

Abstract
B3LYP/6-31G(d) density functional theory has been used to study Diels-Alder reactions of cyclopentadiene with α,β-unsaturated aldehydes and ketones organocatalyzed by MacMillan’s chiral imidazolidinones. Preferred conformations of transition structures and reaction intermediates have been located. The dramatically different reactivities and enantioselectivities exhibited by two similar chiral imidazolidinones are rationalized.
Introduction
The introduction of specific chirality into synthetic targets using metal-free chiral organocatalysts has become a field of great interest in recent years.1 In 2000, MacMillan and coworkers reported the first highly enantioselective Diels-Alder reaction of cyclopentadiene with α,β-unsaturated aldehydes catalyzed by the chiral imidazolidinone 1, derived from phenylalanine (eq 1). The authors demonstrated that the reaction of a variety of aldehydes (R = Me, Pr, i-Pr, Ph, furyl) occurs with good yield and enantioselectivity (>75% yield, endo>90% ee, exo>84% ee).2
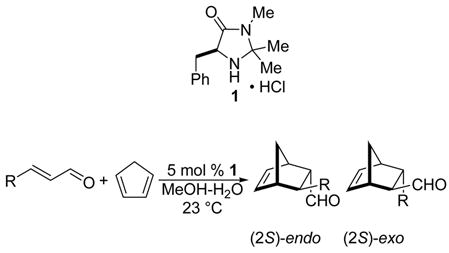 |
(1) |
Catalyst 1 was also successful for 1,3-dipolar cycloaddition reactions between nitrones and and α,β-unsaturated aldehydes,3 alkylation reactions of pyrroles by olefinic aldehydes,4 and α-chlorination of aldehydes.5
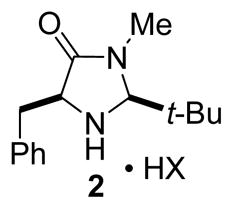
For asymmetric alkylations of indoles, enantioselectivities were improved by using the 2-t-butyl imidazolidinone 2.6 Compound 2 gave also excellent results as enantioselective organocatalyst in 1,4-addition reactions of electron-rich benzenes to α,β-unsaturated aldehydes,7 in the synthesis of butenolides by Mukaiyama-Michael with silyloxyfurans.8
In 2004, MacMillan et al. reported the enantioselective synthesis of (—)-flustramine B, using 6-bromotryptamine and acrolein as starting materials, and compound 2 as organocatalyst, by a cascade addition-cyclization strategy (Scheme 1).9
Scheme 1.

Total synthesis of (—)-flustramine B using catalyst 2.9
More recently, the same research group used imidazolidinone 2 for aldehyde-aldehyde aldol reactions obtaining as major compound the enantiomer of the most favored product in the proline catalyzed process.10 List and coworkers have also obtained good yields and enantioselectivities in asymmetric hydrogenations of α,β-unsaturated aldehydes using amine 2 as organocatalyst.11
One important factor that determines efficiency of imidazolidinone catalysts in their asymmetric enantioselective reactions is the reversible formation of iminium ions from chiral imidazolidinones and α,β-unsaturated carbonyl compounds. Hydrolysis to the final products generally occurs smoothly, and does not have any influence in the chiral center generated in the previous step of the reaction (Scheme 2).
Scheme 2.

