

Abstract
PPARγ is an adipose-selective nuclear hormone receptor that plays a key role in the control of adipocyte differentiation. Previous studies indicated that activation of ectopically expressed PPARγ induces differentiation when cells have ceased growth because of confluence. We show here that ligand activation of PPARγ is sufficient to induce growth arrest in fibroblasts and SV40 large T-antigen transformed, adipogenic HIB1B cells. Cell cycle withdrawal is accompanied by a decrease in the DNA-binding and transcriptional activity of the E2F/DP complex, which is attributable to an increase in the phosphorylation of these proteins, especially DP-1. This effect is a consequence of decreased expression of the catalytic subunit of the serine–threonine phosphatase PP2A. These data suggest an important role for PP2A in the control of E2F/DP activity and a new mode of cell cycle control in differentiation.
Keywords: PPARγ, E2F, DP, protein phosphatase 2A, cell cycle, adipogenesis
Cell differentiation involves the reprogramming of gene expression, resulting in the synthesis of a new set of proteins that characterize a given cell type. In addition, differentiation often correlates with changes in the growth rate or growth potential of a cell. Although some differentiated cell types continue to proliferate, many common forms of differentiation involve the cessation of cell growth and are referred to as terminal differentiation. Because cancer is a disease whose pathology derives largely from inappropriate cell growth, much attention has been given to the notion of stimulating terminal differentiation as an approach to therapy that may have reduced toxicity, at least compared to more conventional forms of chemotherapy (Warrel et al. 1991).
The past several years have seen dramatic advances in our understanding of the transcriptional basis of certain forms of cell differentiation. It is now clear that several proteins of the basic helix–loop–helix (bHLH) family such as MyoD and myogenin play crucial roles in the stimulation of myogenesis (Lassar and Munsterberg 1994). Ectopic expression of these factors also causes the cessation of proliferation in a manner similar to the differentiation of normal myogenic cell lines. Recent data suggest that a key component of the effects of MyoD on cell growth is the expression of the cyclin-dependent kinase (cdk) inhibitors p21 and p27 (Guo et al. 1995; Halevy et al. 1995; Skapek et al. 1995), which play a crucial role in the regulation of the function of pRb-related tumor-suppressor proteins (Weinberg 1995). Another system of differentiation receiving much recent scrutiny is adipogenesis. Several transcription factors are induced in fat cell differentiation [CCAA/enhancer-binding protein-α (C/EBPα), C/EBPβ, peroxisome proliferator-activated receptor-γ (PPARγ), and adipoxcyte determination differentiation dependent factor 1 (ADD1) /sterol regulatory element binding protien 1 (SREBP1)] and strongly influence this process (Samuelsson et al. 1991; Umek et al. 1991; Tontonoz et al. 1993; Freytag et al. 1994; Lin and Lane 1994; Tontonoz et al. 1994a; Wu et al. 1995; Yeh et al. 1995; Kim and Spiegelman 1996). PPARγ has been suggested to play a dominant role because it is induced relatively early, it is selectively expressed in fat tissue, and can evoke a full adipogenic response when expressed at or below the levels seen in adipose tissue in vivo (Tontonoz et al. 1994b). This molecule, a member of the nuclear receptor family, has been shown recently to bind two distinct ligands: the synthetic antidiabetic thiazolidinediones (Forman et al. 1995; Lehmann et al. 1995) and the 15-deoxyΔ12,14 prostaglandin J2 (Forman et al. 1995; Kliewer et al. 1995). Despite the increasing evidence of a central role for PPARγ in adipose development, its relationship to the cessation of cell growth is unclear. Experiments done to date have used mainly ectopic expression of this factor in 3T3 cells, with the application of ligands or activators after cells have ceased growth because of confluence. Hence, it is not clear whether PPARγ has the ability to cause cell cycle withdrawal or is limited to stimulating the differentiation of cells that have already stopped growing.
In this paper, we use ectopically and endogenously expressed PPARγ, along with synthetic thiazolidinedione ligands, to demonstrate that activation of PPARγ is sufficient to cause cell cycle arrest in logarithmically growing cells. This arrest is associated with a dramatic loss of E2F/DP DNA-binding and transcriptional activity, which is a consequence of reduced levels of PP2A. Hence, PPARγ activation illustrates a potentially new mode of cell cycle control.
Results
Activation of PPARγ leads to cell cycle withdrawal
To study the effect of PPARγ activation on cell growth, we used a retrovirus infection system to express PPARγ in NIH-3T3 cells. This system allows us to express ectopic genes in many thousands of cells at relatively equal levels. PPARγ has two isoforms, PPARγ1 and PPARγ2, that have different amino termini formed by alternative splicing (Zhu et al. 1993; Tontonoz et al. 1994a). NIH-3T3 fibroblasts were infected with the retroviral expression vector containing cDNA encoding PPARγ1 or PPARγ2 (NIH–PPARγ), or with the empty vector (NIH-vector) to create stable cell lines. NIH–PPARγ cells expressed approximately one-third the level of endogenous PPARγ mRNA and protein that are observed in differentiated adipocytes (Tontonoz et al. 1994b; Hu et al. 1996).
Exponentially growing cells were treated with a synthetic PPARγ ligand, pioglitazone, which belongs to the class of thiazolidinedione antidiabetic agents (Forman et al. 1995; Lehmann et al. 1995). As shown in Figure 1, treatment with pioglitazone at 5 μm concentration had no obvious effect on cells containing empty vectors. In contrast, this agent had dramatic effects on NIH–PPARγ cells, inhibiting cell proliferation and inducing drastic morphological changes. Starting at ∼48 hr after treatment, increasing numbers of NIH–PPARγ cells changed from an elongated fibroblastic shape to an adipocyte-like morphology, with a round form and accumulation of small drops of lipids within the cytoplasm (Fig. 1, arrow). Time-course studies after pioglitazone treatment showed that the number of NIH–PPARγ cells in ligand-treated plates was reduced by almost 40% relative to controls by 2 days after treatment and by 80% after 5 days with pioglitazone (Fig. 2A,B). The growth of pioglitazone-treated NIH-vector cells decreased by 10% over this period compared to untreated control cells, which may be attributable to the presence of low amount of PPARγ in these cells (data not shown). The addition of 1 μm BRL49653, another synthetic thiozolidinedione ligand for PPARγ (Forman et al. 1995; Lehmann et al. 1995), was found to exert the same degree of inhibition of cell growth of NIH–PPARγ cells (Fig. 2C). No obvious cytotoxic effects were observed at the concentrations used in these compounds.
Figure 1.

Pioglitazone stimulates growth arrest as well as adipose differentiation of NIH-3T3 cells expressing PPARγ ectopically. NIH-3T3 cells were infected with the retrovirus containing PPARγ expression vector (NIH–PPARγ) or with the empty vector (NIH-vector). After selection in puromycin, cells were pooled and cultured with or without pioglitazone (5 μm) for 5 days. Arrow shows a differentiated adipocyte containing lipid drops in the cytoplasm.
Figure 2.



Growth of NIH–PPARγ, NIH-vector, or HIB1B cells in the presence or absence of PPARγ ligands. The same number of NIH–PPARγ, NIH-vector, or HIB1B cells were cultured either in the presence or absence of PPARγ ligands. Cell numbers were determined at the indicated time points. (A) Cumulative growth of cells untreated or treated with 5 μm pioglitazone is shown. (B) Percent decrease of the cell numbers in the pioglitazone-treated plates (solid bars) relative to the untreated plates (open bars) is shown. (C) Exponentially growing cells were treated without or with two members of thiozolidinediones, pioglitazone (5 μm) (solid bars) or BRL49653 (1 μm) (open bars) for 5 days and cell numbers were determined. The effect of ligands on cell growth is represented as percentage decrease in cell numbers in the treated plates relative to untreated control plates.
