | GTACCATCACCCCAGGCTCCCAGACAG | |
| BMP4 | CCTCTTCAACCTCAGCAGCATCC; CACACCCCTCTACCACCAT | |
| CTCC | CACCAGGGCCAGCACGTCAGAATCAGC | |
| Nodal | CCCCACAGGGTTAGGACACTCG; TGCTGAAAGTGCTGTCTGTC | |
| TGCTC | AGAGGGGCGGATGGGGCAGAAGGCAAC | |
Set of primer pairs used for amplification of indicated genes from reverse-transcribed RNA derived from embryoid bodies. Primers are written in 5′ → 3′ orientation. The PCR products were verified by probing with internal primers.
The mesoderm marker brachyury (T) is expressed by day 5 of embryoid body culture, peaks by day 6, and then gradually decreases (Keller et al. 1993). In Smad4 heterozygous embryoid body T was strongly expressed by day 5 and its expression was maintained up to day 12 (Fig. 6E). T expression was virtually absent in the Smad4 homozygous mutants at early time points (days 5 and 7), but was comparable with wild type in all mutant embryoid bodies examined by day 12 (Fig. 6E). This result indicates that Smad4-deficient ES cells have the ability under certain in vitro conditions to undergo mesoderm formation.
Two factors required for mesoderm formation, whose expression prior to gastrulation appears to be either restricted to the ectoderm or the visceral endoderm, are Bmp4 (Mishina et al. 1995; Winnier et al. 1995) and nodal (Varlet et al. 1997), respectively. In Smad4 heterozygous mutant clones, Bmp4 was detected at all stages of the embryoid body growth, whereas nodal was present only at early stages of the culture. In three independent Smad4 homozygous clones, both factors were significantly reduced at early stages (day 5) of the embryoid body cultures. Although later expression of Bmp4 was variable, nodal remained absent in all mutant clones (Fig. 6E). These results suggest that the impaired development of the visceral endoderm may lead to abnormal expression of these factors.
Wild-type visceral endoderm rescues the gastrulation defect of Smad4 mutant embryos
The role of the visceral endoderm in the gastrulation defect of the Smad4-deficient embryo was addressed by tetraploid aggregation experiments. In this procedure, tetraploid embryonic cells, which retain their full potential to contribute only to the extraembryonic tissues, are aggregated with ES cells (Nagy and Rossant 1993). Resulting conceptuses consist of ES cell-derived embryonic tissues with tetraploid-derived extraembryonic tissue. Three Smad4-deficient and one control Smad4 heterozygote ES cell line(s) were aggregated with wild-type tetraploid embryos. Seven days after being transferred into foster mothers, chimeric embryos (corresponding to E8.5–E9.0) were examined. All embryos derived from Smad4 heterozygote ES cells displayed features characteristic of E8.5 embryos (Fig. 7A,B). Seventy-five percent (49/65) of the embryos recovered from their Smad4-deficient clones had distinctive mesoderm-derived tissue, such as somites (Fig. 7C,D). Although two of these embryos developed up to the head-fold stage (Fig. 7C), most chimeric embryos (54%) showed truncations in anterior structures (Fig. 7D). The remaining conceptuses were degenerating embryos often resulting in empty yolk sacs possibly reflecting embryos in resorption. Although embryos recovered from the F9-5 mutant clone were in general less organized than those recovered from the other two mutant ES clones (C8-13) and (C8-24), all expressed Brachyury (Fig. 7E,F) and the somite marker Mox-1 (Fig. 7G,H). Impaired anterior development was confirmed by the absence of a hindbrain specific marker Krox-20, in all mutant ES cell line-derived embryos (Fig. 7G,H). These results confirm that the gastrulation defect in Smad4 mutant embryos was caused by impaired function of the visceral endoderm, and also confirmed the potential of the Smad4-deficient ES cells to form mesodermal tissue as indicated from in vitro experiments (Fig. 6E). The gastrulation rescue in Smad4-deficient embryos by wild-type visceral endoderm uncovered novel roles for Smad4 within the epiblast-derived lineages namely, Smad4 is required for normal anterior cell fate specification.
Figure 7.

