

Abstract
The E2F transcription factor, a heterodimer of E2F and DP subunits, is capable of driving the G1–S transition of the cell cycle. However, mice in which the E2F-1 gene had been disrupted developed tumors, suggesting a negative role for E2F in controlling cell proliferation in some tissues. The consequences of disrupting the DP genes have not been reported. We screened for mutations that disrupt G1–S transcription late in Drosophila embryogenesis and identified five mutations in the dDP gene. Although mutations in dDP or dE2F nearly eliminate E2F-dependent G1–S transcription, S-phase still occurs. Cyclin E has been shown to be essential for S-phase in late embryogenesis, but in dDP and dE2F mutants the peaks of G1–S transcription of cyclin E are missing. Thus, greatly reduced levels of cyclin E transcript suffice for DNA replication until late in development. Both dDP and dE2F are necessary for viability, and mutations in the genes cause lethality at the late larval/pupal stage. The mutant phenotypes reveal that both genes promote progression of the cell cycle.
Keywords: Cell cycle, Drosophila, E2F, DP, DNA replication, S-phase
The regulation of the transition between the G1 gap phase and the S phase is a critical cell cycle control point, because duplication of the DNA commits a cell to division. This transition has been inferred to be controlled both at the post-transcriptional and transcriptional level (Murray and Hunt 1993; Nasymth 1996). In the yeast Saccharomyces cerevisiae the activation of the G1-cyclin (CLN)/CDC28 kinase complex drives the cells into S phase, whereas in mammalian cells activation of the cyclinE/cyclin-dependent kinase 2 (CDK2) complex is crucial for the onset of DNA replication. Thus, phosphorylation of key substrates by these kinase complexes is one regulatory component of the initiation of S-phase. Regulation also appears to occur at the level of transcription, however, because CLN1 and CLN2 and cyclin E transcripts accumulate in G1. In addition, many genes encoding proteins necessary for DNA replication are transcribed at the onset of S-phase.
E2F is a transcription factor whose activity has been linked to the G1–S transition in mammalian cells. This transcription factor is a heterodimer of the E2F protein and the DP protein, and there are several forms of either protein present in mammalian cells (Weinberg 1995). E2F/DP has been shown to activate the transcription of several genes needed for S-phase, and E2F/binding sites are found in the promoters of many other genes (Nevins 1992). In addition, E2F/DP binding may repress the transcription of some of these genes during G1 when the heterodimer is complexed with one of the family of pocket proteins, one member of which is the retinoblastoma protein (pRB); (Weintraub et al. 1992, 1995; Zwicker et al. 1996). E2F/DP is the focal point of a regulatory loop that links both transcriptional and post-transcriptional control of the cell cycle (for review, see Sherr 1996). Phosphorylation of pRB by either the cyclinD/CDK4,6, cyclinE/CDK2, or cyclinA/CDK2 kinases releases pRB, possibly enabling E2F/DP to function as a transcriptional activator. Because the cyclin E gene itself is controlled transcriptionally by E2F/DP, a positive feedback loop ensues (Ohtani et al. 1995; Botz et al. 1996; Geng et al. 1996).
It is not clear whether in vivo E2F/DP acts primarily as a positive or a negative transcription factor or whether it plays alternate predominant roles in different types of tissues. In addition, although the importance of transcriptional control of the onset of S-phase is inferred from its correlation with DNA replication, its significance relative to post-transcriptional control mechanisms is not established. Overexpression of E2F protein in mammalian cell culture drives cells into S phase (Johnson et al. 1993; Shan and Lee 1994; Lukas et al. 1996). However, transgenic mice lacking a functional E2F-1 gene exhibited phenotypes consistent with the E2F transcription factor acting either positively or negatively in different tissues (Field et al. 1996; Yamasaki et al. 1996). In these mice some tissues atrophy, but other tissues develop tumors. The consequences of disrupting the DP genes have not been reported.
Drosophila shares many of the components of the mammalian G1–S regulatory circuitry. Drosophila homologs to E2F, DP, an RB-like protein (RBF), cyclin E, CDK2, cyclin D, CDK4, and the p21 inhibitory protein (dacapo) have been identified (Richardson et al. 1993; Dynlacht et al. 1994; Knoblich et al. 1994; Ohtani and Nevins 1994; Hao et al. 1995; Sauer et al. 1995, 1996; de Nooij et al. 1996; Du et al. 1996a; Finley et al. 1996; Lane et al. 1996). The precise developmental control of the onset of S phase late in Drosophila embryogenesis makes it possible to define mutant phenotypes with high resolution and to infer the primary defect in cell cycle regulation (Smith and Orr-Weaver 1991; Smith et al. 1993). The first 13 divisions in the embryo occur in a rapid S–M cycle that is driven by maternal stockpiles of cell cycle regulators (for review, see Foe et al. 1993; Orr-Weaver 1994). After cellularization and the onset of zygotic gene expression, another three cell cycles take place with a G2 phase during which transcription occurs, but they lack a G1 phase. After mitosis 16, a detectable G1 phase appears for the first time during embryogenesis, and it marks the onset of the endo cell cycle for many of the larval tissues. In these cells S phase alternates with a gap phase, but mitosis does not occur, leading to polyteny. During the latter half of embryogenesis the cells that will form the polytene larval tissues undergo S-phase in a stereotypic developmental pattern. During the same developmental period the nervous system cells continue to go through mitotic divisions.
Cyclin E is essential for S phase late in Drosophila embryogenesis (Knoblich et al. 1994; Sauer et al. 1995). Mutations in cyclin E block DNA replication after mitosis 16, coincident with the first G1 phase. Presumably cyclin E is necessary for S phase earlier in embryogenesis but maternal stockpiles of the protein or transcripts suffice (Richardson et al. 1993). The onset of the requirement for cyclin E correlates with a change in its transcriptional regulation (Knoblich et al. 1994). During the postblastoderm divisions the levels of cyclin E transcript are constitutively high. After mitosis 16 they are down-regulated in all of the tissues except the nervous system, and transcripts accumulate before S-phase in the endo cycle late in embryogenesis. The same transcriptional pattern observed for cyclin E also occurs for several genes encoding replication functions: proliferating cell nuclear antigen (PCNA), ribonucleotide reductase 1 (RNR1), RNR2, and pol α (Duronio and O’Farrell 1994). These observations led to a model in which transcriptional regulation is crucial in controlling the G1–S transition in the endo cycle, with cyclin E being the critical target (Duronio and O’Farrell 1994, 1995; Sauer et al. 1995).
To identify regulatory genes needed for G1–S transcription, we screened for mutants defective in the induction of PCNA transcription late in embryogenesis. We found five mutations in a gene essential for PCNA and RNR2 transcription and demonstrated that these are alleles of the Drosophila DP gene. We show that the dDP gene is essential for viability. Despite the pronounced effect on G1–S transcription, DNA replication still occurs and lethality is late in development. We find that mutations in dE2F give similar phenotypes to those in dDP; E2F/DP-dependent G1–S transcription is disrupted, but S-phase takes place and the animals survive to late larval/early pupal stages.
Results
Screen for genes required for S-phase transcription in the embryo
To recover genes necessary for the transcription of S-phase genes late in Drosophila embryogenesis, we used the trancription pattern as the assay in a genetic screen. The PCNA and RNR2 genes were used because they are transcribed in a pattern that mimicks that observed by bromodeoxyuridine (BrdU) labeling of late Drosophila embryos (Duronio and O’Farrell 1994). The E2F/DP recognition sites in the PCNA promoter are essential for its expression (Yamaguchi et al. 1995). We devised a method for in situ hybridization to detect transcripts on large numbers of independent mutagenized lines. Embryos were collected from lines established from single, mutagenized second chromosomes, aged 8–15 hr, and hybridized in situ to a PCNA riboprobe (see Materials and Methods, Fig. 7, below). This developmental stage was chosen because between 8 and 15 hr there is a G1 phase with a regulated transcriptional program. In addition, S-phase transcription can be observed both in cells undergoing endo cycles as well as in the mitotically dividing cells of the nervous system.
Figure 7.