To analyze whether ligand treatment of cells expressing PPARγ affects progression through a specific cell cycle stage we performed fluorescence-activated cell sorting (FACS) analysis and bromodeoxyuridine (BrdU) incorporation experiments. Ligand treatment led to an accumulation of the cell populations in the G0/G1 phase of cell cycle (data not shown). The percentage of cells undergoing DNA synthesis after 5 days of pioglitazone treatment was determined by the ability of cells to incorporate BrdU. As shown in Table 1, ligand treatment did not change BrdU incorporation rate in NIH-vector cells, but it caused an 80% decrease in the BrdU incorporation rate in NIH–PPARγ and 3T3–F442A preadipocytes after 5 days of treatment.
Table 1.
Activation of PPARγ leads to cell cycle withdrawal in normal NIH–PPARγ cells, in 3T3–F442A preadipocytes, and in transformed HIB1B cells
|
|
Pioglitazone
|
BrdU positive (%)
|
|---|---|---|
| NIH–vector | − | 44 |
| NIH–vector | + | 43 |
| NIH–PPARγ | − | 44 |
| NIH–PPARγ | + | 9 |
| HIB1B | − | 75 |
| HIB1B | + | 11 |
| 3T3–F442A | − | 63 |
| 3T3–F442A | + | 14 |
Cells, cultured on coverslips, were untreated or treated with 5 μm pioglitazone for 5 days and then pulsed with BrdU for 1 hr. Coverslips were fixed and processed as described in Materials and Methods. Cells undergoing DNA synthesis during exposure to BrdU were determined by immunohistochemical staining and considered as BrdU positive. The data represents the average of two independent experiments in which ∼400 cells were counted per sample.
Together these results demonstrate that ligand activation of PPARγ is sufficient to cause cell cycle withdrawal, even in rapidly proliferating cells.
Transcription factor activity is required for PPARγ-mediated cell cycle withdrawal
To determine some of the structural requirements of PPARγ necessary for growth arrest, NIH-3T3 cells were infected with retroviral vectors expressing wild type or various mutant forms of PPARγ. Exponentially growing cells were treated for 5 days with pioglitazone and cell numbers were determined. As shown in Figure 3, ligand activation of both PPARγ1 and PPARγ2 induced a similar growth arrest. We also examined an allele of PPARγ (PPARγ–M1) that lacks the amino-terminal 127 amino acids of PPARγ2. Previous work has shown that this allele is more active than the wild type with respect to the induction of adipogenesis (Tontonoz et al. 1994b). Growth inhibition in NIH-3T3 cells containing PPARγ–M1 was even higher than the cells ectopically expressing wild-type PPARγ1 or PPARγ2. To investigate whether DNA binding and the transcriptional activation domain of PPARγ are required for its effect on cell growth, NIH-3T3 cells were infected with two other mutant forms of PPARγ: PPARγ–M2, containing two point mutations in the DNA-binding domain and a carboxy-end deleted PPARγ–CD, which lacks the activation domain (AF-2) located in the carboxyl-terminal region of all nuclear receptors (Mangelsdorf and Evans 1995). Pioglitazone treatment did not have any affect on cell growth and adipogenesis in NIH–M2 and NIH–CD cells. These results demonstrate that both PPARγ1 and PPARγ2 can stimulate cell cycle withdrawal. These data also suggest that the activity of PPARγ as a DNA-binding protein and transcription factor is required for its effect on cell growth.
Figure 3.

Transcription factor activity of PPARγ is required for its negative regulatory function on cell growth. (Left) Schematic representations of wild-type PPARγ1, PPARγ2, or mutant PPARγ2 cDNAs. NIH–M1 cells contain PPARγ expression vector, which lacks the first 127 amino acids located in the amino terminus. NIH–M2 cells express a PPARγ2 receptor in which cystein residues at the DNA-binding domain at positions 156 and 159 have been changed to serine. NIH–CD cells express a truncated form of PPARγ2, which lacks the conserved carboxyl-terminal transactivation domain. (Right) Effects of pioglitazone treatment on the growth rate of cells expressing wild-type or mutant forms of PPARγ. Cell numbers were determined after 5 days treatment without or with 5 μm pioglitazone. Decrease in the cell number in treated plates was represented as relative change to untreated control plates. The data represent the average of at least three independent experiments.
Ligand activation of PPARγ induces growth arrest in transformed cells
It was also of interest to determine whether activation of PPARγ can induce cell cycle arrest in malignantly transformed cells. We used HIB1B cells, transformed with the SV40 large T antigen (SV40LT), as a model system. These cells, expressing high amounts of PPARγ1, were established from brown fat tumors of transgenic mice that constitutively express SV40LT under the control of the adipocyte-specific aP2 promoter (Ross et al. 1992). Exponentially growing HIB1B cells were treated with pioglitazone and cell numbers and the ability to incorporate BrdU were determined. As shown in Figures 2A,C and 3, PPARγ activation by pioglitazone or BRL49653 strongly repressed the growth of these cells. BrdU incorporation into newly synthesized DNA was also decreased 85% after 5 days of treatment with pioglitazone (Table 1). These results show that PPARγ activation can overcome SV40LT driven transformation and cause cell cycle withdrawal in HIB1B cells.
PPARγ activation leads to a selective loss in the E2F/DP DNA-binding and transcription factor activities
It has been shown that cell proliferation requires the action of cyclins and cdks (Hunter and Pines 1994; Nurse 1994; Sherr 1994; Sherr and Roberts 1995). The levels of the cdk inhibitors p21 and p27 proteins have been shown to be induced by MyoD transcription factors during the differentiation of skeletal muscle cells (Guo et al. 1995; Halevy et al. 1995; Skapek et al. 1995).
We observed no changes in the expression of these cdk inhibitors in NIH–PPARγ or HIB1B cells after pioglitazone treatment, as monitored by immunofluorosence staining, RNA, or protein blots (data not shown). In addition, the induction of cell cycle withdrawal in HIB1B cells upon activation of PPARγ suggests that the function of tumor-suppressor proteins, such as pRb, p107, p130, and p53, are not required for this process, as the transforming activity of SV40LT has been linked to its ability to bind and functionally inactivate these proteins (DeCaprio et al. 1988; Dyson et al. 1989; Ewen et al. 1989; Ludlow et al. 1989; Hannon et al. 1993; Wolf et al. 1995; Zalvide and DeCaprio 1995). In the presence of SV40LT, pRb as well as the related pocket proteins p107 and p130 cannot bind to E2F/DP complexes to repress their transcriptional activity, which is required for cell proliferation. Hence, these data suggested that E2F/DP transcription factors might be a more direct intracellular target for the action of PPARγ to control cell cycle. To test this hypothesis cellular extracts were prepared from HIB1B, NIH–PPARγ, and NIH-vector cells, with or without pioglitazone treatment and subsequently the DNA-binding activity of E2F/DP complexes was analyzed by gel mobility-shift assay. In exponentially growing cells as well as in SV40LT-containing cells, such as HIB1B, much of the E2F/DP complexes are expected to be found as free complexes uninhibited by the pocket proteins pRb, p107, and p130. As shown in Figure 4A, lane 1, we detected only one major complex in extracts of untreated exponentially growing HIB1B cells, which corresponds presumably to the free E2F/DP DNA-binding complex. This complex could bind specifically to the E2F site, as only the wild-type but not a mutated E2F oligonucleotide could compete for binding (Fig. 4A, lanes 3,4).