Wild-type visceral endoderm rescues the gastrulation defect of Smad4-deficient embryos. Whole mount in situ hybridization of E8.5 chimeric embryos generated from tetraploid aggregation experiments. (A,B) Embryos derived from Smad4 heterozygous ES cells were hybridized for T, or Krox-20 (K20) and Mox-1. (C,D) Dissected embryos derived from Smad4 homozygous mutant ES clone, C8-24. (C) Less affected embryo with malformation in headfold region (arrowhead). (D) Representative embryo with anterior truncation and a well developed posterior region with somites (arrow). (E,F) T expression (arrow) in embryos derived from Smad4 homozygous mutant ES clone F9-5 and C8-24, respectively. Embryo derived from clone F9-5 is less organized with abnormal head region (arrowhead). (G,H) Embryos derived from Smad4 homozygous mutant ES clone F9-5 and C8-24 respectively, probed with Krox-20 and Mox-1. Arrowhead points to head region. The arrows in B,G, and H point to Mox-1 expression. (Bar) 20 μm.
Discussion
Smad4 mutant phenotype resembles that of the Bmp4- and Bmpr-I-deficient mice
We have generated a null mutation in the mouse homolog of the tumor suppressor gene DPC4, by homologous recombination in ES cells. Smad4 mutant embryos showed initial signs of growth retardation and impaired differentiation as early as E5.5. E6.5 embryos were small, the morphology of the visceral endoderm was disorganized, and no mesoderm formed. Embryos were resorbed by E8.5. The reduced size of Smad4 mutant embryos was associated with a decrease in ectoderm proliferation rather than an increase in apoptosis.
The phenotype of Smad4 mutant embryos is similar to that reported for some Bmp4 mutant embryos and all BmprI mutants, namely a lack of mesoderm formation and retarded embryonic growth (Mishina et al. 1995; Winnier et al. 1995). Because Smad4 has been shown to be involved in transduction of BMP signals in Xenopus and Drosophila (Massagué et al. 1997), the failure to gastrulate in Smad4 mutant embryos could result from a failure of Smad4-dependent BMP signaling. Smad4-deficient embryos, however, display earlier phenotypic abnormalities as well as abnormal visceral endoderm development, distinguishing them from the Bmpr mutant mice. These differences could be explained by the involvement of Smad4 in other TGFβ-related pathways early in embryogenesis.
The gastrulation defect in Smad4 mutant embryos results from a defective visceral endoderm
The proliferation defect that appears to be responsible for the death of Smad4 embryos could result from two possible mechanisms. One would be an intrinsic inability of mutant ectoderm to respond to growth signals. The other would be a non-cell autonomous defect in which growth signals fail to be secreted from adjacent tissues. Inherent proliferation defect of the ectoderm, leading to lack of mesoderm formation, has been shown in the targeted disruption of the tumor suppressor genes Brca1 and Brca2 (Hakem et al. 1997; Suzuki et al. 1997). The proliferation defect is associated with the deregulation of cell cycle in these embryos as early as E3.5. Unlike Brca1-deficient embryos, however, in vitro proliferation of E3.5 Smad4-deficient blastocysts was normal and all of the cell cycle components examined (p21, p53, cyclin E, cyclin A, and p15) appeared normal (data not shown), indicating that the mechanism underlying the gastrulation defect was different from that of the Brca1 embryo.
The second possibility, although not exclusive to the first, implicates the gastrulation defect of the Smad4-deficient embryo as a non-cell-autonomous defect of the ectoderm. The Smad4 mutant embryos displayed a defective visceral endoderm as assessed by histological analysis and by the reduced Hnf4 expression in vivo and by morphological and molecular analysis of the embryoid body in vitro. Disruption of several genes normally expressed in the visceral endoderm also results in a gastrulation defect. These include evx1, Hβ58, msd, and Hnf4 (Lee et al. 1992; Chen et al. 1994; Holdener et al. 1994; Spyropoulos and Capecchi 1994). The gastrulation defect of Hnf4-deficient embryos can be rescued with wild-type visceral endoderm in tetraploid aggregation experiments (Duncan et al. 1997), confirming the importance of the integrity of the visceral endoderm in mesoderm development. We used the same approach to test whether the gastrulation defect in Smad4 embryos was caused by defective visceral endoderm. On the basis of morphological features and Brachyury and Mox-1 expression, we show that in the presence of wild-type visceral endoderm, E8.5 Smad4 −/− embryos, derived from three independent ES homozygous mutant clones, were able to initiate gastrulation and form somites. These results confirm that the lack of gastrulation in Smad4 −/− embryos was not caused by an intrinsic defect of the ectoderm, but rather the consequence of the impaired function of the visceral endoderm.
