

Abstract
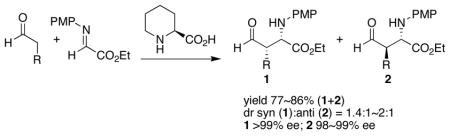
Mannich reactions between aldehydes and N-p-methoxyphenyl-protected α-imino ethyl glyoxylate have been performed using (S)-pipecolic acid as catalyst. The reactions give both syn- and anti-products (dr = 1.4–2:1) with high enantioselectivities (>98% ee). In contrast, (S)-proline-catalyzed reactions give mainly syn-products with high enantioselectivities. Computational studies reveal that the energetic preference between the transition structures involving the s-cis-enamine and the s -trans-enamine is smaller for the pipecolic acid as compared to proline, yielding the (2S,3R)-anti- and the (2S,3S)-syn Mannich product, in nearly equal amounts.
Preformed enamines of both five-membered pyrrolidine and six-membered piperidine rings have been used as nucleophiles in many reactions.1,2 For the reactions involving in situ-generated enamines, pyrrolidine-based catalysts have been extensively examined.3,4,5 One of the most effective routes for the synthesis of enantiomerically enriched α- and β-amino acid derivatives is pyrrolidine derivative-catalyzed Mannich-type reactions of an aldehyde donor. (S)-Proline and various (S)-proline-derivatives give the syn-product (2S,3S)-1 as the major product with high diastereo- and enantioselectivity (Scheme 1).4 The six-membered analog, pipecolic acid, has received little attention as a catalyst for asymmetric reactions, and has proven ineffective for aldol reactions involving acetone as donor.3a,6 Here we report the experimental and computational investigation of (S)-pipecolic acid-catalyzed Mannich reaction between aldehydes and N-p-methoxyphenyl (N -PMP)-protected α-imino ethyl glyoxylate.4
Scheme 1.

Mechanism of the (S)-proline-catalyzed Mannich reaction of aldehydes with α-imino ethyl glyoxylate.
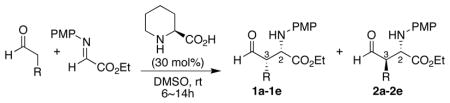
Pipecolic acid catalyzed Mannich reactions provide both the syn-product 1 and anti-product 2 in good yields (Table 1). The reaction rates were similar to that of proline-catalyzed reactions under the same conditions. The enantioselectivities of the syn-product (2S,3S)-14 and anti-product (2S,3R)-24,6 were both typically greater than 98% ee. The diastereomeric ratio of syn-product 1 to anti-product 2 ranged from 2:1 to 1:1, regardless of the bulkiness of the aldehyde substituent (Table 1). The insensitivity of the diastereoselectivity to steric bulk is in sharp contrast to the proline-catalyzed reactions, in which a bulky R groups often led to excellent enantio- and diastereoselectivity.4 (S)-Pipecolic acid catalysis provides a route to highly enantiomerically pure products of both diastereomers.
Table 1.
(S)-Pipecolic acid-catalyzed Mannich reaction of aldehydes and α-imino ethyl glyoxylate to afford products syn-1a-1e and anti-2a-2e.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Product | Yield (%) | dr syn(1):anti(2) |
ee (%)
|
|
| syn(1) | anti(2) | |||||
| 1 | Me | 1a+2a | 80 | 2.0:1 | >99 | >99 |
| 2 | i-Pr | 1b+2b | 82 | 1.4:1 | >99 | 98 |
| 3 | n-Bu | 1c+2c | 83 | 1.5:1 | >99 | >99 |
| 4 | n-Pent | 1d+2d | 86 | 1.4:1 | >99 | >99 |
| 5 |
|
1e+2e | 77 | 1.6:1 | >99 | >99 |
Typical conditions: To a solution of N-PMP-protected α-imino ethyl glyoxylate (0.5 mmol) and aldehyde (1.0 mmol) in anhydrous DMSO (5 mL), (S)-pipecolic acid (0.15 mmol) was added, and the mixture was stirred for 6–14 h at room temperature. The diastereomeric ratio was determined without purification by 1H NMR. The enantiomeric excess was determined by chiral-phase HPLC analysis.