Figure 4.
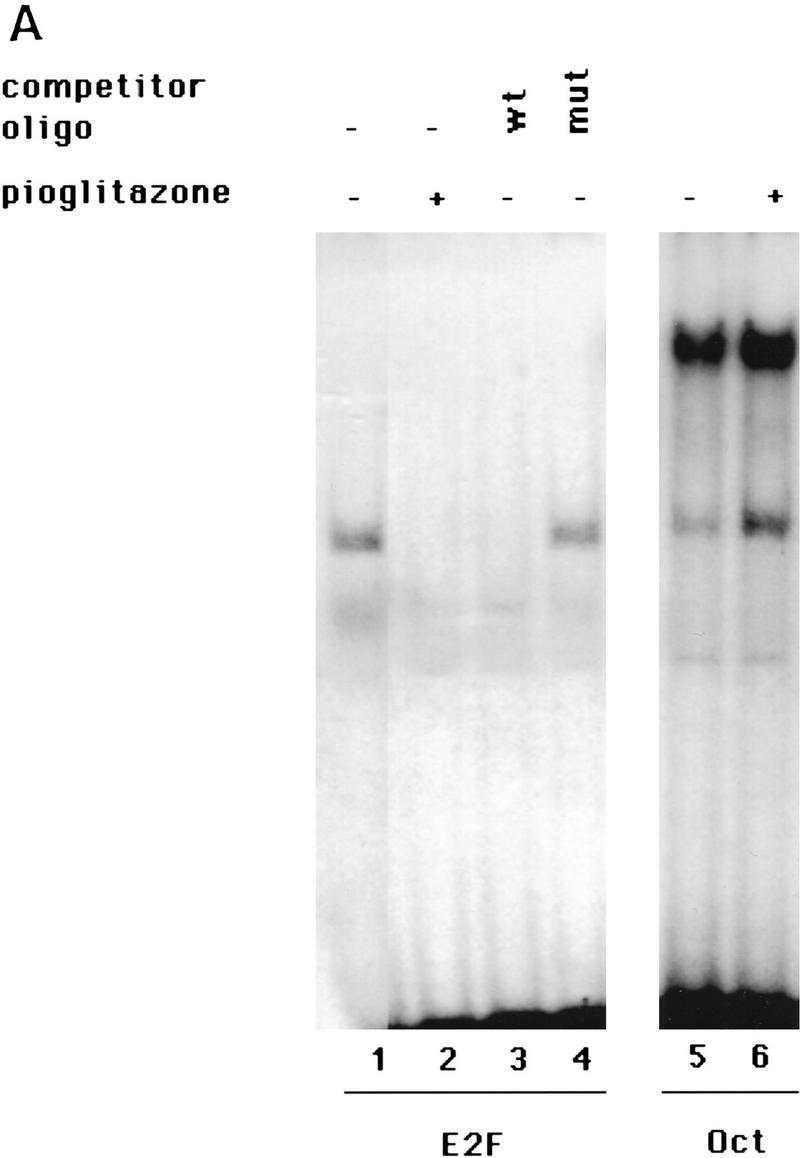

PPARγ-induced growth arrest is accompanied by a strong decrease in the E2F/DP DNA-binding activity. (A) HIB1B cells were treated as described in the legend to Fig. 2. Whole cell extracts were prepared and electrophoretic mobility-shift assays were performed by using E2F or Oct oligonucleotides as probe (see Materials and Methods). For competition experiments a 100-fold molar excess of unlabeled wild-type (wt) or mutant (mut) oligonucleotides were used. (B) HIB1B cells were treated for various times with 5 μm pioglitazone. Whole cell extracts were prepared and electrophoretic mobility-shift assays were performed with E2F probe. (C) HIB1B cells were transfected transiently with a luciferase reporter gene linked to three tandemly repeated E2F-binding sites. All transfections included a plasmid directing the expression of β-galactosidase under the control of β-actin promoter. Twenty-four hours after transfection, cells were cultured for an additional 48 hr in the presence or absence of 5 μm pioglitazone. Cells were harvested and assayed for luciferase activity, which was normalized for variable transfection efficiencies by correcting for β-galactosidase activity. Decrease in the luciferase activity in pioglitazone-treated cells is shown as a change relative to untreated control plates. Results are the average of six independent transfection experiments.
Interestingly, PPARγ activation by pioglitazone strongly reduced the DNA-binding activity of E2F/DP in extracts from the HIB1B cells (Fig. 4A, lane 2). Assays of the same extracts for DNA-binding activity of an unrelated control transcription factor Oct revealed that the binding to this element did not decline after treatment with pioglitazone (Fig. 4A, lanes 5,6). We also performed similar experiments with NIH-vector and NIH–PPARγ cells and a selective loss of E2F/DP-binding activity was observed only in NIH–PPARγ cells after pioglitazone treatment (data not shown).
The time-course of down-regulation of E2F/DP DNA-binding activity was investigated by treating HIB1B cells with pioglitazone for different times, cellular extracts were prepared, and then DNA-binding assays were performed. As shown in Figure 4B, E2F/DP DNA binding was clearly reduced by 2 days of pioglitazone treatment and nearly disappeared by 5 days of treatment.
The relationship between PPARγ activation and the endogenous E2F/DP transcription factor activity was studied in HIB1B cells that were transfected transiently with a luciferase reporter plasmid linked to three copies of an E2F-binding site. As shown in Figure 4C activation of PPARγ with pioglitazone led to a 50% decrease in the luciferase activity after a period of 48 hr. This effect of PPARγ activation on the endogenous E2F/DP transcription factor activity is in a good general agreement with the decrease of E2F/DP DNA-binding activity, as well as the decrease in cell growth at this time. These results demonstrate that PPARγ-mediated cell cycle withdrawal is accompanied by a reduction in the E2F/DP DNA-binding and transcription factor activities.
The identity of proteins in the E2F/DP complex was investigated with specific antibodies. Figure 5 shows that addition of two different antibodies specific for DP-1 causes a loss in the major E2F/DP complex (lanes 3,4) and a “supershift” is seen with the A33 antibody (lane 3). This suggests that DP-1 is the major DP component in these complex. In contrast, addition of an antibody specific for E2F-1 did not cause any change in the amount or mobility of this complex (Fig. 5, lane 7), suggests that E2F-1 is not a major component here. Because there are at least five members of the E2F family, the identity of the E2F component of this complex will require further study.
Figure 5.

DP-1 is a major component of the endogenous E2F/DP DNA-binding activity. Extracts from exponentially growing HIB1B cells were incubated with preimmune serum (PI) or with the various antibodies as indicated here and then subjected to electrophoretic mobility-shift assays using a 32P-labeled E2F probe. The respective antibodies were as follows: K-20, an α-DP-1 polyclonal antibody (Santa Cruz sc-610x); A33, a rabbit polyclonal antiserum raised against DP-1; α-E2F-1, a mouse monoclonal antibody (Santa Cruz sc-251x); sc-525, an unrelated control antibody prepared against the GADD45 protein (Santa Cruz). Arrowhead shows the supershift.