Because Smad4 is thought to act as a critical downstream regulator in TGFβ-related signaling pathways, these results imply that there is an intrinsic requirement for TGFβ-related signaling for normal development of the visceral endoderm. Exactly which TGFβ family member, acting at what time and emanating from which tissue, is involved in visceral endoderm development is unclear. It has been assumed that the gastrulation defect in Bmp4- and BmprI-deficient embryos results from a failure of BMP response in the epiblast (Mishina et al. 1995; Winnier et al. 1995). In light of our results, however, those assumptions might bear reconsideration. It would be of interest, for instance, to perform tetraploid aggregation experiments with BmprI-deficient ES cells to determine whether the impaired signaling response is intrinsic to the epiblast or to the visceral endoderm.
The defective visceral endoderm of Smad4 mutant embryos could lead to growth and gastrulation defects in the epiblast by its failure to provide either general nutritional requirements for the embryo or specific inductive signals for gastrulation and patterning. Genetic analysis in the mouse showed that at least two TGFβ-related proteins, Bmp4 and nodal, are required for gastrulation. Whereas the expression of Bmp4 appears to be restricted to the ectoderm, the expression of nodal is restricted to the visceral endoderm prior to gastrulation. Interestingly, both factors were reduced or absent during early in vitro differentiation of Smad4-deficient embryoid bodies compared with wild type. Concurrent with the reduction of these factors, was the absence of the mesoderm marker, Brachyury, suggesting that the deficiency of these factors (and possibly others) could be responsible for the absence of gastrulation in the Smad4 mutant mice. It is not yet clear whether Bmp4 and nodal are direct targets of Smad4, or whether the reduced level of serum proteins, Hnf4, Bmp4, and nodal expression in the visceral endoderm is a consequence of the inability of the Smad4-deficient embryo to develop to the stage at which these genes are normally expressed.
Multifunctional role of Smad4
Given the nature of Smad4 as a central signaling molecule of several TGFβ-related pathways, it was anticipated that later stages of embryonic development would also be affected by the loss of Smad4. Most of the mutant embryos rescued by wild-type visceral endoderm had anterior truncations. Although few rescued embryos (<5%) developed abnormal headfolds, most did not express the hindbrain marker Krox-20, confirming the absence of anterior structures. These results imply that post-gastrulation, Smad4 is also required for anterior-posterior axis patterning. Interestingly, when the gastrulation defect in nodal-deficient blastocysts is rescued by the inclusion of wild-type ES cells, rostral neural structures fail to develop (Varlet et al. 1997). Because the visceral endoderm of these chimeric embryos is nodal-deficient, these results imply that the restricted nodal expression found in the visceral endoderm is critical for anterior development at later stages. In our tetraploid aggregation experiment with Smad4-deficient embryos, the visceral endoderm is of wild type origin, thus providing the nodal required for anterior development. Rescued Smad4-deficient embryos, however, still exhibited anterior defects, suggesting that Smad4 could be involved in the downstream response to nodal signaling in the epiblast-derived tissue. The defect is more pronounced in Smad4 than in nodal-deficient embryos, suggesting that Smad4 could be involved in the downstream response of other factors involved in anterior specification.
Defects in the formation of the anterior neural axis have also been described in embryos carrying loss-of-function mutations in the genes HNF3β (Ang and Rossant 1994; Weinstein et al. 1994), Lim1 (Shawlot and Behringer 1995), and Otx2 (Acampora et al. 1995; Matsuo et al. 1995). Together with Smad4, they might participate in a common pathway regulating anterior specifications. Recently, functional interaction between Smad4, Smad2, and Fast1, a member of the HNF3β family (winged-helix transcription factor), has been shown (Chen et al. 1996, 1997).
At this stage, it is difficult to establish a parallel between the embryonic functions of Smad4 and its role as a tumor suppressor in adult. A combined approach involving tissue-specific gene inactivation as well as biochemical analysis on Smad4-deficient cell lines should elucidate the roles of Smad4 in adult tissue.