This unusual change in diastereoselectivity upon increase in the ring size of the catalyst caused us to investigate these reactions computationally. The enamine of propionaldehyde, N-PMP-protected α-imino methyl gloxylate and the transition structures leading to the four possible stereoisomeric products for both proline and pipecolic acid were calculated at the HF level of theory with 6-31G(d) basis set.7 We have previously used density functional theory to study related organocatalytic reactions.8 However, HF/6-31G(d) was used over B3LYP/6-31G(d) in this study for rapidly computing the stereoselectivity.
The s-cis- or s-trans-enamine attack on the re or si face of the imine acceptor is the stereo- and rate-determining step of this reaction. Four possible diastereomeric transition structures are possible that allow for intramolecular proton transfer. The four lowest energy transition structures involving (S)-proline and (S)-pipecolic acid are shown in Figure 1.
Figure 1.

Transition structures for the C-C bond formation of the (S)-proline and (S)-pipecolic acid-catalyzed Mannich reaction between propionaldehyde and N-PMP-protected α-imino methyl gloxylate. S-trans-si transition structures TS-(S,S)-4 and TS-(S,S)-8 give rise to product 1a, while TS-(R,S)-6 and TS-(R,S)-10 gives rise to 2a.
The syn-product 1 arises from the s-trans-si transition state and the anti-product 2 from the s-cis-si. The conformations of the proline enamine were previously discussed as anti- and syn-enamine. 8 These notations are changed to s-trans- and s-cis-enamine, respectively, in this work to distinguish from anti and syn diastereoselective products. In the proline-catalyzed reaction, the computed energy difference between these transition structures, TS-(S,S)-4 and T S - (R,S)-6, is 1.0 kcal/mol. The corresponding energy difference for the pipecolic acid-catalyzed reaction between TS-(S,S)-8 and TS-(R,S)-10 is only 0.2 kcal/mol. This decrease in energetic difference reflects the experimentally observed decrease in diastereoselectivity for the pipecolic acid-catalyzed reaction.
The computed selectivities arising from the relative energies for all transition structures are summarized in Table 2. There is an excellent agreement between the computed stereoselectivity and the observed product ratios.
Table 2.
Comparison of the experimentally observed product ratios involving reaction of propionaldehyde with N-PMP-protected α-imino ethyl glyoxylate with computed stereoselectivities based on transition state theory predictions involving N-PMP-protected α-imino methyl glyoxylate.
| Entry | Catalyst | Type | dr syn(1):anti(2) |
ee (%) syn (anti) |
|---|---|---|---|---|
| 1 | Proline | Exp. | 3:1 | >99 |
| 2 | Proline | Computed | 3.5:1 | 97 |
| 3 | Pipecolic acid | Exp. | 2:1 | >99 (>99) |
| 4 | Pipecolic acid | Computed | 1.4:1 | 93 (96) |
The facial re or si selectivity of the imine acceptor is governed by the necessity for intramolecular proton transfer and minimization of steric interactions between the imine and the reactive enamine. The (E)-imine is more stable than the (Z)-imine. Combined with the observation that transition structures involving intramolecular proton transfer are favored, the re-face attacks necessitate substantial eclipsing of the imine and enamine (TS-(S,R)-3, TS-(S,R)-7, TS-(R,R)-5, and TS-(R,R)-9, Figure 1). Consequently, as shown in Figure 1, the s-trans-re and s-cis-re transition structures are higher in energy by >1 kcal/mol than the s-trans-si or s-cis-si transition structure, for both proline and pipecolic acid.