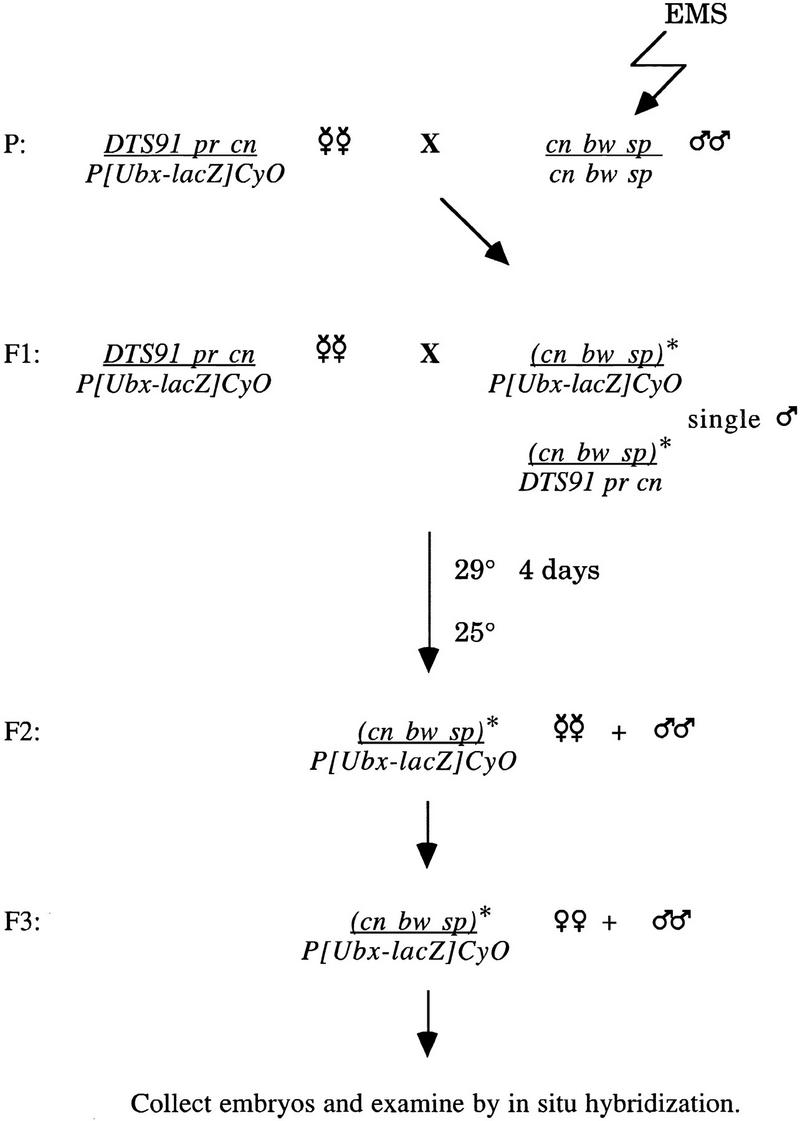
Screen for mutations that disrupt G1–S transcription. Isogenized cn bw sp flies were mutagenized with EMS. The DTS91 chromosome was used to select for flies bearing the mutagenized chromosome over a balancer expressing lacZ. From the F3 adults, 8- to 15-hr embryos were collected and hybridized in situ with PCNA and lacZ riboprobes. Embryos that did not express lacZ were homozygous for the mutagenized second chromosome. F3 lines that gave rise to progeny with aberrant PCNA expression were maintained.
From 3010 mutated second chromosome lines, seven lines were recovered that failed to express the PCNA transcript at normal levels in 8- to 15-hr embryos. In all of these lines PCNA transcript was reduced in both the mitotic and endo cycle tissues. As a secondary test, the RNR2 transcript was also affected in the mutant embryos. Complementation tests showed that the mutants fell into two complementation groups. One group, containing two alleles, showed a reduction in PCNA and RNR2 transcript levels. The second group of five mutations had a stronger reduction in PCNA or RNR2 transcripts to nearly undetectable levels late in embryogenesis. This latter complementation group was subjected to further analysis.
Mutations in the Drosophila DP gene
The expression of PCNA and RNR2 in the strong complementation group was almost identical to that described previously for mutations in the dE2F gene (Fig. 1) (Duronio et al. 1995). The one distinction was that in the new complementation group there was a higher level of RNR2 transcript uniformly present throughout the epidermis. This was a weak signal that gave the appearance of a higher background in the homozygous mutant embryos (data not shown).
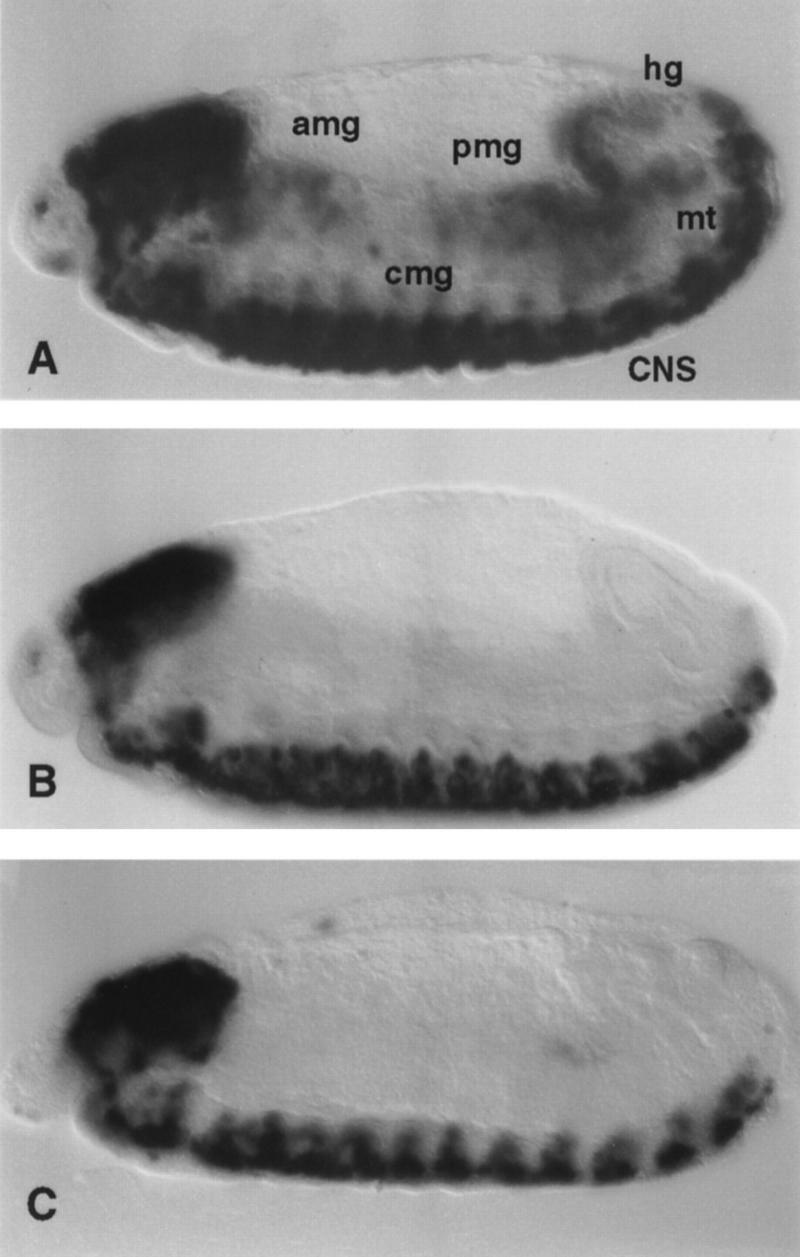
Figure 1.
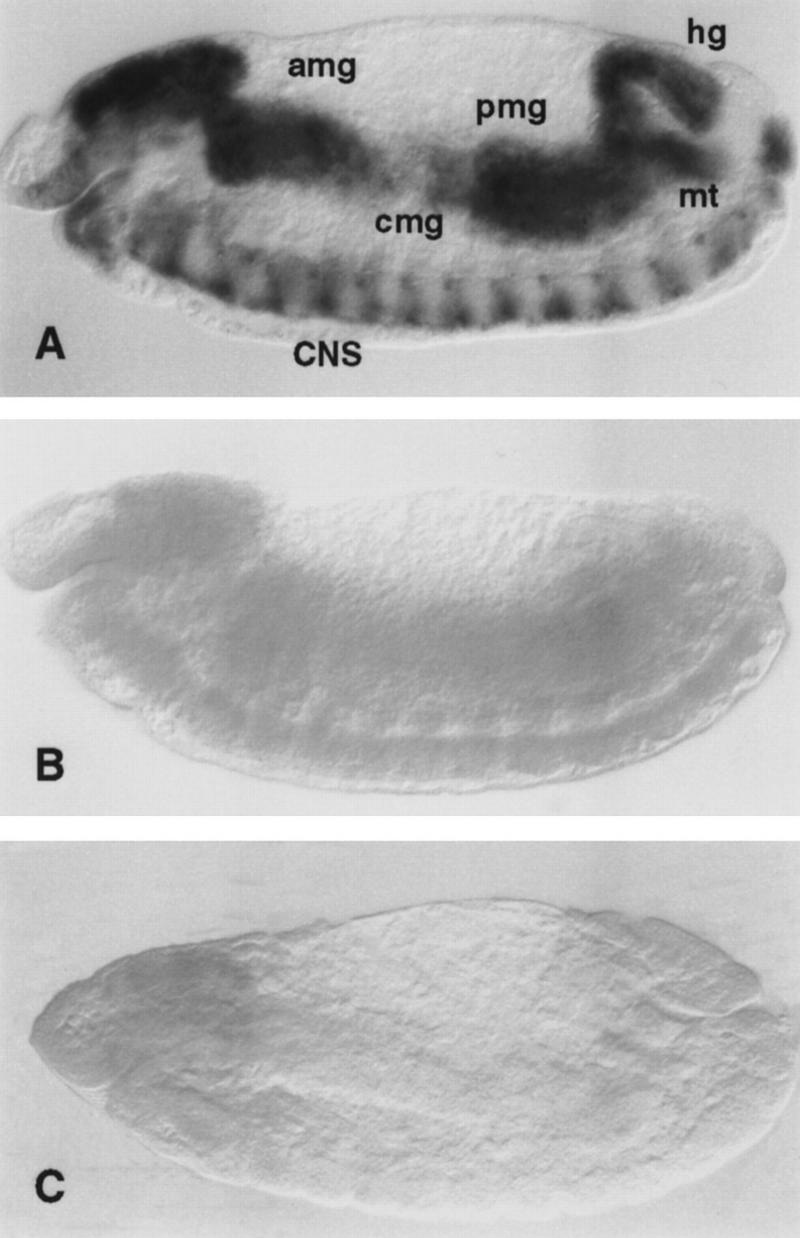
PCNA expression in dDP and dE2F mutant embryos. PCNA transcripts were detected by whole-mount in situ hybridization. (A) In wild-type 10.5-hr embryos PCNA is expressed in the mitotically proliferating central nervous system (CNS), in the endoreplicating anterior midgut (amg), central midgut (cmg), posterior midgut (pmg), hindgut (hg), and malpighian tubules (mt). (B) In 10.5-hr homozygous dDPa4 mutant embryos PCNA is expressed at greatly reduced levels in the CNS and is not detectable in the endodomains. (C) dE2F91 homozygous 10.5-hr embryos show a similar phenotype. In some embryos a low level of PCNA expression can be seen in the CNS, but expression is absent in the endodomains.