PP2A restores the E2F/DP DNA-binding activity in extracts of pioglitazone-treated cells
It has been reported that serine phosphorylation of E2F and DP proteins can inhibit their DNA-binding activities (Dynlacht et al. 1994; Krek et al. 1994, 1995; Xu et al. 1994; Kitagawa et al. 1995). The mechanism leading to a decrease in the DNA-binding activity of E2F/DP in pioglitazone-treated cells was explored by performing mixed extract experiments. Interestingly, the addition of extracts from untreated cells to the extracts prepared from pioglitazone-treated cells drastically restored E2F/DP DNA-binding activity (Fig. 6A, lane 3). The intensity of the complex was even higher than that of the corresponding complex formed in untreated cellular extracts (Fig. 6A, cf. lane 1 with lane 3), which may be attributable to increased gene expression of DP-1 in pioglitazone-treated cells (data not shown). However, when the phosphatase inhibitor okadaic acid (OA) was added to the extracts before mixing, the material from untreated cells was not able to restore the DNA-binding activity of E2F/DP in pioglitazone-treated HIB1B extracts (Fig. 6A, lane 4). Because OA has been shown to inhibit the activities of serine–threonine phosphatases PP2A and PP1A, this suggested that one of these enzymes may be responsible for this activity. To distinguish between these two enzymes we used the inhibitor protein 2 (IP-2), which at nanomolar concentrations inhibits the PP1A activity but has no effect on PP2A. The addition of IP-2 at 1 μg concentration did not inhibit the restoration of E2F/DP activity in mixing experiments (data not shown). This result indicates that the activity deficient in pioglitazone-treated cells and restored by untreated cells is probably PP2A. Therefore, we treated extracts with purified PP2A catalytic subunit (PP2Ac) and the E2F/DP DNA-binding activity was determined. Treatment with PP2Ac had no effect on the E2F/DP DNA-binding activity in extracts of untreated cells (Fig. 6A, lane 5), but restored the endogenous E2F/DP activity in extracts of pioglitazone-treated HIB1B cells (Fig. 6A, lane 6). These data strongly suggest that PPARγ-induced decrease in the activity of PP2Ac plays a critical role in the reduction of E2F/DP binding in pioglitazone-treated HIB1B cells.
Figure 6.

PP2Ac is able to restore decreased E2F/DP DNA binding activity in pioglitazone-treated cells. (A) Whole cell extracts prepared from untreated (lane 1) or pioglitazone (5 μm)-treated (lane 2) HIB1B cells were used either directly or first mixed in the absence (lane 3) or presence (lane 4) of 3 nm okadaic acid (OA) and then used for gel mobility-shift assays to determine E2F/DP DNA-binding activity. Extracts from ligand untreated (lane 5) or treated (lane 6) HIB1B cells were treated with 0.5 unit of PP2Ac (GIBCO BRL) for 30 min at room temperature and electrophoretic mobility-shift assays were carried out by using E2F oligonucleotide as probe. The data shown represent the results of a single experiment. Exactly identical results were obtained in another independent experiment. (B) Calyculin A treatment leads to a decrease in the E2F/DP DNA-binding activity. HIB1B cells were treated with 5 μm pioglitazone or with 0.1 nm calyculin A for 3 days. Whole cell extracts were prepared from HIB1B cells treated as described above, and gel mobility-shift assays were performed to determine the DNA-binding activity of endogenous E2F/DP.
Previous data have shown that inhibition of the PP2A activity disrupts cell proliferation in various organisms (Kinoshita et al. 1990; Mayer-Jaekel et al. 1993; Das et al. 1994; You and Bird 1995 and references therein), but the mechanism of this effect is not clear. To investigate whether direct pharmacological manipulation of PP2A activity could regulate E2F/DP activity, HIB1B cells were treated with either pioglitazone (3 days) or calyculin A (1 day). Calyculin A treatment caused a dramatic loss of E2F/DP DNA binding (Fig. 6B, lane 3), illustrating that direct modulation of PP2A activity can regulate E2F/DP DNA binding.
Ligand activation of PPARγ induces phosphorylation of the DP-1 in cells
The ability of PP2A to reverse the changes in DNA binding of E2F/DP caused by PPARγ suggested that phosphorylation events may be linked causally to ligand activation of this receptor. To study this HIB1B cells untreated or treated with pioglitazone were labeled with [32P]orthophosphate and immunoprecipitation experiments were carried out by using the anti-DP-1 antibody A33. As shown in Figure 7A pioglitazone treatment resulted in a dramatic increase in the phosphorylation of DP-1 (lanes 6,7). This is attributable to a change in the relative phosphorylation state, as ligand treatment did not cause any change in amount of 35S-labeled DP-1 (lanes 3,4). To determine whether PP2Ac could act directly on the phosphorylated form of DP-1, the immunoprecipitates were treated with the catalytic subunit of PP2A for 20 min. The complete loss of phosphorylation after PP2Ac treatment (lane 6) indicates that PPARγ-mediated phosphorylation of DP-1 occurs mainly on the serine–threonine residues. This result also suggests that PP2A action on E2F/DP is likely to be direct.
Figure 7.

Activation of PPARγ induces phosphorylation of DP-1 but not E2F-1. HIB1B cells with or without pioglitazone treatment for 3 days were metabolically labeled with [32P]orthophosphate or with [35S]methionine. Whole cell lysates were prepared and then subjected to immunoprecipitation using preimmune serum (PI), antiserum specific for DP-1, or antibody prepared for HA. (A) DP-1 immunoprecipitates were boiled in 1% SDS, and 32P-labeled DP-1 was reimmunoprecipitated with DP-1 antibody. Immunoprecipitates prepared from PPARγ-activated cells were either tested directly (lane 4) or after treatment with PP2Ac for 20 min at room temperature (lane 5). Lane 1 shows 35S-labeled in vitro translated DP-1. (B) Lysates of metabolically labeled HIB1B cells ectopically expressing HA–E2F-1 were subjected to preimmuneserum (PI) or anti-HA antibody MAb 12CA5. 35S-Labeled, in vitro-translated HA–E2F-1 is shown in lane 1. Samples were analyzed by SDS–gel electrophoresis under reducing conditions. Mobilities of DP-1 and E2F-1 are indicated by arrows.
Although the identity of the endogenous E2F species in these experiments is unknown, we examined whether ectopically expressed E2F-1 could become phosphorylated upon ligand treatment. HIB1B cells were infected with a retroviral vector encoding a hemagglutinin (HA)-tagged version of E2F-1. No significant changes were observed in either amount or the phosphorylation of E2F-1 in pioglitazone-treated cells (Fig. 7B).
Activation of PPARγ decreases the expression of PP2Ac protein
The decreased PP2A activity that is responsible for the inhibition of binding of E2F/DP complexes to DNA could be regulated through changes at the transcriptional or post-transcriptional levels. This is an especially important question for components of the cell cycle machinery, as many are known to be controlled through protein degradation (Hochstrasser 1995). Therefore, we determined the protein and mRNA levels of the 36-kD catalytic subunit of PP2A in pioglitazone-treated and untreated cells. The treatment with this drug caused a significant decrease in the amount of PP2Ac protein in NIH–PPARγ as well as in HIB1B cells (Fig. 8A). The same treatment with pioglitazone did not change the amount of PP2Ac protein in NIH-vector cells (Fig. 8A, top). In the same experiments we also performed Northern analysis to test whether the decrease of the catalytic subunit of the PP2A occurs at the mRNA level. As shown in Figure 8A (middle) activation of PPARγ did not lead to any change in the amount of RNA of this protein in NIH–PPARγ, HIB1B, or NIH-vector cells. These results indicate that PPARγ regulates the expression of the PP2Ac mainly at the post-transcriptional level.
Figure 8.