The re or si attack on the enamine is determined by whether the s-cis or s-trans enamine conformer is favored in the transition state. In the case of proline, the transition structures involving the s-trans-enamine are favored over those that involve the s-cis-enamine. The latter involves distortions of the developing iminium from planarity to accommodate proton transfer. Thus proline provides the syn-product (2S,3S)-1 as the major product.8
This differentiation is weakened in the case of pipecolic acid. The piperidine ring has different steric interactions with the s-trans- or s-cis-enamines than the pyrrolidine ring of proline. The relatively rigid piperidine ring holds the carboxylic acid more rigidly than the more flexible pyrrolidine. This alters electrostatic interactions with the ester of the iminoglyoxylate and with the protonated imine. These differences allow the imine to react via both the s-trans- and s-cis-enamine, giving rise to roughly equal amounts of both syn-product (2S,3S)-1 and anti-product (2S,3R)-2.
The (S)-pipecolic acid-catalyzed Mannich reactions of aldehydes affords ready access to both syn- and anti-products with high enantioselectivities. In contrast, proline-catalyzed reactions yield primarily the syn product. Work is under way to further develop anti-selective Mannich catalysts based on these discoveries.9,10
Supplementary Material
Acknowledgments
We are grateful to the Skaggs Institute for Chemical Biology, the National Institute of General Medical Sciences, and the National Institutes of Health for financial support of this research. This research was facilitated through the Partnerships for Advanced Computational Infrastructure (PACI) through the support of the National Science Foundation. The computational analyses were performed on the National Science Foundation Terascale Computing System at the Pittsburgh Supercomputing Center (PSC), the UCLA Academic Technology Services (ATS) Hoffman Beowulf cluster, and the California Nano Systems Institute Itanium cluster.
Footnotes
Supporting Information Available. Experimental procedures, characterization of compound, cartesian coordinates, energies, thermodynamic corrections for all reported structures, full authorship of Gaussian and Q-Chem.
References
- 1.Kempf B, Hampel N, Ofial AR, Mayr H. Chem Eur J. 2003;9:2209. doi: 10.1002/chem.200204666. [DOI] [PubMed] [Google Scholar]
- 2.(a) Stork G, Brizzolara A, Landesman H, Szmuszkovicz J, Terrell R. J Am Chem Soc. 1963;85:207. [Google Scholar]; (b) Hickmott PW. Tetrahedron. 1982;38:1975. [Google Scholar]
- 3.(a) Sakthivel K, Notz N, Bui T, Barbas CF., III J Am Chem Soc. 2001;123:5260. doi: 10.1021/ja010037z. [DOI] [PubMed] [Google Scholar]; (b) Notz W, Tanaka F, Barbas CF., III Acc Chem Res. 2004;37:580. doi: 10.1021/ar0300468. [DOI] [PubMed] [Google Scholar]; (c) Northrup AB, Mangion IK, Hettche F, MacMillan DWC. Angew Chem Int Ed. 2004;43:2152. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]; (d) Suri JT, Ramachary DB, Barbas CF., III Org Lett. 2005;7:1383. doi: 10.1021/ol0502533. [DOI] [PubMed] [Google Scholar]; (e) Allemann C, Gordillo R, Clemente FR, Cheong PHY, Houk KN. Acc Chem Res. 2004;37:558. doi: 10.1021/ar0300524. [DOI] [PubMed] [Google Scholar]