A homolog to the mammalian DP protein, the other subunit of the E2F transcription factor, was identified in Drosophila (dDP) and mapped to the second chromosome. Mutations in the dDP gene had not been identified (Hao et al. 1995). Given the similarity between the dE2F phenotype and that of our mutants, we tested whether a deficiency known to delete the dDP gene also uncovered our complementation group. All five mutations were lethal in trans to each of the three deletions that remove dDP [Df (2R)vg-B (Hao et al. 1995); Df (2R)vg-33 and Df (2R)vg-56 (R. Duronio, pers. comm.)]. Furthermore, in situ hybridization of embryos transheterozygous for the mutation and the Df(2R)vg-B deficiency gave the mutant phenotype, undetectable PCNA expression in late embryos.
To confirm that the mutations mapped to the dDP gene, we sequenced the dDP gene from two of the mutant lines. Because the mutations cause pupal lethality (see below) we were able to isolate genomic DNA from larvae transheterozygous for the mutation and a deficiency. A region of 1400 bp that encompasses the regions of the protein conserved with mammals was amplified by PCR from mutant larval genomic DNA, and the PCR product was sequenced directly. The sequenced region contains several important motifs, including the DNA-binding region, the DEF box that is predicted to be required for DP/E2F heterodimerization, and three other highly homologous regions named DP-conserved box 1 (DCB1), DCB2, and negatively charged box (NCB) (Fig. 2) (Dynlacht et al. 1994; Hao et al. 1995). To recognize polymorphisms between our strains and those used for the published sequence, we also sequenced the same region from transheterozygous adults from two unrelated lines recovered from the screen. Within the region we sequenced there are six introns ranging in size from 56 to 68 bp (Fig. 2). There is a polymorphism in our strains, changing the histidine at position 275 to a leucine. This histidine residue is conserved between humans, mice, and Xenopus, and is present in the DCB2 box (Dynlacht et al. 1994; Hao et al. 1995). However, it is not essential given that strains containing a leucine at this position have a wild-type phenotype .
Figure 2.
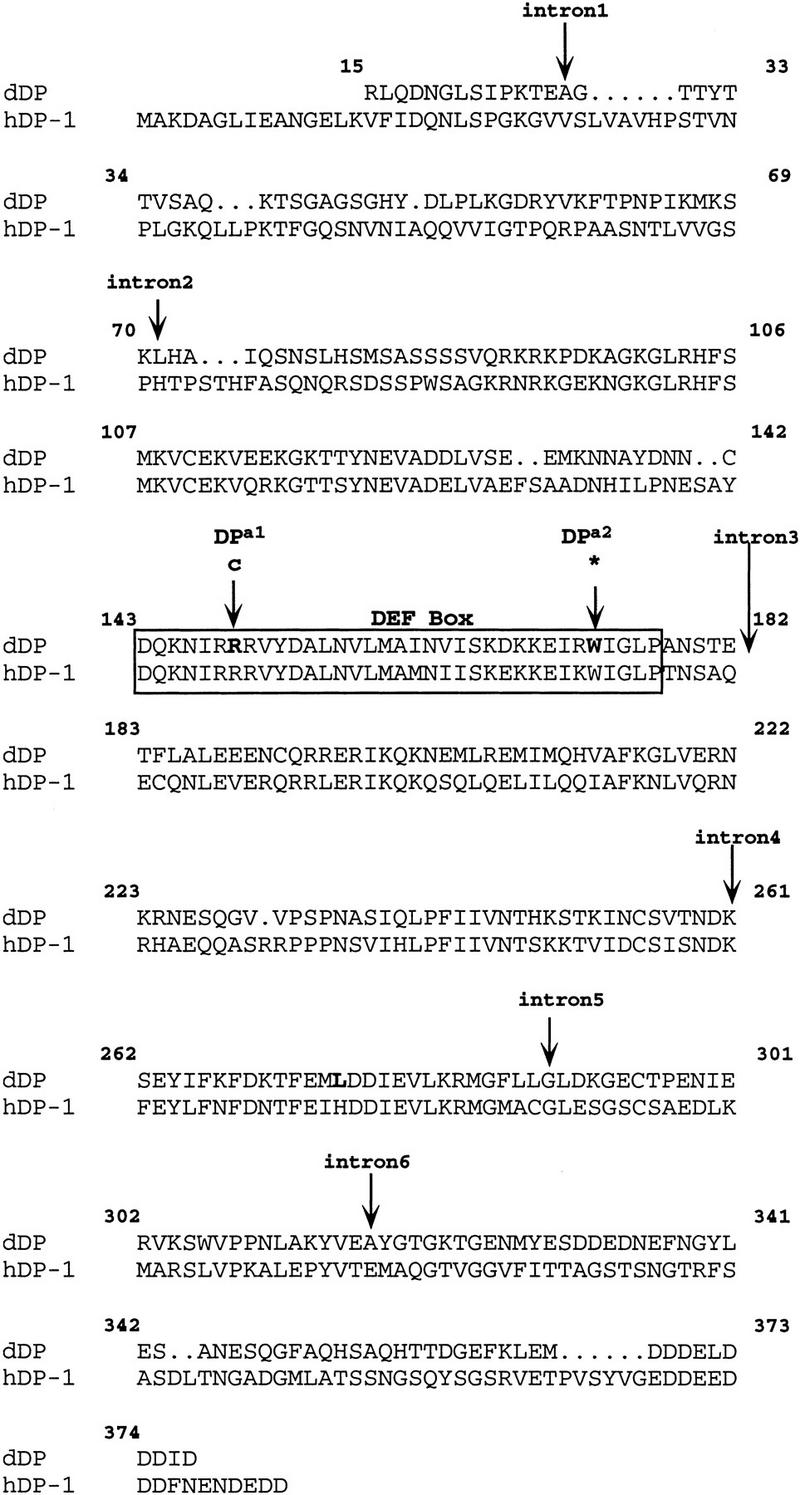
Amino acid changes in the dDPa1 and dDPa2 mutant lines. The wild-type Drosophila dDP sequence from a 1400-bp region PCR amplified from genomic DNA is compared with the human DP-1 sequence. The amino acid numbering of the Drosophila sequence is that of Dynlacht et al. (1994). Amino acid changes in dDPa1 and dDPa2 are indicated, and both of these changes occur in the DEF box (outlined). The region of genomic DNA sequenced contains six small introns: the first, in amino acid 28, is 57 bp; the second, in amino acid 71, is 63 bp; the third, after amino acid 182, is 68 bp; the fourth, in amino acid 261, is 60 bp; the fifth, in amino acid 289, is 63 bp; and the sixth, in amino acid 317, is 56 bp. The sequence shown does not include the first 14 amino acids of the amino terminus reported by Dynlacht et al. (1994). The polymorphism present between our sequence and that of Dynlacht et al., is shown in boldface type.
We found that both of the mutations were associated with codon changes within the dDP open reading frame (ORF) (Fig. 2). The first mutation is a C → T nucleotide transition that changes Arg149 to a Cys. This residue lies within the DEF box in a block of amino acids that is highly conserved between DP and E2F, and it is conserved between Drosophila, mice, and humans (Dynlacht et al. 1994; Hao et al. 1995). With sequence confirmation that the mutation disrupted the dDP ORF, this allele was named dDPa1. The second mutation is a G to A nucleotide transition that converts Trp173 to a stop codon. The resulting truncated protein would lack part of the DEF box as well as the three highly conserved domains, DCB1, DCB2, and NCB. This allele was named dDPa2. The molecular data combined with the genetic complementation data on the five mutations confirm that this complementation group is the Drosophila dDP gene. Consequently, the other mutations have been designated dDPa3, dDPa4, and dDPa5.
The role of dDP and dE2F in G1–S progression
In mammalian cells, the E2F transcription factor triggers the expression of essential S-phase genes, and the transcription of these genes correlates with progression into S phase. In Drosophila, mutations in the dE2F gene were reported to block the G1–S transition and eliminate detectable DNA replication late in embryogenesis (Duronio et al. 1995). We tested whether the mutations in dDP also would block entry into S phase.