Activation of PPARγ leads to decreased PP2Ac but not mRNA. (A) Exponentially growing NIH–PPARγ, NIH-vector, or HIB1B cells were cultured in the absence or presence of 5 μm pioglitazone for 5 days. Total RNA or whole cell lysates were then prepared. For Northern analysis total RNA (10 μg) was blotted to nylon membrane and hybridized with 32P-labeled mouse PP2Ac prepared by RT-PCR from NIH-3T3 cells. An equivalent amount of intact RNA was run in each lane as indicated as 28S RNA. The lysates were equalized for protein content and processed for Western blotting with anti-PP2Ac antibody. (B) HIB16B cells were treated for various times with 5 μm pioglitazone. Lysates were prepared and processed for Western blotting.
A dramatic decrease in the expression level of PP2Ac protein can be observed within 48 hr of pioglitazone treatment of HIB1B cells (Fig. 8B). Because a decrease in E2F/DP DNA-binding activity can be observed first 48 hr after pioglitazone treatment (see Fig. 4B), this is consistent with the finding that down-regulation of PP2Ac can regulate E2F/DP DNA-binding activity.
Discussion
We show here that activation of either isoform of PPARγ, PPARγ1, or PPARγ2, is sufficient to induce growth arrest, as well as to initiate adipogenesis in exponentially growing fibroblast cell lines. It is important to note that these cell lines express approximately one-third the level of PPARγ mRNA and protein that are seen in cultured adipocytes or adipose tissue. Thus, it is very likely that PPARγ does indeed play an important role in cell cycle withdrawal during adipogenesis in vivo. In addition to the effects observed with ectopic expression of PPARγ in fibroblastic cells, we have observed essentially identical results with ligand activation of endogenous PPARγ in murine HIB1B cells, an aggressively growing brown fat cell line derived from tumors produced through the transgenic expression of SV40LT (Ross et al. 1992).
It has been shown that ectopic expression of C/EBPα, which is also induced in adipogenesis, can stimulate cell cycle withdrawal (Umek et al. 1991; Freytag et al. 1994; Lin and Lane 1994). However, two pieces of evidence suggested that this factor alone is probably not responsible for cell cycle withdrawal. First, when expressed at or slightly above adipocyte levels with retroviruses, cell growth continues at a normal rate (Tontonoz et al. 1994b). Second, the induction of C/EBPα occurs relatively late in the process of differentiation in established preadipocyte lines such as 3T3-L1 or 3T3-F442A, well after most cell cycle withdrawal has already taken place (Cao et al. 1991; Tontonoz et al. 1994b). On the other hand, physiological levels of C/EBPα cooperate dramatically with PPARγ in promoting adipogenesis (Tontonoz et al. 1994b; Hu et al. 1995; Brun et al. 1996) and it is entirely possible that these two factors cooperate to bring about a final halt to cell growth.
Ligand activation of PPARγ leads to a progressive decrease in the DNA-binding activity of E2F/DP transcription factors. The members of E2F/DP transcription factors are of particular interest because of the critical role they play in cell growth. The E2F-binding sequence has been identified in the promoters of genes, the products of which are associated with entry into S-phase and DNA synthesis (La Thangue 1994). To date, five members of the E2F (Helin et al. 1992; Kaelin et al. 1992; Shan et al. 1992; Ivey-Hoyle et al. 1993; Lees et al. 1993; Beijersbergen et al. 1994; Ginsberg et al. 1994) and three members of the DP (Girling et al. 1993; Ormondroyd et al. 1995; Wu et al. 1995; Zhang and Chellappan 1995) families have been reported. Overexpression of the members of the E2F or DP family has been shown to be sufficient to allow initiation of S-phase (Johnson et al. 1993; Beijersbergen et al. 1994; Logan et al. 1994, 1995; Qin et al. 1994; Shan and Lee 1994; Lukas et al. 1996) and to lead to the neoplastic transformation of rat embryo fibroblasts (Beijersbergen et al. 1994; Ginsberg et al. 1994; Johnson et al. 1994; Singh et al. 1994; Jooss et al. 1995; Xu et al. 1995), as well as to block terminal differentiation of megakaryocytes (Guy et al. 1996). Furthermore, expression of dominant negative mutant DP-1 has been reported to block cell cycle progression in G1 (Wu et al. 1996). The time-course study shown here (Fig. 4C) demonstrates that the down-regulation of E2F/DP occurs within 48 hr after treatment with pioglitazone and correlates well with the inhibition of the endogenous transcription factor activity of E2F/DP as determined in transfection assays. This time course is also in agreement with the reduction in cell growth, which becomes significant ∼2 days after adding ligand to the cells containing PPARγ. This temporal correlation along with the crucial role played by the E2F/DP complex in cell growth strongly suggests that at least part of this mechanism is responsible for causing the PPARγ-containing cells to exit the cell cycle.
The in vivo and in vitro experiments presented here show that PPARγ-induced loss of E2F/DP DNA-binding activity is attributable to an increase in the phosphorylation of this complex, in large measure to a decrease in the protein level of PP2A catalytic subunit. In vivo labeling experiments reveal that activation of PPARγ leads to an increase in the phosphorylation of DP-1, which can be reversed specifically by the serine–threonine phosphatase PP2Ac. Because DP-1 is a major component of the E2F/DP complex in HIB1B cells, as shown in Figure 5, our observation taken together with the previous reports about the negative regulation of DP-1 activity on serine–threonine phosphorylation could provide an explanation how the DNA-binding activity of E2F/DP decreases in cells treated with pioglitazone. Furthermore, these data also suggest that PP2A plays a crucial role in maintaining E2F/DP transcription factors in the un(der)phosphorylated state at certain key residues. The identity of these key residues and the protein kinases that modify them remain to be determined. Cyclin A–CDK2 has been reported previously to phosphorylate and reduce the DNA-binding activity of E2F/DP complexes (Dynlacht et al. 1994; Xu et al. 1994; Krek et al. 1995). Because cyclin A–CDK2 activity remains high in PPARγ-activated cells (data not shown), this complex must be considered a candidate. However, antibody neutralization experiments have revealed recently that E2F-4 is a major component of the E2F/DP complex in HIB1B cells (data not shown). Because E2F-4, unlike E2F-1, E2F-2, and E2F-3 does not contain a cyclin-binding motif (Krek et al. 1995), it is tempting to speculate that either another kinase is involved in the phosphorylation of E2F/DP complex in PPARγ-activated cells or a new mechanism of cyclin recruitment is implied.
The very important role that E2F/DP complexes have been shown to play in cell cycle progression strongly suggests that the PPARγ-linked decrease in the DNA-binding activity of these factors is linked causally to the differentiation-related cell cycle withdrawal. Could the regulation of E2F/DP activity by PP2A have a general importance beyond adipogenesis in differentiation-linked cell cycle control? There is indirect evidence suggesting that this might indeed be the case. For example, many immortalized cell lines of different cellular lineages established from transgenic animals expressing SV40LT still retain the ability to undergo terminal differentiation and cell cycle withdrawal (Hanahan 1988, 1989). This would suggest the presence of an alternative mechanism in those cells that control cell growth by regulating E2F/DP activity, rather than the function of tumor-suppressor proteins. On the other hand, differentiation-inducing agents have been reported to induce a decrease in the DNA-binding activity of E2F/DP during the differentiation process of several other cell types; post-translational modifications have been implicated in some cases (La Thangue and Rigby 1987; Hara et al. 1993; Melamed et al. 1993; Kranenburg et al. 1995).