- 4.(a) Cordova A, Watanabe S, Tanaka F, Notz W, Barbas CF., III J Am Chem Soc. 2002;124:1866. doi: 10.1021/ja017833p. [DOI] [PubMed] [Google Scholar]; (b) Notz W, Tanaka F, Watanabe S, Chowdari NS, Turner JM, Thayumanuvan R, Barbas CF., III J Org Chem. 2003;68:9624. doi: 10.1021/jo0347359. [DOI] [PubMed] [Google Scholar]; (c) Chowdari NS, Ramachary DB, Barbas CF., III Synlett. 2003:1906. [Google Scholar]; (d) Chowdari NS, Suri JT, Barbas CF., III Org Lett. 2004;6:2507. doi: 10.1021/ol049248+. [DOI] [PubMed] [Google Scholar]
- 5.(a) List B. J Am Chem Soc. 2000;122:9336. [Google Scholar]; (b) List B, Pojarliev P, Biller WT, Martin HJ. J Am Chem Soc. 2002;124:827. doi: 10.1021/ja0174231. [DOI] [PubMed] [Google Scholar]; (c) Cordova A, Notz W, Zhong G, Betancort JM, Barbas CF., III J Am Chem Soc. 2002;124:1842. doi: 10.1021/ja017270h. [DOI] [PubMed] [Google Scholar]; (d) Hayashi Y, Tsuboi W, Ashimine I, Urushima T, Shoji M, Sakai K. Angew Chem, Int Ed. 2003;42:3677. doi: 10.1002/anie.200351813. [DOI] [PubMed] [Google Scholar]; (e) Hayashi Y, Tsuboi W, Shoji M, Suzuki N. J Am Chem Soc. 2003;125:11208. doi: 10.1021/ja0372513. [DOI] [PubMed] [Google Scholar]; (f) Notz W, Watanabe S, Chowdari NS, Zhong G, Betancort JM, Tanaka F, Barbas CF., III Adv Synth Cat. 2004;346:1131. [Google Scholar]; (g) Kumaragurubaran N, Juhl K, Zhuang W, Bogevig A, Jorgensen KA. J Am Chem Soc. 2002;124:6254. doi: 10.1021/ja026412k. [DOI] [PubMed] [Google Scholar]; (h) Brown SP, Brochu M, Sinz CJ, MacMillan WC. J Am Chem Soc. 2003;125:10808. doi: 10.1021/ja037096s. [DOI] [PubMed] [Google Scholar]
- 6.(S)-Pipecolic acid catalyzes the Morita-Baylis-Hillman reaction. Aroyan CE, Vasbinder MM, Miller SJ. Org Lett. 2005;7:3849. doi: 10.1021/ol0513544.
- 7.Q-Chem and Gaussian series of programs were used in this work. See supporting information for full authorships. Frisch MJ, et al. Gaussian03. Gaussian, Inc; Wallingford CT: 2004. Kong J, et al. Q-Chem, Version 2.0. Q-Chem, Inc; Export, PA: 2000.
- 8.(a) Bahmanyar S, Houk KN. J Am Chem Soc. 2001;123:12911. doi: 10.1021/ja011714s. [DOI] [PubMed] [Google Scholar]; (b) Bahmanyar S, Houk KN. Org Lett. 2003;5:1249. doi: 10.1021/ol034198e. [DOI] [PubMed] [Google Scholar]; (c) Bahmanyar S, Houk KN, Martin HJ, List B. J Am Chem Soc. 2003;125:2475. doi: 10.1021/ja028812d. [DOI] [PubMed] [Google Scholar]; (c) Cheong PHY, Houk KN, Warrier JS, Hanessian S. Adv Synth Cat. 2004;346:1111. [Google Scholar]; (d) Cheong PHY, Houk KN. J Am Chem Soc. 2004;126:13912. doi: 10.1021/ja0464746. [DOI] [PubMed] [Google Scholar]; (e) Cheong PHY, Houk KN. Synthesis. 2005;9:1533. [Google Scholar]
- 9.An organocatalyst derived from chiral aminosulfonamide has been shown to be anti-selective. Kano T, Yamaguchi Y, Tokuda O, Maruoka K. J Am Chem Soc. 2005;127:16408. doi: 10.1021/ja056008w.
- 10.Mitsumori S, Zhang H, Cheong PH-Y, Houk KN, Tanaka F, Barbas CF., III J Am Chem Soc. 2005 doi: 10.1021/ja056984f. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.