To determine whether dDP is required for G1–S progression, we analyzed BrdU incorporation after the first G1 phase in Drosophila embryogenesis. Normally this G1 phase is followed by polytene replication in an invariant tissue-specific pattern in the embryo (Fig. 3A) (Smith and Orr-Weaver 1991). Embryos (8–15 hr) were collected from dDP mutant lines and labeled with BrdU during a 40-min interval. To our surprise we detected BrdU incorporation in embryos homozygous for mutant alleles of dDP, and the spatiotemporal pattern of BrdU incorporation in these embryos was normal (Fig. 3B).
Figure 3.

S-phase occurs in dDP and dE2F mutants. DNA replication was assayed by BrdU and shown for 10.5-hr embryos labeled for either 40 min (A–C,H), 20 min (D), or 10 min (E–G) followed by antibody staining detected by the horseradish peroxidase (HRP) reaction. The darkly staining cells are those that incorporated BrdU during the pulse. The central nervous system (CNS), anterior midgut (amg), central midgut (cmg), posterior midgut (pmg), malpighian tubules (mt), and hindgut (hg) incorporate BrdU. The characteristic replication pattern can be seen for the 40-min BrdU pulse for dE2F7172/+ embryos (A) which have a wild-type phenotype, homozygous mutant dDPa4 (B) and dE2F91 (C) embryos, for the 20-min pulse in the homozygous dE2F7172 (D), and for the 10-min BrdU pulse for dE2F7172/+ (E) embryos. The 10-min BrdU pulse for the dDPa4 and dE2F91 mutants shows reduced labeling in both the mitotic (CNS) and polytene cells (F,G), but replication is still detected. (H) There is no detectable replication in the endodomains (arrows) of a cyclin EPZ5 homozygous mutant after a 40-min BrdU pulse.
The normal replication observed in the dDP mutants prompted us to examine DNA synthesis in the dE2F91 null allele as well as the dE2F7172 mutant. The dE2F91 mutation is a stop codon early in the dE2F coding sequence (Duronio et al. 1995). The dE2F7172 mutation is also likely to be a null because dE2F protein is not detectable in homozygous embryos (Asano et al. 1996). Furthermore, the failure to observe protein in these mutant embryos implies that maternal pools of the E2F protein do not persist in late embryos. Strikingly, DNA replication was observed in embryos mutant for either of the dE2F alleles (Fig. 3C,D), although the intensity of BrdU incorporation was slightly diminished. As a control, we repeated the 40-min BrdU pulse on embryos homozygous for cyclin EPZ5. As expected, BrdU incorporation was not detectable in the polytene tissues of cyclin E mutant embryos (Fig. 3H, arrows). We conclude that although the bursts of E2F-dependent G1–S transcription are not evident in dDP and dE2F mutant embryos, DNA replication still occurs.
Although DNA replication takes place in dDP and dE2F mutant embryos, two observations suggest that the rate of replication is slowed. First, if a 10-min pulse of BrdU labeling was used, BrdU incorporation was reduced in both polytene and neural tissues of homozygous dE2F or dDP embryos (Fig. 3F,G), whereas replication was normal with this short pulse in heterozygous controls (Fig. 3E). The previous studies that concluded DNA replication was undetectable in dE2F mutant embryos used a short pulse of BrdU labeling (Duronio et al. 1995). However, even with a 10-min BrdU pulse the level of labeling in dE2F and dDP mutants is much higher than that obtained in the endodomains with a 40-min pulse of the cyclin E mutant (Fig. 3F–H). Second, in the dE2F mutants the developmental time of the onset of the later polytene S phases appeared delayed. Replication persisted in the anterior and posterior midgut at a developmental stage when only the central portion of the midgut normally replicates. In the mutant embryos the second round of polytene replication in the central midgut was not observed (data not shown), suggesting it was delayed until after cuticle deposition when antibody detection is no longer possible.
Developmental phenotypes of dE2F and dDP
To evaluate whether the E2F/DP transcription factor functions to promote entry into S phase or suppress hyperproliferation, we determined the furthest developmental point reached by analyzing the lethal phase of both the dE2F and the dDP mutants. Because S-phase occurs during embryogenesis in dDP and dE2F mutants, we did not expect the mutations to cause embryonic lethality, although the dE2F mutations were described previously as being embryonic lethal (Duronio et al. 1995). We tested whether the dDP and dE2F mutants were embryonic lethal by scoring whether the embryos hatched as first instar larvae. For all five of the dDP alleles heterozygous mutant/+ females were crossed to Df/+ males. At least 300 eggs were collected and scored, but no embryonic lethality of the dDP alleles in trans to the deficiency was observed. In the same way, we tested the dE2F91 and dE2F7172 alleles in trans to a deficiency that uncovers dE2F. The alleles were also examined in trans to each other. In each experiment 500 eggs were collected and scored, and there was no embryonic lethality. We tested dE2F91 homozygous embryos and found the homozygous chromosome to cause embryonic lethality. However, this is clearly the result of other mutations on the chromosome, as both the dE2F91 and dE2F7172 in trans to a deficiency or each other are not embryonic lethal.
We examined the role of dE2F in larval and pupal development to determine whether the gene was essential and whether the dE2F mutations caused any defects in polytenization or cell proliferation. In Drosophila most of the larval cells do not undergo mitosis after completion of embryogenesis but grow from polytene replication. However, the imaginal cells that will form the adult body remain diploid. Thus, defects in the endo cell cycle producing polytene cells would result in small larvae, whereas defects in proliferation would be manifest after pupation when the imaginal cells differentiate to produce the adult body. Larvae and pupae mutant for dE2F were identified by the absence of a dominant marker (see Materials and Methods). Approximately 240 total larvae were examined, and 63% of the dE2F91/dE2F7172 survived to third instar. Of the third instar larvae, 54% initiated pupation. Because the mutant animals die as late larvae or early pupae, dE2F is clearly essential for development. The late lethality of the dE2F mutant is consistent with the high levels of dE2F protein observed in wild-type second and third instar larval extracts (Brook et al. 1996).
Although the dE2F mutant animals survive through larval life, we observed a dramatic delay in larval growth. It took between 288 and 432 hr for the dE2F mutant larvae to pupate, compared to 120 hr for heterozygous sibling controls. Five days after egg laying (AEL) the dE2F mutant larvae were very sluggish and much smaller in size than their wild-type counterparts (Fig. 4A,B). The polytene salivary gland and diploid imaginal discs could not be identified in the 5-day-old dE2F mutant larvae, presumably because they were so small. The brains were also greatly reduced in size as compared to wild type (Fig. 4 E,F). The size of the dE2F mutant larvae increases over time (Fig. 4B,C), and the internal tissues approached wild-type size (Fig. 4F,G; data not shown). Therefore, DNA replication can occur during this larval period, but it is slow. Replication in the absence of dE2F is further evidenced by the formation of banded polytene salivary gland chromosomes in some of the 12- to 18-day larvae (Fig. 4H). Although the polytene chromosomes from the dE2F mutant larvae were smaller and more fragile than normal they were clearly visible. Thus, we conclude that S phase occurs in the absence of dE2F, but dE2F is necessary for timely replication and growth.
Figure 4.

dE2F is required for wild-type growth in mitotic and polytene tissues. (A–C) All the larvae are at the same magnification. (A) dE2F7172/+ larva at 5 days AEL. (B) dE2F91/dE2F7172 mutant larva 5 days AEL. (C) dE2F91/dE2F7172 mutant larva 13 days AEL with two posterior melanotic pseudotumors. (D) A magnification of posterior pseudotumors (arrows) shown in C. (E–G) All the brains are at the same magnification. (E) Brain of a dE2F7172/+ larva at 5 days AEL. (F) Brain of a dE2F91/dE2F7172 mutant larva at 5 days AEL. (G) Brain of a dE2F91/dE2F7172 mutant larva at 9 days AEL. (H) Polytene chromosomes from the salivary glands of a dE2F91/dE2F7172 mutant larva at 13 days AEL.
In addition to the growth delay, the dE2F mutant larvae had another striking phenotype, melanotic pseudotumors were formed (Fig. 4C,D). Melanotic tumors are groups of cells within the larvae that are recognized by the immune system and encapsulated in melanized cuticle (Sparrow 1978; Watson et al. 1991). We refer to them as pseudotumors to emphasize that they are not necessarily the consequence of hyperproliferation but can be abnormal cells recognized by the immune cells. Small pseudotumors were first observed in the dE2F mutants 7 days AEL, and these early pseudotumors grew and darkened as the larvae aged. In the dE2F mutants that initiated pupation, numerous additional small pseudotumors formed.
We compared the lethal phenotype of the dDP alleles over a deficiency to that of the dE2F mutations (Table 1). Unlike the dE2F mutants, the larval growth of the dDP mutants was not delayed dramatically. The lethality was largely pupal, and some melanotic pseudotumors were observed in the early pupae. The dDP alleles ranged in the severity of the phenotype they produced (Table 1). The strongest phenotype was observed with dDPa5, which resulted in late larval lethality when in trans to a deficiency. The dDPa2 and dDPa4 mutants in trans to a deficiency survived to the pupal stage. We think that the pupal stage is the lethal phase, and that the dDPa5 mutant is unusual. The dDPa5 larval lethality is either attributable to another mutation on the chromosome that enhances the phenotype, or because this allele is antimorphic. The phenotype of the other alleles in trans to dDPa5 was stronger than in trans to the deficiency.