Materials and methods
Cell culture
The E14K ES cells derived from 129/Ola mice were maintained on a monolayer of mitomycin-C-treated embryonic fibroblasts in Dulbecco’s modified Eagle medium (DMEM) supplemented with 15% FCS (Hyclone), leukemia inhibitory factor, sodium pyruvate, l-glutamine, and β-mercaptoethanol. Primary embryonic fibroblasts were maintained in DMEM supplemented with 5% FCS (Sigma) and β-mercaptoethanol.
Targeting vector and generation of Smad4-deficient ES cells
A 129/J mouse genomic library was screened with a mouse DPC-4 probe containing exons 8 and 9. Both arms of the targeting construct were derived from a 12-kb phage clone and subcloned into the ploxPneo vector (Puri et al. 1995) containing two loxP sites flanking the neomycin gene. The expression of the neomycin gene was under the control of the PGK promoter and was in reverse orientation relative to the Smad4 gene expression. Five independent electroporations were performed in 5 × 106 ES cells with 20 μg of targeting vector linearized with NotI by use of a Bio-Rad Gene Pulsar (0.34 kV, 250 mF). Cells were selected in 180 μg/ml of active G418 (Sigma) and G418 resistant colonies were pooled and analyzed by PCR for homologous recombination. Two rounds of PCR, 30 cycles each, were performed with sense primers in the neo gene and antisense primers in the genomic region, flanking the short-arm of the targeting vector (see Fig. 1). The primers used for the primary PCR were (sense; antisense, respectively) 5′-CGAAGGGGCCACCAAAGAACG-3′; 5′-TACTTTGCCGTGGTGGTGCTC-3′ and 5′-CCGGTGGATGTGGAATGTGTG-3′; 5′-CCTGCCGTCTGTTGAATGTGC-3′ for the nested PCR.
Generation of Smad4 mutant mice
Chimeric mice were produced by microinjection of Smad4 heterozygous ES cells into E3.5 C57BL6/J blastocysts and transferred to CD1 pseudopregnant foster mother. Chimeric males were mated with C57BL6/J females (Jackson Laboratories) and germ-line transmission of the mutated allele was verified by PCR and Southern blot analysis of tail DNA from agouti coat color F1 offspring. F2 and F3 offspring from heterozygous intercrosses were used for most of the analysis presented in this manuscript. Similar results were obtained when heterozygous males were backcrossed four generations into C57BL6/J inbred mice and to CD1 outbred mice. Embryos and yolk sacs were genotyped by lysing cells into PCR-compatible buffer (50 mm KCl, 10 mm Tris at pH 8.3, 2 mm MgCl2, 0.01% gelatin, 0.45% NP-40, 0.45% Tween 20, 100 μg/ml of proteinase K). The primers detecting the wild type locus were within deleted intronic sequences (see Fig. 1) and are as follows: (sense; anti-sense) 5′-CCTGTGGCCTGCTCTCTTCTC-3′; 5′-GGACAGGCAGTGGAGGATAGG-3′ for the primary PCR and 5′-AGCTTGTCTTTTAGGTGATTG-3′; 5′-GGAAGAACTTATGATTAGGAA-3′ for the nested PCR. The presence of the mutated allele was identified by use of the same primers used for screening homologous recombination in the ES cells (see above; Fig. 1A).
Molecular and biochemical analysis
Southern blot analysis was performed on 15 μg of digested DNA separated on a 0.6% agarose gel transferred according to published protocol (Sambrook et al. 1989). Probes were radiolabeled by use of [32P]dCTP with a random priming kit (Amersham) and hybridization was performed in Church and Gilbert solution. Total mRNA was extracted from cells with Trizol (GIBCO) following the recommended protocol from the manufacturer. Ten micrograms of RNA was separated on a 1% agarose gel and processed as described elsewhere (Sambrook et al. 1989). Western blot analysis was performed on 100 μg of total protein lysed in PLC buffer (50 mm HEPES at pH 7.5, 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1.5 mm MgCl2, 1 mm EGTA, 10 mm sodium pyrophosphate, 100 mm NaF, 10 μg/ml of aprotinin, 10 μg/ml of leupeptin, 1 mm PMSF, 1 mm Na3VO4), separated on a 7% SDS-polyacrylamide gel and electroblotted. The membrane was then hybridized for 16 hr at 4°C with a rabbit polylclonal α-Dpc-4 antibody diluted 1:1000, followed by HRP-conjugated donkey anti-rabbit immunoglobulin and ECL (Amersham).