Finally, it is worth noting that as differentiation-linked cell cycle withdrawal can be driven by a reduction in PP2A activity, this molecule may be an attractive target for chemotherapeutic intervention in cancer. As noted in previous publications, PP2A inhibitors can cause cell cycle withdrawal. What may be noteworthy here is that this proposed mechanism, direct regulation of E2F/DP by PP2Ac, ought to work even in the absence of the function of tumor suppressors such as pRb, p107, and p130 or p53. Because many cancers may have genetic damage at these loci, direct pharmacological manipulation of PP2A or regulation of its activity through activation of PPARγ in tumors expressing this factor may have special promise.
Materials and methods
Cell culture, transfections, and plasmids
Preparation of the PPARγ2, PPARγ1, PPARγ–M2, PPARγ–M1 viral expression vectors (Tontonoz et al. 1994a,b) and 3xwt-E2F-Luciferase construct (Krek et al. 1993) were described previously. The PPARγ2–CD cDNA (encoding amino acids 1–494) was amplified from the PPARγ2 cDNA by PCR and inserted into the pBabe-Puro retroviral expression vector.
Stable cell lines expressing wild-type or mutant forms of PPARγ were derived as described (Tontonoz et al. 1994b). BOSC23 cells were cultured in 90-mm dishes and transfected at 80% confluence by calcium–phosphate coprecipitation with 10 μg of pBabe-derived expression vector as described (Pear et al. 1993). Forty-eight hours after transfection viral supernatants were collected and NIH-3T3 cells were infected at 50% confluence with equal titers of recombinant virus. The supernatants were applied to the cells in Dulbecco’s modified Eagle medium (DMEM) containing 10% cosmic calf serum (Hyclone) and 4 μg/ml of polybrene. Twenty-four hours after infection, cells were split and plated in DMEM containing 10% calf serum and 2 μg/ml of puromycin to select infected cells. NIH-3T3 cell lines infected with empty vector or with viral expression vectors containing wild-type or mutant forms of PPARγ cDNA as well as HIB1B and 3T3-F442A cell lines were cultured in DMEM containing 10% cosmic calf serum. Pioglitazone (5-[4-[2-(5-ethyl-2-pyridyl)-ethoxy]benzyl]-2,4-thiazolidinedione) (Upjohn) was dissolved in DMSO and used in cell culture experiments.
HIB1B cells were transfected transiently with lipofectAMINE following the protocol provided by the supplier (GIBCO BRL). Briefly, a mixture of E2F luciferase reporter plasmid, β-galactosidase expression vector, and 20 μl of lipofectAMINE in 0.6 ml of DMEM was incubated at room temperature for 45 min and then diluted to 3 ml with DMEM. Cells per 60-mm dish were washed with DMEM and diluted DNA–lipid mixture was added. Five hours after incubation at 37°C, the cells were refed with DMEM containing 10% calf serum. Twenty-four hours after transfection cells were treated with or without pioglitazone for an additional 48 hr. Luciferase (de Wet et al. 1987) and β-galactosidase (Maniatis et al. 1989) experiments were performed as described previously.
RNA and protein analysis
Total RNA was isolated from cultured cells by guanidine isothiocyanate extraction (Chirgwin et al. 1979). RNA, denatured in formamide and formaldehyde, was electrophoresed through formaldehyde-containing agarose gels as described (Maniatis et al. 1989). cDNA probes for mouse PP2A catalytic subunit was prepared by RT–PCR. First-strand DNA was prepared from mouse NIH-3T3 cell total RNA (10 μg) using the cDNA Cycle Kit (Invitrogen). The sequences of the PCR primers used were 5′-ATGGACGAGAAGTTGTTCACCAGG-3′ and 5′-TTACAGGAAGTAAGTCTGGGGTAC-3′. PCR products were isolated after electrophoresis through an agarose gel and labeled with [α-32P]dCTP (6000 Ci/mmole) by the random-priming method (Feinberg and Vogelstein 1984) to a specific activity of at least 109 cpm/μg. For Western blot analysis cellular extracts were prepared as described (Maniatis et al. 1989) and blotted with a polyclonal anti-PP2A catalytic subunit antibody (Upstate Biotech, Inc.).
Electrophoretic mobility-shift assays
Whole cell extracts were prepared and electrophoretic mobility-shift assays were performed as described previously (Shirodkar et al. 1992). The double-stranded oligonucleotides used as a labeled DNA probe included the E2F-binding site 5′-ATTTAAGTTTCGCGCCCTTTCTCAA-3′ (Cao et al. 1992) or Oct-binding site 5′-TGTCGAATGCAAATCACTAGAA-3′ (Scheidereit et al. 1988). The mutant E2F oligonucleotide was 5′-ATTTAAGTTTCGATCCCTTTCTCAA-3′ (Cao et al. 1992). For competition experiments 100-fold molar excess of unlabeled oligonucleotides were added before the addition of labeled probe. The reaction products were separated in a 4% polyacrylamide gel run in 0.25× TBE (22.5 mm Tris-borate and 0.5 mm EDTA). The gel was dried, and autoradiography with intensifying screen was performed.
BrdU incorporation experiments
BrdU incorporation experiment was performed as described in the protocol provided by the supplier (Boehringer Mannheim Biochemical). Briefly, cells grown on coverslips were labeled with 10 μm BrdU for 1 hr. Samples were washed and fixed with ethanol–glycine buffer and incubated with anti-BrdU monoclonal antibody. After incubation with anti-mouse-immunoglobulin-alkaline phosphatase followed by the substrate reaction, bound anti-BrdU antibody was visualized by light microscopy.
Metabolic labeling and immunoprecipitations
HIB16B cells were labeled metabolically with [35S]methionine (1 mCi/ml) or [32P]orthophosphate (2 mCi/ml) for 4 hr. Cells were lysed in a buffer containing 50 mm Tris HCl (pH 7.4), 150 mm NaCl, 5 mm EDTA, 1% Tween 20, 1 mm sodium orthophosphate, 10 mm NaF, 0.5 mm PMSF, 2 μg/ml of aprotinin, 2 μg/ml of leupeptin. Proteins were precipitated with antibodies as indicated and Sepharose CL-4B–protein A beads (Pharmacia). Immunocomplexes were washed four times. For immune reprecipitation experiments, washed immunobeads were boiled for 10 min in 50 μl of 50 mm Tris HCl (pH 7.4), 1% SDS, and 1 mm DTT, followed by addition of 0.5 ml of lysis buffer, and incubated with DP-1 antibody at 4°C.
For in vitro translation of DP-1 and HA–E2F-1[35S]methionine-labeled translation products were prepared with the TNT lysate system (Promega) according to the manufacturer’s instructions.
Acknowledgments
We thank W.G. Kaelin, Jr. and E. Flemington for helpful discussions and critical reading of this manuscript. We thank P. Tontonoz for providing wild-type and mutant forms of PPARγ expression vectors, E. Flemington for providing plasmids and A33 DP-1 antibody. S.A. was supported by fellowships from the Swiss National Fonds, the CIBA Research Foundation, and the American Heart Association Massachussets Affiliate. This work was supported by the National Institutes of Health grant DK31405 to B.M.S.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Bruce_Spiegelman@dfci.harvard.edu; FAX (617) 632-4655.