Table 1.
Developmental phenotypes of dDP mutants
| Genotype
|
Lethal phasea
|
Phenotype
|
|---|---|---|
| dDPa1/Df(2R)vg-B | adult flies 11%–20%; adults in pupal case 80%–89% | rough eyes, incomplete wing vein, thin and short bristles, etched tergites, more female flies than males, females and males sterile, females lay eggs with thin chorions |
| dDPa3/Df(2R)vg-B | adults in pupal case | rough eyes, etched tergites, thin and short bristles |
| dDPa2/Df(2R)vg-B dDPa4/Df(2R)vg-B | adults in pupal case 50%; pupae 50% adults in pupal case 50%; pupae 50% | kidney-shaped rough eyes, thin and short bristles on thorax and head, severe abdominal defects |
| dDPa5/Df(2R)vg-B | late larval lethal |
Lethal phase is the furthest developmental stage reached by the dDP mutants.
Approximately half of the dDPa2/Df pupae reached adulthood in the pupal case (Table 1). These adults struggled to eclose but ultimately died. Organisms dissected from the pupal case had essentially normal heads and thoraxes. However, their abdominal defects were severe. This is informative as the head and thorax are derived from imaginal discs, whereas the abdomen arises from the abdominal histoblast nests. The imaginal discs proliferate during larval stages, but the abdominal histoblast nests proliferate during pupal development. Thus, pupal lethality may result from a defect in abdomen formation that occurs during pupal development.
The weakest allele, dDPa1, was semilethal in trans to a deficiency (Table 1). The recovered adults had rough eyes and wing vein defects. These phenotypes are diagnostic of compromised mitotic proliferation. They also had thin and short bristles that indicate a defect in polytene replication, because the cells that give rise to the bristle shaft and socket endoreplicate (Lees and Waddington 1942). We conclude that dDP, like dE2F, provides an essential function for the development of the organism. The eye and bristle defects indicate that dDP is required for normal development in both mitotic and endo cycle cells.
Genetic interaction between dDP and dE2F
To demonstrate that the late lethality and developmental phenotypes associated with the dDP mutants were attributable to loss of dDP function rather than a synthetic effect with other loci, we tested whether the lethality could be rescued by expressing the wild-type dDP gene under a heat shock promoter (hsp). We introduced an hsp70–dDP transgene into the dDPa1 background (Table 2) (Duronio et al. 1996). At 25°C we observed a partial rescue of the dDPa1/Df lethality attributable to basal expression of dDP from the hsp70 promoter. The recovered dDPa1/Df; hs–dDP/+ males were fertile. The recovered adults, however, did have rough eyes, wing vein, and bristle defects and were female sterile. The effect of induced dDP expression (37°C heat treatment) on the dDP mutants was even more dramatic (Table 2). The lethality of dDPa1/Df was completely rescued, and all the developmental phenotypes were suppressed. The rescue confirms that the pupal lethality, male and female sterility, rough eye, wing vein, and bristle defects are all attributable to loss of dDP function.
Table 2.
Rescue of dDP mutants by overexpression of dDP and dE2F
| Genotype of recovered adults
|
Recovered/expected
|
||
|---|---|---|---|
| 25°C
|
37°C
|
||
|
11%a (males and females sterile) | 0%d | |
|
42%b (males fertile; females sterile) | 100 %e (males fertile; females semifertile) | |
|
37%c (males and females sterile) | 18%f (males and females sterile) | |
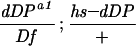
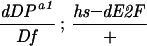
w; Df(2R)vg-56/CyO females were mated to w; dDPa1/CyO; P[w+;hs–dDP]/+ or w; dDPa1/CyO; P[w+;hs–dE2F]+ males at 25°C. Eggs collected from the crosses were either allowed to develop at 25°C or received heat shock treatments at 37°C.
The recovered adults of the indicated genotype were expected to be one-quarter of the CyO progeny according to Mendelian ratios (see Materials and Methods):
9/324;
34/324;
20/218;
0/184;
51/184;
12/266. The numerator is the number of recovered adults of the indicated genotype. The denominator is the number of the CyO progeny. To calculate the percentage, the ratios were multiplied by 4.
Because ectopic expression of dE2F and dDP in the Drosophila eye results in excess cell proliferation (Asano et al. 1996; Du et al. 1996b), we examined the effect of dDP mutations on eye development in more detail. We used scanning electron microscopy (SEM) to analyze the eyes of dDPa1/dDPa2; hs–dDP/+ with and without heat shock. The eyes of dDP mutant flies carrying the uninduced hs–dDP transgene were rough (Fig. 5B). The reduced size of the eye, missing, and disorganized ommatidia, as well as stunted, missing, and disorganized bristles suggested a proliferation defect in the dDP mutant (Fig. 5B). Strikingly, flies of the same genotype that underwent daily heat shock treatments had normal eyes (Fig. 5A,C). dDPa1 in trans to a deficiency resulted in the same rough eye phenotype as observed for dDPa1/dDPa2 flies at 25°C, and the rough eye was rescued by induced ectopic dDP expression (data not shown). Because the dDPa2 allele exhibits the same strength phenotype as a deficiency, it behaves genetically as a null allele.
Figure 5.

Overexpression of dDP rescues the eye phenotype of dDP mutants. Shown are SEMs of Drosophila compound eyes at 200× magnification. The genotypes are: (A) Df(2R)vg-56/+ . (B) dDPa2/dDPa1 with a hs–dDP transgene raised at 25°C. The reduced size of the eye and aberrant morphology are indicative of a proliferation defect in the dDP mutant. (C) dDPa2/dDPa1 with a hs–dDP transgene raised at 25°C with daily heat shocks at 37°C. Overexpression of dDP completely rescues the size and morphology of the dDP mutant eye.
Having shown that heat shock dDP rescues the dDP mutants we defined the developmental period during which ectopic dDP expression is capable of rescuing the lethality of the dDP mutants. Females heterozygous for dDPa2 were mated to heterozygous dDPa1 males carrying the hs–dDP transgene. The heat shock regimen began immediately after the 24-hr egg collection was completed or 5–6 days later (late larval/early pupal life). Both treatments yielded the same results, 100% rescue of dDPa1/dDPa2 mutant animals. Thus, the late lethality of dDP mutants is not a manifestation of a defect in the early development of the organism, but rather it stems from defects in larval/pupal life.
We tested whether the lethality of dDP mutants was a result of disrupting the E2F/DP heterodimer by asking whether overexpression of dE2F could suppress the dDP mutant phenotype. The dE2F transgene under the inducible heat shock promoter was crossed into the dDPa1 and dDPa2 background. The dDPa1 allele was chosen because this mutation changes a single conserved residue in the E2F/DP heterdimerization domain (Girling et al. 1993; Hao et al. 1995). The dDPa2 mutation truncates the protein and is predicted to delete regions necessary for E2F binding, providing a useful comparison for the dDPa1 mutation. Mild overexpression of dE2F at 25°C resulted in significant rescue of the dDPa1/Df mutant phenotype (Table 2). At 37°C the rescue conferred by the ectopic expression of dE2F is dampened (Table 2). This may result either from high levels of ectopic dE2F expression being detrimental (Asano et al. 1996), or it may be attributable to fewer dDPa1/Df adult escapers arising at 37°C. In contrast to the effect on dDPa1, overexpression of dE2F provided no rescue of the dDPa2 mutants (data not shown). The ability of ectopic dE2F to rescue phenotypes in the dDPa1 but not the dDPa2 mutants suggests that despite the alteration in the DEF box of the dDPa1 allele, the mutant DP protein is still capable of binding E2F. This observation also indicates that the dDP phenotypes are a result of a failed E2F/DP transcription factor activity.
Genetic interactions between the E2F/DP heterodimer and cyclin E
We investigated whether dDP is necessary for cyclin E expression. In embryos that were dDPa1/Df, cyclin E expression was not detectable in the endodomains but present in the central nervous system (CNS) (Fig. 6A,B), a pattern comparable to that seen previously in homozygous dE2F91 mutant embryos (Fig. 6C) (Duronio and O’Farrell 1995). Because the levels of cyclin E transcripts are so much higher in the nervous system than in the endodomains, it is difficult to assess whether the transcript levels are reduced in the mutants in the CNS.
Figure 6.
cyclin E expression in dDP and dE2F mutant embryos. cyclin E transcripts were detected by whole-mount in situ hybridization. (A) In wild-type 10.5-hr embryos cyclin E is expressed at high levels in the mitotically proliferating CNS and at low levels in the endodomains. (The endodomains are labeled as in Fig. 1). (B) In dDPa1/Df 10.5-hr mutant embryos cyclin E is expressed at high levels in the CNS but is undetectable in the endodomains. (C) dE2F91 homozygous mutant 10.5-hr embryos show the same phenotype. cyclin E is expressed at high levels in the CNS but is undetectable in the endodomains.