Histological analysis
Deciduae from females plugged in a 2-hr mating period were isolated in ice-cold PBS at E5.5 and E6.5, fixed overnight in 4% paraformaldehyde at 4°C, dehydrated, and embedded in paraffin. Sections (6 μm-thick) were cut and stained with hematoxylin and eosin.
Radioactive in situ hybridization
Deciduae were isolated in ice-cold PBS at E6.5, and processed as for histological analysis. The probes used were Smad4, Mash-2 (Guillemot et al. 1994), and HNF4 (Chen et al. 1994). Probes were labeled with [33P]UTP and processed according to described protocols (Hui and Joyner 1993).
Immunohistochemistry
Deciduae were isolated in ice-cold PBS at E6.5, fixed in 4% paraformaldehyde for 3 hr, dehydrated, embedded in wax, and sectioned at 6 μm. The anti-T polyclonal antiserum was used at a 1:500 dilution, following the described protocols (Hakem et al. 1996).
TUNEL assay for visualization of apoptosis
Deciduae were processed as for immunohistochemistry. TUNEL reaction to detect incorporation of digoxygenin–dUTP mediated by terminal transferase was carried out on sectioned embryos with a TUNEL kit (Boehringer Mannheim), following manufacturers instructions.
BrdU labeling of embryos
BrdU labeling of cells was performed according to the protocol described elsewhere (Hayashi et al. 1988). BrdU (100 mg/gram of body weight) was injected intraperitoneally into pregnant females at E6.5. The females were sacrificed 1 hr after injection, the uteri were removed, and the decidual swellings were fixed in 4% paraformaldehyde at 4°C overnight and processed for immunohistochemistry. The sections were incubated with an anti-BrdU monoclonal antibody (Boehringer Mannheim) at a 1:10 dilution. Staining was performed according to the protocol described by Mishina (Mishina et al. 1995).
Differentiation of ES cells in embryoid bodies
Embryoid bodies were obtained as described elsewhere (Doetschman et al. 1985; Robertson, 1987). Briefly, ES cells were cultured on 0.1% gelatin-coated tissue culture dishes and then transferred onto bacterial dishes in the absence of LIF; this was counted as day 0 of embryoid body culture. For histological analysis, embryoid bodies were fixed for 16 hr in 4% paraformaldehyde and 6 μm-paraffin sections stained with hematoxylin and eosin. The expression patterns of various genes in the wild-type and mutant embryoid bodies were compared by semiquantitative RT–PCR analysis. One microgram of total RNA was reverse transcribed with the RT–PCR kit (Clontech). For all genes analyzed, 5 μl (except for G3PDH, 0.5 μl) of the cDNA reaction was used for PCR amplification. The PCR amplification of the cDNA remained linear after 30 cycles (data not shown). The PCR products were separated on a 2% agarose gel and Southern blot analysis performed by use of radioactively end-labeled internal primers as probes (Sambrook et al. 1989). Primers used for PCR amplification and internal probing are listed in Table 2. Controls for DNA contamination in the RNA preparation were performed for all sets of primers with the identical procedure as for the RT–PCR, but without reverse transcriptase.
Production of ES cell-derived embryos by tetraploid aggregation
E1.5 embryos at the two-cell stage were flushed from the oviduct of CD1 females and subjected to electrofusion. Successfully fused embryos were cultured overnight in embryo culture medium in 5% CO2 at 37°C. ES cell-tetraploid chimeric embryos were produced by aggregation as described elsewhere (Nagy and Rossant 1993).
Whole mount in situ hybridization
Dissected embryos were fixed and processed for in situ hybridization as described (de la Pompa et al. 1997). The following antisense RNA probes were used: Brachyury (Herrmann 1991), Mox-1 (Candia et al. 1992), and Krox-20 (Wilkinson et al. 1989).