References
- Beijersbergen RL, Kerkhoven RM, Zhu L, Carlee L, Mathijs FM, Bernards R. E2F-4, a new member of the E2F gene family, has oncogenic activity and associates with p107 in vivo. Genes & Dev. 1994;8:2680–2690. doi: 10.1101/gad.8.22.2680. [DOI] [PubMed] [Google Scholar]
- Brun RP, Tontonoz P, Forman BM, Ellis R, Chen J, Evans RM, Spiegelman BM. Differential activation of adipogenesis by multiple PPAR isoforms. Genes & Dev. 1996;10:974–984. doi: 10.1101/gad.10.8.974. [DOI] [PubMed] [Google Scholar]
- Cao L, Faha B, Dembski M, Tsai LH, Harlow E, Dyson N. Independent binding of the retinoblastoma protein and p107 to the transcription factor E2F. Nature. 1992;355:176–179. doi: 10.1038/355176a0. [DOI] [PubMed] [Google Scholar]
- Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes & Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Das A, Gale M, Carter V, Parsons M. The protein phosphatase inhibitor okadaic acid induces defects in cytokinesis and organellar genome segregation in Trypanosoma brucei. J Cell Sci. 1994;107:3477–3483. doi: 10.1242/jcs.107.12.3477. [DOI] [PubMed] [Google Scholar]
- de Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: Structure and expression in mammalian cells. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Flores O, Lees JA, Harlow E. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes & Dev. 1994;8:1772–1786. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- Dyson N, Buchkovich K, Whyte P, Harlow E. The cellular 107K protein that binds to adenovirus E1A also associates with the large T antigens of SV40 and JC virus. Cell. 1989;58:249–255. doi: 10.1016/0092-8674(89)90839-8. [DOI] [PubMed] [Google Scholar]
- Ewen ME, Ludlow JW, Marsilio E, DeCaprio JA, Millikan RC, Cheng SH, Paucha E, Livingston DM. An N-terminal transformation-governing sequence of SV40 large T antigen contributes to the binding of both p110Rb and a second cellular protein, p120. Cell. 1989;58:257–267. doi: 10.1016/0092-8674(89)90840-4. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1984;137:266–267. doi: 10.1016/0003-2697(84)90381-6. [DOI] [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- Freytag SO, Paielli DL, Gilbert JD. Ectopic expression of the CCAAT/enhancer-binding protein alpha promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes & Dev. 1994;8:1654–1663. doi: 10.1101/gad.8.14.1654. [DOI] [PubMed] [Google Scholar]
- Ginsberg D, Vairo G, Chittenden T, Xiao ZX, Xu G, Wydner KL, DeCaprio JA, Lawrence JB, Livingston DM. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes & Dev. 1994;8:2665–2679. doi: 10.1101/gad.8.22.2665. [DOI] [PubMed] [Google Scholar]
- Girling R, Partridge JF, Bandara LR, Burden N, Totty NF, Hsuan JJ, La Thangue NB. A new component of the transcription factor DRTF1/E2F. Nature. 1993;365:468. doi: 10.1038/365468d0. [DOI] [PubMed] [Google Scholar]
- Guo K, Wang J, Andres V, Smith RC, Walsh K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Zhou W, Kaufman S, Robinson MO. E2F-1 blocks terminal differentiation and causes proliferation in transgenic megakaryocytes. Mol Cell Biol. 1996;16:685–693. doi: 10.1128/mcb.16.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lasser AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Dissecting multistep tumorigenesis in transgenic mice. Annu Rev Genet. 1988;22:479–519. doi: 10.1146/annurev.ge.22.120188.002403. [DOI] [PubMed] [Google Scholar]
- ————— Transgenic mice as probes into complex systems. Science. 1989;246:1265–1275. doi: 10.1126/science.2686032. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Demetrick D, Beach D. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes & Dev. 1993;7:2378–2391. doi: 10.1101/gad.7.12a.2378. [DOI] [PubMed] [Google Scholar]
- Hara E, Okomoto S, Nakada S, Taya Y, Sekiya S, Oda K. Protein phosphorylation required for the formation of E2F complex regulates N-myc transcription during differentiation of human embryonal carcinoma cells. Oncogene. 1993;8:1023–1032. [PubMed] [Google Scholar]
- Helin K, Lees JA, Vidal M, Dyson N, Harlow E, Fattaey A. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell. 1992;70:337–350. doi: 10.1016/0092-8674(92)90107-n. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin, proteosomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- Hu E, Tontonoz P, Spiegelman BM. Transdifferentiation of myoblasts by the adipogenic transcription factors PPAR gamma and C/EBP alpha. Proc Natl Acad Sci. 1995;92:9856–9860. doi: 10.1073/pnas.92.21.9856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPAR-gamma. Science. 1996;274:2100–2103. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- Ivey-Hoyle M, Conroy R, Huber HE, Goodhart PJ, Oliff A, Heimbrook DC. Cloning and characterization of E2F-2, a novel protein with the biochemical properties of transcription factor E2F. Mol Cell Biol. 1993;13:7802–7812. doi: 10.1128/mcb.13.12.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Cress WD, Jakoi L, Nevins JR. Oncogenic capacity of the E2F1 gene. Proc Natl Acad Sci. 1994;91:12823–12827. doi: 10.1073/pnas.91.26.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jooss K, Lam EW, Bybee A, Girling R, Muller R, La Thangue NB. Proto-oncogenic properties of the DP family of proteins. Oncogene. 1995;10:1529–1536. [PubMed] [Google Scholar]
- Kaelin WG, Jr, Krek W, Sellers WR, DeCaprio JA, Ajchenbaum FA, Fuchs CS, Chittenden T, Li Y, Farnham PJ, Blanar MA, et al. Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell. 1992;70:351–364. doi: 10.1016/0092-8674(92)90108-o. [DOI] [PubMed] [Google Scholar]
- Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes & Dev. 1996;10:1096–1107. doi: 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- Kinoshita N, Ohkura H, Yanagida M. Distinct, essential roles of type 1 and 2A protein phosphatases in the control of the fission yeast cell division cycle. Cell. 1990;63:405–415. doi: 10.1016/0092-8674(90)90173-c. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Higashi H, Suzuki-Takahashi I, Segawa K, Hanks SK, Taya Y, Nishimura S, Okuyama A. Phosphorylation of E2F-1 by cyclin A-cdk2. Oncogene. 1995;10:229–236. [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, de Groot RP, Van der Eb AJ, Zantema A. Differentiation of P19 EC cells leads to differential modulation of cyclin-dependent kinase activities and to changes in the cell cycle profile. Oncogene. 1995;10:87–95. [PubMed] [Google Scholar]
- Krek W, Livingston DM, Shirodkar S. Binding to DNA and the retinoblastoma gene product promoted by complex formation of different E2F family members. Science. 1993;262:1557–1560. doi: 10.1126/science.8248803. [DOI] [PubMed] [Google Scholar]
- Krek W, Ewen ME, Shirodkar S, Arany Z, Kaelin WG, Jr, Livingston DM. Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell. 1994;78:161–172. doi: 10.1016/0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- Krek W, Xu G, Livingston DM. Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell. 1995;83:1149–1158. doi: 10.1016/0092-8674(95)90141-8. [DOI] [PubMed] [Google Scholar]
- La Thangue NB. DP and E2F proteins: Components of a heterodimeric transcription factor implicated in cell cycle control. Curr Opin Cell Biol. 1994;6:443–450. doi: 10.1016/0955-0674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- La Thangue NB, Rigby PW. An adenovirus E1A-like transcription factor is regulated during the differentiation of murine embryonal carcinoma stem cells. Cell. 1987;49:507–513. doi: 10.1016/0092-8674(87)90453-3. [DOI] [PubMed] [Google Scholar]