The cyclin E mutant phenotype is more severe than that of the dE2F and dDP mutants, although cyclin E transcription requires the E2F/DP transcription factor. In contrast to the near-normal pattern of replication seen in dE2F and dDP mutant embryos, DNA replication was not detected in the endodomains of embryos homozygous for cyclin EPZ5 (see Fig. 3H). In this cyclin E allele S-phase continued in the CNS in late embryos (Fig. 1H). No embryonic lethality resulted from the cross of cyclin EPZ5 heterozygous mothers to fathers heterozygous for a cyclin E deficiency, rather the cyclin EPZ5/Df transheterozygotes died as early first instar larvae. Because BrdU incorporation is not seen in cyclin E mutant embryos in the endodomains, this shows that the embryonic endo cycles are not required for hatching.
We tested whether persistent maternal levels or low levels of zygotic expression of cyclin E transcript accounted for the viability of dE2F mutant larvae. In contrast to the late larval/early pupal lethality normally seen with dE2F91/dE2F7172, reducing the maternal dosage of cyclin E dramatically enhanced the lethality. This resulted in early larval lethality, with only 5% of the dE2F mutant larvae surviving until the second to small third instar larval stage. Reducing the dosage of the cyclin E gene in the father also enhanced the dE2F lethality, but to a lesser extent. In this cross 30% of the cyclin EPZ5/+;dE2F91/dE2F7172 progeny survived until the second to third instar larval stage. The pronounced effect of reducing the gene dosage in the mother indicates that persistent maternal pools of cyclin E transcript or protein permit viability of dE2F mutant animals. In addition, zygotically provided transcripts also contribute, but we cannot distinguish whether this is attributable to persistence of constitutively expressed transcripts or cyclic transcription below our detection limit.
Discussion
From a genetic screen to identify mutants that fail to undergo the S-phase transcriptional program during embryogenesis we recovered five mutations in the Drosophila DP gene. The mutant phenotype reveals that dDP, like dE2F, is required for viability in Drosophila and that the gene has an essential role in vivo. The mutant phenotypes of dDP and dE2F provide insight into the functions these genes provide in controlling the cell cycle during development.
The E2F/DP transcription factor promotes S phase in vivo
Analysis of the activity of E2F in mammalian cell culture showed that the transcription factor was capable of driving cells into S-phase when overexpressed and that this could lead to apoptosis (Johnson et al. 1993; Shan and Lee 1994; Heibert et al. 1995; Lukas et al. 1996). This observation was consistent with results demonstrating that E2F could activate the transcription of a number of genes needed for the G1–S transition and the onset of S-phase. However, it was also shown that when complexed with pRB the E2F transcription factor could repress the transcription of some of these genes in G1 (Weintraub et al. 1992, 1995; Zwicker et al. 1996). In transgenic mice in which the E2F-1 gene was disrupted tumors developed (Field et al. 1996; Yamasaki et al. 1996). These latter two observations raised the possibility that in some tissues the E2F transcription factor could act primarily as a tumor suppressor to repress progression of the cell cycle.
The phenotypes of the Drosophila E2F and DP mutants reveal that the E2F transcription factor indeed plays a significant role as an activator of S phase in vivo. It was surprising that mutations in either gene have almost no effect on BrdU incorporation during embryogenesis. However, the rate of nucleotide incorporation and DNA replication appears to be slower in both mutants, and the slow larval growth rate in dE2F mutants is particularly striking. Because larval growth occurs by increasing ploidy of the larval tissues by the G–S endo cycle, these phenotypes reflect a compromised ability to undergo S phase in the mutants.
In addition to the effects on polytene tissues, mitotically proliferating cells are affected dramatically by mutations in both dE2F and dDP. In the dE2F mutants that pupariate, either a very small adult body is formed inside the pupal case or an adult body is not formed at all. This is consistent with a defect in proliferation that reduces the number of imaginal disc cells and abdominal histoblasts that give rise to adult structures. Previously, it was observed that mutations in dE2F reduce the rate of cell growth in clones in the eye (Brook et al. 1996). The interpretation that mutations in the dE2F and dDP genes disrupt the diploid cell cycle in the imaginal cells is supported by experiments demonstrating that overexpressing dE2F and dDP induced S-phase and excess cell proliferation in the eye (Asano et al. 1996; Du et al. 1996b).
The defects in diploid larval cells most likely arise from aberrant cell proliferation caused by the dE2F and dDP mutations. Although we do not know the cell cycle stage affected, the demonstrated effects on polytene larval cells imply that it is the G1–S transition. Because the imaginal discs are present in dE2F mutants yet fail to differentiate to produce adult tissues, it is also likely that E2F has a role in post-mitotic differentiation. Such a role was demonstrated previously by clonal analysis in the eye disc (Brook et al. 1996). A failure in differentiation of the imaginal cells could be a contributing factor in the dDP mutant phenotypes as well.
Although the mutant phenotypes suggest that the predominant function of the E2F transcription factor in Drosophila is to promote progression of the cell cycle, two observations raise the possibility that it may have an inhibitory role in some tissues. First, in dDP mutant embryos a low level of RNR2 transcripts is present in the epidermis, and it may result from a failure to repress transcription. Second, melanotic pseudotumors develop in the dE2F and dDP mutants. We refer to these as pseudotumors because they arise in Drosophila larvae when the lamellocytes of the immune system recognize aberrant cells, surround them, and secrete a cuticle that melanizes and becomes black (Sparrow 1978). Thus, the clearest cause of melanotic tumors is an alteration of the cell surface. In the dE2F and dDP mutants this could result from perturbation of the differentiation program or apoptosis. There is evidence in Drosophila suggesting that overproliferation may contribute to melanotic tumor formation (Watson et al. 1991, 1992, 1994; Bryant et al. 1993). Although it is possible that the melanotic pseudotumors in the dE2F and dDP mutants result from hyperproliferation, we did not observe overproliferation in the larval imaginal tissues. Apoptosis may occur in some tissues in the dE2F and dDP mutants, leading to melanotic tumors. Further analysis will be required to determine whether E2F represses the cell cycle in some developmental contexts and to distinguish between these potential mechanisms for the formation of melanotic pseudotumors in the mutants.
Does dE2F have a function that is independent of dDP?
Although both the dDP and dE2F mutants show late lethality, there are differences in the phenotypes resulting from mutations in the two genes. The dE2F mutant animals have slower larval growth than dDP mutants, and the dDP mutants develop further as pupae. We think that the difference in strength of the phenotypes reflects the biological system rather than strength of the dE2F and dDP alleles, because we analyzed null alleles of both genes. The dE2F91 mutation is a truncation after amino acid 31 (Duronio et al. 1995), and dDPa2 is a truncation at the end of the DEF box. In mammalian cells truncation of DP after the DEF box abolishes both the E2F and DNA-binding ability (Wu et al. 1996). Given the high degree of conservation between the mammalian and Drosophila proteins, dDPa2 is likely to ablate DP activity.
One explanation for the less severe phenotype of the dDP mutations is that there is a redundant activity for dDP but not for dE2F. This could be because there are additional family members in the Drosophila genome, and these may complement the dDP defect better than that of the dE2F mutants. Alternatively, the maternal stockpiles of the dDP gene may persist longer than those of dE2F. DP protein is more easily detected in embryo extracts than E2-F and may be present at higher levels (N. Dyson, pers. comm.).
Another explanation for the more severe effects exhibited by the dE2F mutants is that dE2F plays biological roles independent of dDP. Possibly the dE2F protein can act as a homodimer. The rate of larval growth is influenced greatly by nutritional signals (Poodry and Woods 1990). One possibility is that dE2F links the endo and mitotic cell cycles to nutritional input, and it does so without requiring dDP function.
Significance of E2F/DP-dependent G1–S transcription
There is a clear correlation between the E2F/DP-dependent transcriptional activation of genes whose products are necessary for DNA replication and the onset of S phase. The implication was that this relationship was causal and that the cyclic transcription of these genes, some of which like cyclin E are known to be key regulatory genes, was necessary for normal S phase. The striking observation from the Drosophila dDP and dE2F mutants is that although cyclic transcription of cyclin E, PCNA, and RNR2 is not detectable, S phase still occurs. Although we cannot exclude the possibility that cyclic transcription of these genes occurs at a low level driven by maternal pools of dDP and dE2F, the bursts of transcription that normally precede S phase are not essential for the G1–S transition. In these mutants the cell cycle may be driven by basal levels of transcripts and post-transcriptional regulation. The maternal pools of components of the replication machinery can persist until late in development, as evidenced by the fact that mutations in PCNA and MCM2 cause late larval lethality (Henderson et al. 1994; Treisman et al. 1995).
The precise developmental control exercised over the cell cycle in Drosophila permits the in vivo role of cell cycle regulators to be evaluated. The ability to visualize the G1–S transcriptional program during embryogenesis enabled us to recover mutations in dDP. Although the mutant phenotypes reveal that dE2F and dDP promote progression of the cell cycle, they reveal a distinction between the effect on E2F/DP-dependent G1–S transcription and the onset of S phase.
Materials and methods
Fly strains
The cn bw sp and DTS 91 strains were provided by R.Lehmann (Skirball Institute, New York University Medical Center, New York, NY). The Ubx–lacZ CyO balancer chromosome has been described previously (McCall et al. 1994). The cyclin EPZ5allele and the cyclin E deficiencies Df(2L)TE116(R)GW1 and Df(2L)TE116(R)GW3 were acquired from J. Roote (University of Cambridge, UK).