Acknowledgments
We thank Ivan del Barco for help with the whole mount in situ hybridizations; Vuk Stambolic for critical reading of the manuscript, Kate Sullivan for help generating the Smad4 antibody, and Fillio Billia for molecular and cell cycle analysis in mutant blastocysts. C.S. is the recipient of a fellowship from the Medical Research Council of Canada.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tmak@oci.utoronto.ca; FAX (416) 204-5300.
References
- Acampora D, Mazan S, Lallemand Y, Avantaggiato V, Maury M, Simeone A, Brulet P. Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development. 1995;121:3279–3290. doi: 10.1242/dev.121.10.3279. [DOI] [PubMed] [Google Scholar]
- Ang SL, Rossant J. HNF-3 beta is essential for node and notochord formation in mouse development. Cell. 1994;78:561–574. doi: 10.1016/0092-8674(94)90522-3. [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Signal transduction by members of the transforming growth factor-beta superfamily. Cytokine Growth Factor Rev. 1996;7:327–339. doi: 10.1016/s1359-6101(96)00042-1. [DOI] [PubMed] [Google Scholar]
- Baker JC, Harland RM. A novel mesoderm inducer, Madr2, functions in the activin signal transduction pathway. Genes & Dev. 1996;10:1880–1889. doi: 10.1101/gad.10.15.1880. [DOI] [PubMed] [Google Scholar]
- Candia AF, Hu J, Crosby J, Lalley PA, Noden D, Nadeau JH, Wright CV. Mox-1 and Mox-2 define a novel homeobox gene subfamily and are differentially expressed during early mesodermal patterning in mouse embryos. Development. 1992;116:1123–1136. doi: 10.1242/dev.116.4.1123. [DOI] [PubMed] [Google Scholar]
- Chen WS, Manova K, Weinstein DC, Duncan SA, Plump AS, Prezioso VR, Bachvarova RF, Darnell JE., Jr Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes & Dev. 1994;8:2466–2477. doi: 10.1101/gad.8.20.2466. [DOI] [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner for MAD proteins in TGF-beta signalling. Nature. 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Conlon FL, Lyons KM, Takaesu N, Barth KS, Kispert A, Herrmann B, Robertson EJ. A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development. 1994;120:1919–1928. doi: 10.1242/dev.120.7.1919. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Wakeham A, Correia KM, Samper E, Brown S, Aguilera RJ, Nakano T, Honjo T, Mak TW, Rossant J, Conlon RA. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997;124:1139–1148. doi: 10.1242/dev.124.6.1139. [DOI] [PubMed] [Google Scholar]
- Derynck R, Gelbart WM, Harland RM, Heldin CH, Kern SE, Massagué J, Melton DA, Mlodzik M, Padgett RW, Roberts AB, Smith J, Thomsen GH, Vogelstein B, Wang XF. Nomenclature: Vertebrate mediators of TGFbeta family signals. Cell. 1996;87:173. doi: 10.1016/s0092-8674(00)81335-5. [DOI] [PubMed] [Google Scholar]
- Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: Formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- Duncan SA, Manova K, Chen WS, Hoodless P, Weinstein DC, Bachvarova RF, Darnell Jr JE. Expression of transcription factor HNF-4 in the extraembryonic endoderm, gut, and nephrogenic tissue of the developing mouse embryo: HNF-4 is a marker for primary endoderm in the implanting blastocyst. Proc Natl Acad Sci. 1994;91:7598–7602. doi: 10.1073/pnas.91.16.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SA, Nagy A, Chan W. Murine gastrulation requires HNF-4 regulated gene expression in the visceral endoderm: Tetraploid rescue of Hnf-4(−/−) embryos. Development. 1997;124:279–287. doi: 10.1242/dev.124.2.279. [DOI] [PubMed] [Google Scholar]
- Graff JM, Bansal A, Melton DA. Xenopus Mad proteins transduce distinct subsets of signals for the TGF beta superfamily. Cell. 1996;85:479–487. doi: 10.1016/s0092-8674(00)81249-0. [DOI] [PubMed] [Google Scholar]
- Guillemot F, Nagy A, Auerbach A, Rossant J, Joyner AL. Essential role of Mash-2 in extraembryonic development. Nature. 1994;371:333–336. doi: 10.1038/371333a0. [DOI] [PubMed] [Google Scholar]
- Hahn SA, Hoque AT, Moskaluk CA, da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, Kern SE. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996;56:490–494. [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, Firpo E, Hui CC, Roberts J, Rossant J, Mak TW. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Elia A, Potter J, Mak TW. Partial rescue of Brca1 (5-6) early embryonic lethality by p53 or p21 null mutation. Nature Genet. 1997;16:298–302. doi: 10.1038/ng0797-298. [DOI] [PubMed] [Google Scholar]