- Lassar A, Munsterberg A. Wiring diagrams: Regulatory circuits and the control of skeletal myogenesis. Curr Opin Cell Biol. 1994;6:432–442. doi: 10.1016/0955-0674(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Lees JA, Saito M, Vidal M, Valentine M, Look T, Harlow E, Dyson N, Helin K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol. 1993;13:7813–7825. doi: 10.1128/mcb.13.12.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkinson WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Lin FT, Lane MD. CCAAT/enhancer binding protein alpha is sufficient to initiate the 3T3-L1 adipocyte differentiation program. Proc Natl Acad Sci. 1994;91:8757–8761. doi: 10.1073/pnas.91.19.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan TJ, Jordan KL, Hall D. Altered shape and cell cycle characteristics of fibroblasts expressing the E2F1 transcription factor. Mol Biol Cell. 1994;5:667–678. doi: 10.1091/mbc.5.6.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow JW, DeCaprio JA, Huang CM, Lee WH, Paucha E, Livingston DM. SV40 large T antigen binds preferentially to an underphosphorylated member of the retinoblastoma susceptibility gene product family. Cell. 1989;56:57–65. doi: 10.1016/0092-8674(89)90983-5. [DOI] [PubMed] [Google Scholar]
- Lukas J, Petersen BO, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol Cell Biol. 1996;16:1047–1057. doi: 10.1128/mcb.16.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Mayer-Jaekel RE, Ohkura H, Gomes R, Sunkel E, Baumgartner S, Hemmings BA, Glover DM. The 55 kd regulatory subunit of Drosophila protein phosphatase 2A is required for anaphase. Cell. 1993;72:621–633. doi: 10.1016/0092-8674(93)90080-a. [DOI] [PubMed] [Google Scholar]
- Melamed D, Tiefenbrun N, Yarden A, Kimichi A. Interferons and interleukin-6 suppress the DNA-binding activity of E2F in growth-sensitive hematopietic cells. Mol Cell Biol. 1993;13:5255–5265. doi: 10.1128/mcb.13.9.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P. Ordering S phase and M phase in the cell cycle. Cell. 1994;79:547–550. doi: 10.1016/0092-8674(94)90539-8. [DOI] [PubMed] [Google Scholar]
- Ormondroyd E, de la Luna S, La Thangue NB. A new member of the DP family, DP-3, with distinct protein products suggests a regulatory role for alternative splicing in the cell cycle transcription factor DRTF1/E2F. Oncogene. 1995;11:1437–1446. [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Kaelin WG, Jr, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SR, Choy L, Graves RA, Fox N, Solevjeva V, Klaus S, Ricquier D, Spiegelman BM. Hibernoma formation in transgenic mice and isolation of a brown adipocyte cell line expressing the uncoupling protein gene. Proc Natl Acad Sci. 1992;89:7561–7565. doi: 10.1073/pnas.89.16.7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson L, Stromberg K, Vikman K, Bjursell G, Enerback S. The CCAAT/enhancer binding protein and its role in adipocyte differentiation: Evidence for direct involvement in terminal adipocyte development. EMBO J. 1991;10:3787–3793. doi: 10.1002/j.1460-2075.1991.tb04948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidereit C, Cromlish JA, Gerster T, Kawakami K, Balmaceda C, Currie RA, Roeder RG. A human lymphoid-specific transcription factor that activates immunoglobulin genes is a homeobox protein. Nature. 1988;336:551–557. doi: 10.1038/336551a0. [DOI] [PubMed] [Google Scholar]
- Shan B, Lee WH. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol. 1994;14:8166–8173. doi: 10.1128/mcb.14.12.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Zhu X, Chen PL, Durfee T, Yang Y, Sharp D, Lee WH. Molecular cloning of cellular genes encoding retinoblastoma-associated proteins: Identification of a gene with properties of the transcription factor E2F. Mol Cell Biol. 1992;12:5620–5631. doi: 10.1128/mcb.12.12.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. G1 phase progression: Cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Shirodkar S, Ewen M, DeCaprio JA, Morgan J, Livingston DM, Chittenden T. The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell. 1992;68:157–166. doi: 10.1016/0092-8674(92)90214-w. [DOI] [PubMed] [Google Scholar]
- Singh P, Wong SH, Hong W. Overexpression of E2F-1 in rat embryo fibroblasts leads to neoplastic transformation. EMBO J. 1994;13:3329–3338. doi: 10.1002/j.1460-2075.1994.tb06635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Spicer DB, Lassar AB. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Kim JB, Graves RA, Spiegelman BM. ADD1: A novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol. 1993;13:4753–4759. doi: 10.1128/mcb.13.8.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARγ: Tissue-specific regulator of an adipocyte enhancer. Genes & Dev. 1994a;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994b;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Umek RM, Friedman AD, McKnight SL. CCAAT-enhancer binding protein: A component of a differentiation switch. Science. 1991;251:288–292. doi: 10.1126/science.1987644. [DOI] [PubMed] [Google Scholar]
- Warrel RP, Fankel SR, Miller WH, Scheinberg DA, Itri LM, Hittleman WN, Vyas R, Andreef M, Tafuri A, Jakubowski A, Gabrilove J, Gordon MS, Dmitrovsky E. Differentiation therapy of acute promyelocytic leukemia with tretinoin. N Engl J Med. 1991;324:1385–1393. doi: 10.1056/NEJM199105163242002. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Wolf DA, Hermeking H, Albert T, Herzinger T, Kind P, Eick D. A complex between E2F and the pRb-related protein p130 is specifically targeted by the Simian virus 40 large T antigen during cell transformation. Oncogene. 1995;10:2067–2078. [PubMed] [Google Scholar]
- Wu CL, Zukerberg LR, Ngwu C, Harlow E, Lees JA. In vivo association of E2F and DP family proteins. Mol Cell Biol. 1995;15:2536–2546. doi: 10.1128/mcb.15.5.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-L, Classon M, Dyson N, Harlow E. Expression of dominant-negative mutant DP-1 blocks cell cycle progression in G1. Mol Cell Biol. 1996;16:3698–3706. doi: 10.1128/mcb.16.7.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Xie Y, Bucher NLR, Farmer SR. Conditional ectopic expression of C/EBPβ in NIH-3T3 cells induces PPARγ and stimulates adipogenesis. Genes & Dev. 1995;9:2350–2365. doi: 10.1101/gad.9.19.2350. [DOI] [PubMed] [Google Scholar]
- Xu G, Livingston DM, Krek W. Multiple members of the E2F transcription factor family are the products of oncogenes. Proc Natl Acad Sci. 1995;92:1357–1361. doi: 10.1073/pnas.92.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica-Worms H. Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol Cell Biol. 1994;14:8420–8431. doi: 10.1128/mcb.14.12.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WC, Cao Z, Classon M, McKnight SL. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes & Dev. 1995;9:168–181. doi: 10.1101/gad.9.2.168. [DOI] [PubMed] [Google Scholar]
- You J, Bird RC. Selective induction of cell cycle regulatory genes cdk1 (p34cdc2), cyclins A/B, and the tumor suppressor gene Rb in transformed cells by okadaic acid. J Cell Physiol. 1995;164:424–433. doi: 10.1002/jcp.1041640223. [DOI] [PubMed] [Google Scholar]
- Zalvide J, DeCaprio JA. Role of pRb-related proteins in simian virus 40 large-T-antigen-mediated transformation. Mol Cell Biol. 1995;15:5800–5810. doi: 10.1128/mcb.15.10.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chellappan SP. Cloning and characterization of human DP2, a novel dimerization partner of E2F. Oncogene. 1995;10:2085–2093. [PubMed] [Google Scholar]
- Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J Biol Chem. 1993;268:26817–26820. [PubMed] [Google Scholar]