The dE2F91 and dE2F7172 alleles have been described previously (Duronio et al. 1995). The P[w+, hsp70–dE2F] and P[w+, hsp70–dDP] strains were provided by N. Dyson (Duronio et al. 1996). The deficiencies uncovering the dDP genes Df(2R)vg-33, Df(2R)vg-56, and Df(2R)vg-B have been described previously (Morgan et al. 1938; Lasko and Pardue 1988; Williams and Bell 1988) and were provided by R. Duronio and the Bloomington stock center. The deficiency uncovering dE2F, Df(3R)e-BS2 was obtained from the Bloomington Stock Center.
Genetic screen
Isogenized cn bw sp homozygous males were fed 35 mm ethylmethane sulfonate (EMS) in a 1% sucrose solution for a 24-hr period (Fig. 7). Immediately after feeding, these males were crossed to virgin females heterozygous for a chromosome carrying a dominant temperature-sensitive mutation DTS91 pr cn and a CyO balancer carrying a lacZ reporter gene under the control of the Ubx promoter P[Ubx–lacZ] CyO. Single male progeny from this cross with the genotypes cn bw sp/P[Ubx–lacZ] CyO and cn bw sp/DTS91 pr cn were collected and mated individually to DTS91 pr cn/P[Ubx–lacZ] CyO virgin females. To facilitate collection of the desired cn bw sp /P[Ubx–lacZ]CyO virgin females and male progeny, the vials were placed at the restrictive temperature for the DTS91 mutation, 29°C, for 3 days. The adults were removed, and the vials were incubated at 29°C for an additional day before being moved to 25°C. cn bw sp/P[Ubx–lacZ] CyO virgin female and male progeny from this cross were collected and mated to establish mutant lines. Embryos (8–15 hr) were collected from these lines and hybridized in situ with a PCNA riboprobe. To distinguish between embryos homozygous for the cn bw sp mutagenized chromosome and their heterozygous cn bw sp/Ubx–lacZ siblings, the embryos were hybridized simultaneously with a lacZ riboprobe.
In situ hybridization
In situ hybridization was carried out essentially as described previously (Tautz and Pfeifle 1989). Multiwell baskets were used to perform in situ hybridization on large numbers of independent samples. Hybridization was carried out at 65°C. Digoxygenin-labeled antisense RNA probes were made as described in the Boehringer Mannheim kit. The RNR2 clone used to make probe was obtained by PCR amplification of Oregon R genomic DNA using degenerate primers (Duronio and O’Farrell 1994). The PCR products were cloned into Bluescript KS. cyclin E probes were made from the E41-1 clone obtained from H. Richardson (Richardson et al. 1993). The PCNA probe was made from a full-length cDNA isolated in this laboratory by J. M. Axton from the library generated by N. Brown (Brown and Kafatos 1988). lacZ probes were made from the pC4β-galactosidase plasmid (Thummel et al. 1988).
Sequence analysis of the dDP alleles
Trans-heterozygous dDP/Df(2R)vg-B third instar larvae were collected from a cross of dDP/T(2;3)TSTL14 and Df(2R)vg-B/T(2;3)TSTL14 adults by selecting non-Tb larvae. T(2;3)TSTL14 is a translocation between the balancers SM5 and TM6B that carries the dominant larval/pupal marker Tubby (Tb) (Gatti and Goldberg 1991). To recognize polymorphisms between our strains and the published sequence, we isolated DNA from adults that were transheterozygous for two unrelated lines created in our screen. Genomic DNA was isolated from these larvae and adults and a 1400-bp region encompassing approximately amino acids 15–377 (Dynlacht et al. 1994) was amplified by PCR. Because two different 5′ ends have been described for the dDP gene (Dynlacht et al. 1994; Hao et al. 1995), the amplified region contained the conserved domains but not the 5′ end of the gene. The amino acid numbering is that of Dynlacht et al. (1994). Primer sequences were 5′-CTTTAGTCAGATGGGCAGTCAAG-3′ and 5′-CTGTAACTAACTCGACTACCAC-3′. Ten to fifteen separate PCR reactions were pooled and sequenced directly in the Whitehead sequencing facility using fluorescence automated sequencing. Primers for sequencing were spaced at ∼300-bp intervals.
BrdU labeling
Embryos (8–15 hr) were labeled with BrdU according to a protocol obtained from Rolf Bodmer, a modified version of Bodmer et al. (1989). After permeabilization, the embryos were incubated with BrdU for either 10, 20, or 40 min at room temperature. The fixed embryos were hydrolyzed in 2 n HCl for 70 min. The 70-min acid treatment is the key difference from the published protocol and provides better detection of the label. The balancers used to maintain the dDP and dE2F mutant lines were marked with a Ubx–lacZ transgene. An anti-β-galactosidase antibody (Promega) was used in conjunction with the anti-BrdU antibody to distinguish the 25% homozygous mutant embryos from their siblings. The antibody staining was detected using the horseradish peroxidase histochemical reaction (Bodmer et al. 1989).
Lethal phase
Embryonic lethality was assessed by mating dDP/+ females to Df(2R)vg-B/CyO males. The parents were allowed to mate at 25°C for several days to reduce the number of unfertilized eggs in the collections. The eggs were collected on apple juice plates at 25°C and counted. The number of eggs that hatched was recorded 24 hr later. The same procedure was carried out for alleles of dE2F. The furthest developmental stage reached by the dDP alleles in trans to each other or in trans to Df(2R)vg-B was determined by generating stocks with the dDP alleles and the deficiency over T(2;3)TSTL14 (Gatti and Goldberg 1991). The dE2F alleles were balanced over TM6B to determine the lethal phase.
To test for the effect of reducing the dosage of cyclin E on the viability of dE2F mutants two reciprocal crosses were done. The effect of zygotic expression alone was examined by crossing dE2F7172/TM6B females to cycEPZ5; dE2F91/TSTL males. The effect of the maternal plus the zygotic contribution was tested by the reciprocal cross.
Heat shock rescue experiments
w; Df(2R)vg-56/CyO females were mated to w; dDPa1/CyO; P[w+;hs–dDP]/+ or w; dDPa1/CyO; P[w+;hs–dE2F]/+ males at 25°C (Duronio et al. 1996). All the progeny from the cross were counted and placed into genotypic classes. To test for rescue of dDPa1/dDPa2 transheterozygotes, dDPa2/CyO females were crossed to w; dDPa1/CyO; P[w+; hs–dDP]/TM3, Sb males at 25°C. No dDPa1/dDPa2 adults were observed that lacked the hsdDP gene.
For the heat shock treatments, embryos were collected (from the crosses shown above) for 24 hr at 25°C in vials. The heat shock treament was administered by placing the vials in a 37°C air incubator for 1 hr and then the vials were returned to 25°C. The heat shocks were initiated either immediately after the completion of a 24-hr collection or after the eggs were allowed to develop at 25°C for 5–6 days. In either case, subsequent heat shocks were delivered two times daily.
Acknowledgments
We thank R. Duronio and N. Dyson for providing stocks and communicating results before publication. Members of R. Lehmann’s laboratory gave helpful advice on the genetic screen. We thank L. Ziaugra of the Whitehead Institute Sequencing Facility for sequence analysis. J. Lopez provided help with polytene chromosome squashes, and H. LeBlanc assisted in the brain dissections. A. Amon, R. Beijersbergen, S. Bickel, N. Dyson, J. Lees, J. Lopez, and W. Lundberg provided critical comments on the manuscript. I.R. was supported by a National Science Foundation predoctoral fellowship. A.W. was supported by a National Institutes of Health predoctoral training grant. The initial stages of this work were supported by National Institutes of Health grant GM-39341 to T. O-W.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
The GenBank accession no. for the sequence reported here is AF011362.
Footnotes
E-MAIL weaver@wi.mit.edu; FAX (617) 258-9872.