- Hata A, Lo RS, Wotton D, Lagna G, Massagué J. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. 1997;388:82–87. doi: 10.1038/40424. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Koike M, Matsutani M, Hoshino T. Effects of fixation time and enzymatic digestion on immunohistochemical demonstration of bromodeoxyuridine in formalin-fixed, paraffin-embedded tissue. J Histochem Cytochem. 1988;36:511–514. doi: 10.1177/36.5.3282006. [DOI] [PubMed] [Google Scholar]
- Herrmann BG. Expression pattern of the brachyury gene in whole-mount Twis/Twis mutant embryos. Development. 1991;113:913–917. doi: 10.1242/dev.113.3.913. [DOI] [PubMed] [Google Scholar]
- Holdener BC, Faust C, Rosenthal NS, Magnuson T. msd is required for mesoderm induction in mice. Development. 1994;120:1335–1346. doi: 10.1242/dev.120.5.1335. [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Haerry T, Abdollah S, Stapleton M, O’Connor MB, Attisano L, Wrana JL. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: The extra-toes J mutation contains an intragenic deletion of the Gli3 gene. Nature Genet. 1993;3:241–246. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley DM. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes & Dev. 1994;8:133–146. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- Kispert A, Hermann BG. The Brachyury gene encodes a novel DNA binding protein. EMBO J. 1993;12:4898–4899. doi: 10.1002/j.1460-2075.1993.tb06179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar M, Liu F, Hata A, Doody J, Massagué J. The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes & Dev. 1997;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- Lagna G, Hata A, Hemmati-Brivanlou A, Massagué J. Partnership between DPC4 and SMAD proteins in TGF-beta signalling pathways. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Radice G, Perkins CP, Costantini F. Identification and characterization of a novel, evolutionarily conserved gene disrupted by the murine H beta 58 embryonic lethal transgene insertion. Development. 1992;115:277–288. doi: 10.1242/dev.115.1.277. [DOI] [PubMed] [Google Scholar]
- Liu F, Hata A, Baker JC, Doody J, Carcamo J, Harland RM, Massagué J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Macias-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFbeta receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Massagué J, Hata A, Liu F. TGF-β signalling through the Smad pathway. Trends Cell Biol. 1997;7:187–192. doi: 10.1016/S0962-8924(97)01036-2. [DOI] [PubMed] [Google Scholar]
- Matsuo I, Kuratani S, Kimura C, Takeda N, Aizawa S. Mouse Otx2 functions in the formation and patterning of rostral head. Genes & Dev. 1995;9:2646–2658. doi: 10.1101/gad.9.21.2646. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes & Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- Mortensen RM, Conner DA, Chao S, Geisterfer-Lowrance AA, Seidman JG. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatake M, Takagi Y, Osada H, Uchida K, Mitsudomi T, Saji S, Shimokata K, Takahashi T, Takahashi T. Somatic in vivo alterations of the DPC4 gene at 18q21 in human lung cancers. Cancer Res. 1996;56:2718–2720. [PubMed] [Google Scholar]
- Nagy A, Rossant J. Production of a complete ES cell derived fetus. In: Joyner AL, editor. Gene targeting: A practical approach. Oxford, UK: IRL Press; 1993. pp. 147–179. [Google Scholar]
- Oh SP, Li E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes & Dev. 1997;11:1812–1826. doi: 10.1101/gad.11.14.1812. [DOI] [PubMed] [Google Scholar]
- Puri MC, Rossant J, Alitalo K, Bernstein A, Partanen J. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. EMBO J. 1995;14:5884–5891. doi: 10.1002/j.1460-2075.1995.tb00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery LA, Twombly V, Wharton K, Gelbart WM. Genetic screens to identify elements of the decapentaplegic signaling pathway in Drosophila. Genetics. 1995;139:241–254. doi: 10.1093/genetics/139.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson EJ. Embryo-derived stem cell lines. In: Robertson EJ, editor. Teratocarcinoma and embryonic stem cells: A practical approach. Oxford, UK: IRL Press; 1987. pp. 71–112. [Google Scholar]
- Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–1734. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Savage C, Das P, Finelli AL, Townsend SR, Sun CY, Baird SE, Padgett RW. Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor beta pathway components. Proc Natl Acad Sci. 1996;93:790–794. doi: 10.1073/pnas.93.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero RA, Jr, Meltzer PS, Hahn SA, Kern SE. DPC4 gene in various tumor types. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- Sekelsky JJ, Newfeld SJ, Raftery LA, Chartoff EH, Gelbart WM. Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics. 1995;139:1347–1358. doi: 10.1093/genetics/139.3.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawlot W, Behringer RR. Requirement for Lim1 in head-organizer function. Nature. 1995;374:425–430. doi: 10.1038/374425a0. [DOI] [PubMed] [Google Scholar]
- Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87–93. doi: 10.1038/40431. [DOI] [PubMed] [Google Scholar]
- Soudais C, Bielinska M, Heikinheimo M, MacArthur CA, Narita N, Saffitz JE, Simon MC, Leiden JM, Wilson DB. Targeted mutagenesis of the transcription factor GATA-4 gene in mouse embryonic stem cells disrupts visceral endoderm differentiation in vitro. Development. 1995;121:3877–3888. doi: 10.1242/dev.121.11.3877. [DOI] [PubMed] [Google Scholar]
- Spyropoulos DD, Capecchi MR. Targeted disruption of the even-skipped gene, evx1, causes early postimplantation lethality of the mouse conceptus. Genes & Dev. 1994;8:1949–1961. doi: 10.1101/gad.8.16.1949. [DOI] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Hakem R, Elia A, Yoshida R, Mo R, Nishina H, Chuang T, Wakeham A, Itie A, Koo W, Billia P, Ho A, Fukumoto M, Hui CC, Mak TW. Brca2 is required for embryonic cellular proliferation in the mouse. Genes & Dev. 1997;11:1242–1252. doi: 10.1101/gad.11.10.1242. [DOI] [PubMed] [Google Scholar]
- Taraviras S, Monaghan AP, Schutz G, Kelsey G. Characterization of the mouse HNF-4 gene and its expression during mouse embryogenesis. Mech Dev. 1994;48:67–79. doi: 10.1016/0925-4773(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Thomsen GH. Xenopus mothers against decapentaplegic is an embryonic ventralizing agent that acts downstream of the BMP-2/4 receptor. Development. 1996;122:2359–2366. doi: 10.1242/dev.122.8.2359. [DOI] [PubMed] [Google Scholar]
- Varlet I, Collignon J, Robertson EJ. nodal expression in the primitive endoderm is required for specification of the anterior axis during mouse gastrulation. Development. 1997;124:1033–1044. doi: 10.1242/dev.124.5.1033. [DOI] [PubMed] [Google Scholar]
- Wall NA, Hogan BL. TGF-beta related genes in development. Curr Opin Genet Dev. 1994;4:517–522. doi: 10.1016/0959-437x(94)90066-c. [DOI] [PubMed] [Google Scholar]
- Weinstein DC, Ruiz i Altaba A, Chen WS, Hoodless P, Prezioso VR, Jessell TM, Darnell Jr JE. The winged-helix transcription factor HNF-3 beta is required for notochord development in the mouse embryo. Cell. 1994;78:575–588. doi: 10.1016/0092-8674(94)90523-1. [DOI] [PubMed] [Google Scholar]
- Wiersdorff V, Lecuit T, Cohen SM, Mlodzik M. Mad acts downstream of Dpp receptors, revealing a differential requirement for dpp signaling in initiation and propagation of morphogenesis in the Drosophila eye. Development. 1996;122:2153–2162. doi: 10.1242/dev.122.7.2153. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Bhatt S, Chavrier P, Bravo R, Charnay P. Segment-specific expression of a zinc-finger gene in the developing nervous system of the mouse. Nature. 1989;337:461–464. doi: 10.1038/337461a0. [DOI] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes & Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L. MAD-related proteins in TGF-beta signalling. Trends Genet. 1996;12:493–496. doi: 10.1016/s0168-9525(96)30109-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng X, We R, Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF- beta response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]