References
- Asano M, Nevins JR, Wharton RP. Ectopic E2F expression induces S-phase and apoptosis in Drosophila imaginal discs. Genes & Dev. 1996;10:1422–1432. doi: 10.1101/gad.10.11.1422. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Carretto R, Jan YN. Neurogenesis of the peripheral nervous system in Drosophila embryos: DNA replication patterns and cell lineages. Neuron. 1989;3:21–32. doi: 10.1016/0896-6273(89)90112-8. [DOI] [PubMed] [Google Scholar]
- Botz J, Zerfass-Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen-Durr P. Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol. 1996;16:3401–3409. doi: 10.1128/mcb.16.7.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook A, Xie J-E, Du W, Dyson N. Requirements for dE2F function in proliferating cells and in post-mitotic differentiating cells. EMBO J. 1996;15:3676–3683. [PMC free article] [PubMed] [Google Scholar]
- Brown NH, Kafatos F. Functional cDNA libraries from Drosophila embryos. J Mol Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- Bryant, P.J., K.L. Watson, R.W. Justice, and D.F. Woods. 1993. Tumor suppressor genes encoding proteins required for cell interactions and signal transduction in Drosophila. Development (Suppl.) 239–249. [PubMed]
- de Nooij JC, Letendre MA, Hariharan IK. A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell. 1996;87:1237–1247. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- Du W, Vidal M, Xie J-E, Dyson N. RBF, a novel RB-related gene that regulates E2F activity and interacts with cyclin E in Drosophila. Genes & Dev. 1996a;10:1206–1218. doi: 10.1101/gad.10.10.1206. [DOI] [PubMed] [Google Scholar]
- Du W, Xie J-E, Dyson N. Ectopic expression of dE2F and dDP induces cell proliferation and death in the Drosophila eye. EMBO J. 1996b;15:3684–3692. [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH. Developmental control of a G1-S transcriptional program in Drosophila. Development. 1994;120:1503–1515. doi: 10.1242/dev.120.6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Developmental control of the G1 to S transition in Drosophila: cyclin E is a limiting downstream target of E2F. Genes & Dev. 1995;9:1456–1468. doi: 10.1101/gad.9.12.1456. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH, Xie J-E, Brook A, Dyson N. The transcription factor E2F is required for S-phase during Drosophila embryogenesis. Genes & Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, Brook A, Dyson N, O’Farrell PH. E2F-induced S phase requires cyclin E. Genes & Dev. 1996;10:2505–2513. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Brook A, Dembski M, Yenush L, Dyson N. DNA-binding and trans-activation properties of Drosophila E2F and DP proteins. Proc Natl Acad Sci. 1994;91:6359–6363. doi: 10.1073/pnas.91.14.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field SJ, Tsai F-Y, Kuo F, Zubiaga AM, Kaelin WG, Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Finley RL, Thomas BJ, Zipursky SL, Brent R. Isolation of Drosophila cyclin D, a protein expressed in the morphogenetic furrow before entry into S phase. Proc Natl Acad Sci. 1996;93:3011–3015. doi: 10.1073/pnas.93.7.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foe VE, Odell GM, Edgar BA. Mitosis and morphogenesis in the Drosophila embryo: Point and counterpoint. In: Bate M, Martinez-Arias A, editors. The development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 149–300. [Google Scholar]
- Gatti M, Goldberg ML. Mutations affecting cell division in Drosophila. Methods Cell Biol. 1991;35:543–586. doi: 10.1016/s0091-679x(08)60587-7. [DOI] [PubMed] [Google Scholar]
- Geng Y, Eaton EN, Picon M, Roberts JM, Lundberg AS, Gifford A, Sardet C, Weinberg RA. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- Girling R, Partridge JF, Bandara LR, Burden N, Totty NF, Hsuan JJ, La Thangue NB. A new component of the transcription factor DRTF1/E2F. Nature. 1993;362:83–87. doi: 10.1038/362083a0. [DOI] [PubMed] [Google Scholar]
- Hao XF, Alphey L, Bandara LR, Lam E, Glover D, LaThangue NB. Functional conservation of the cell cycle-regulating transcription factor DRTF1/E2F and its pathway of control in Drosophila melanogaster. J Cell Sci. 1995;108:2945–2954. doi: 10.1242/jcs.108.9.2945. [DOI] [PubMed] [Google Scholar]
- Heibert SW, Packham G, Strom DK, Haffner R, Oren M, Zambetti G, Cleveland JL. E2F-1:DP-1 induces p53 and overrides survival factors to trigger apoptosis. Mol Cell Biol. 1995;15:6864–6874. doi: 10.1128/mcb.15.12.6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson DS, Banga SS, Grigliatti TA, Boyd JB. Mutagen sensitivity and suppression of position-effect variegation result from mutations in mus209, the Drosophila gene encoding PCNA. EMBO J. 1994;13:1450–1459. doi: 10.1002/j.1460-2075.1994.tb06399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77:107–120. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, Vaessin H. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell. 1996;87:1225–1235. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- Lasko PF, Pardue ML. Studies of the genetic organization of the vestigial microregion of Drosophila melanogaster. Genetics. 1988;120:495–502. doi: 10.1093/genetics/120.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees AD, Waddington CH. The development of the bristles in normal and some mutant types of Drosophila melanogaster. Proc Royal Soc London Series B. 1942;131:87–110. [Google Scholar]
- Lukas J, Petersen BO, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol Cell Biol. 1996;16:1047–1057. doi: 10.1128/mcb.16.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall K, O’Connor MB, Bender W. Enhancer traps in the Drosophila bithorax complex mark parasegmental domains. Genetics. 1994;138:387–399. doi: 10.1093/genetics/138.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TH, Bridges C, Schultz C. Constitution of the germinal material in relation to heredity. Carnegie Inst Washington Year Book. 1938;37:304–309. [Google Scholar]
- Murray A, Hunt T. The cell cycle: An introduction. New York, NY: Freeman; 1993. [Google Scholar]
- Nasymth K. At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- Nevins JR. E2F: A link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- Ohtani K, Nevins JR. Functional properties of a Drosophila homolog of the E2F1 gene. Mol Cell Biol. 1994;14:1603–1612. doi: 10.1128/mcb.14.3.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL. Developmental modification of the Drosophila cell cycle. Trends Genet. 1994;10:321–327. doi: 10.1016/0168-9525(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Poodry CA, Woods DF. Control of the developmental timer for Drosophila pupariation. Wilhelm Roux’s Arch Dev Biol. 1990;199:219–227. doi: 10.1007/BF01682081. [DOI] [PubMed] [Google Scholar]
- Richardson HE, O’Keefe LV, Reed SI, Saint R. A Drosophila G1-specific cyclin E homolog exhibits different modes of expression during embryogenesis. Development. 1993;119:673–690. doi: 10.1242/dev.119.3.673. [DOI] [PubMed] [Google Scholar]
- Sauer K, Knoblich JA, Richardson H, Lehner CF. Distinct modes of cyclin E/cdc2c kinase regulation and S-phase control in mitotic and endoreduplication cycles of Drosophila embryogenesis. Genes & Dev. 1995;9:1327–1339. doi: 10.1101/gad.9.11.1327. [DOI] [PubMed] [Google Scholar]
- Sauer K, Weigmann K, Sigrist S, Lehner CF. Novel members of the cdc2-related kinase family in Drosophila: cdk4/6, cdk5, PFTAIRE, and PITSLRE kinase. Mol Biol Cell. 1996;7:1759–1769. doi: 10.1091/mbc.7.11.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Lee W-H. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol. 1994;14:8166–8173. doi: 10.1128/mcb.14.12.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Smith AV, King JA, Orr-Weaver TL. Identification of genomic regions required for DNA replication during Drosophila embryogenesis. Genetics. 1993;135:817–829. doi: 10.1093/genetics/135.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AV, Orr-Weaver TL. The regulation of the cell cycle during Drosophila embryogenesis: The transition to polyteny. Development. 1991;112:997–1008. doi: 10.1242/dev.112.4.997. [DOI] [PubMed] [Google Scholar]
- Sparrow JC. Melanotic “tumours”. In: Ashburner M, Wright T W, editors. The genetics and biology of Drosophila. 2b. London, UK: Academic Press; 1978. pp. 277–313. [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Thummel CS, Boulet AM, Lipshitz HD. Vectors for Drosophila P element-mediated transformation and tissue culture transfection. Gene. 1988;74:445–456. doi: 10.1016/0378-1119(88)90177-1. [DOI] [PubMed] [Google Scholar]
- Treisman JE, Follette PJ, O’Farrell PH, Rubin GM. Cell proliferation and DNA replication defects in a Drosophila MCM2 mutant. Genes & Dev. 1995;9:1709–1715. doi: 10.1101/gad.9.14.1709. [DOI] [PubMed] [Google Scholar]
- Watson KL, Johnson TK, Denell RE. Lethal (1) aberrant immune response mutations leading to melanotic tumor formation in Drosophila melanogaster. Dev Genet. 1991;12:173–187. doi: 10.1002/dvg.1020120302. [DOI] [PubMed] [Google Scholar]
- Watson KL, Konrad KD, Woods DF, Bryant PJ. Drosophila homolog of the human S6 ribosomal proteins is required for tumor suppression in the hematopoietic system. Proc Natl Acad Sci. 1992;89:11302–11306. doi: 10.1073/pnas.89.23.11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, K.L., R.W. Justice, and P.J. Bryant. 1994. Drosophila in cancer research: The first fifty tumor suppressor genes. J. Cell Sci. (Suppl.) 18: 19–33. [DOI] [PubMed]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Weintraub SJ, Prater CA, Dean DC. Retinoblastoma protein switches the E2F site from positive to negative element. Nature. 1992;358:259–261. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- Weintraub SJ, Chow KNB, Luo RX, Zhang SH, He S, Dean DC. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- Williams JA, Bell JB. Molecular organization of the vestigial region in Drosophila melanogaster. EMBO J. 1988;7:1355–1363. doi: 10.1002/j.1460-2075.1988.tb02951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-L, Classon M, Dyson N, Harlow E. Expression of dominant-negative mutant DP-1 blocks cell cycle progression in G1. Mol Cell Biol. 1996;16:3698–3706. doi: 10.1128/mcb.16.7.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Hayashi Y, Matsukage A. Essential role of E2F recognition sites in regulation of the proliferating cell nuclear antigen gene promoter during Drosophila development. J Biol Chem. 1995;270:25159–25165. doi: 10.1074/jbc.270.42.25159. [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- Zwicker J, Liu N, Engeland K, Lucibello FC, Muller R. Cell cycle regulation of E2F site occupation in vivo. Science. 1996;271:1595–1597. doi: 10.1126/science.271.5255.1595. [DOI] [PubMed] [Google Scholar]