Activation of the carbonyl compound by iminium ion formation.
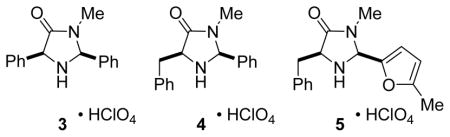
Surprisingly, catalysts 1 and 2 were found to be unsuccessful in the Diels-Alder cycloaddition reactions of cyclopentadiene with 4-hexen-3-one (<30% yield, 0% ee). Higher reaction rates and moderate enantioselectivity could be obtained by using the cis-2,5-diphenylamine 3 (88% yield, 21:1 endo:exo, 47% ee). Introduction of a benzyl group at C2 of the imidazolidinone ring (catalyst 4) provided a higher enantioselectivity (83% yield, 23:1 endo:exo, 82% ee). The best results were obtained with amine 5 which possesses a 5-methyl-2-furyl group at C2 (89% yield, 25:1 endo:exo, 90% ee).12
MacMillan et al. explained the enantioselectivities according to a model of the iminium ion 6 calculated with MM3, depicted in Figure 1.12,13 The trans-iminium isomer 6 will be energetically disfavored on the basis of nonbonding interactions between the benzyl and CH2-• (green) substituents, and the calculated cis-iminium isomer 6 will be selectively exposed to cycloaddition at the Si-face.
Figure 1.
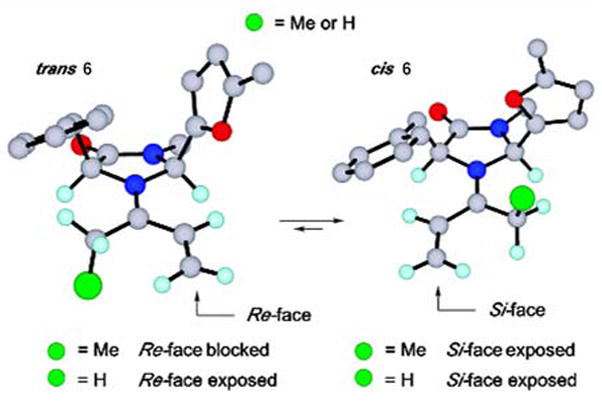
Computed MM conformations of iminium ion 6, derived from amine 5 and 4-hexen-3-one.13 Reprinted with permission from J. Am. Chem. Soc. 2002, 124, 2458–2460. Copyright 2002 American Chemical Society.
In this paper we present a detailed theoretical study of the Diels-Alder cycloaddition reactions of cyclopentadiene with (E)-crotonaldehyde and 4-hexen-3-one catalyzed by chiral imidazolidinones 1 and 5 in order to explain the differences observed in reaction rates and enantioselectivities.
Our research group have demonstrated that hybrid density functional theory14 can be a powerful tool to predict enantioselectivities in asymmetric organocatalyzed reactions.15–17 In a previous work the theoretical study of enantioselective alkylation reactions of pyrroles and indoles organocatalyzed chiral imidazolidinones have been reported.16 In this article, the authors rationalized theoretically the structural factors that govern the different observed enantioselectivities when either catalyst 1 or 2 was employed.
With the aim to determine quantitatively the catalytic effect produced by secondary amines as organocatalysts, the transition structures corresponding to the uncatalyzed Diels-Alder reactions between cyclopentadiene with α,β-unsaturated aldehydes and ketones have been also computed.
Computational Methods
All structures were computed using the functional B3LYP14a–c and the 6-31G(d)14d–f basis sets as implemented in Gaussian 98.18 All energy minima and transition structures were characterized by frequency analysis. Reported gas phase energies are electronic energies plus zero point vibrational energy corrections, scaled by 0.9806.19 The energies computed for structures in solvent (water ε = 78.39, benzene ε = 2.247) include the electronic energy at the B3LYP/6-31G(d) level of theory plus the solvation energy calculated with the CPCM solvation model20 as implemented in Gaussian 0321 at the HF/6-31G(d)//B3LYP/6-31G(d) level of theory, with the UAKS cavity model model. Both the electrostatic and non electrostatic components of the energy have been considered. Solvation energies of a representative group of neutral, anionic, and cationic molecules, are computed relatively well with this methodology.22
Results and Discussion
Uncatalyzed Diels-Alder Reactions with α,β-unsaturated aldehydes and ketones
Figure 2 shows the s-trans- and s-cis-(E)-crotonaldehyde conformers, and their corresponding endo and exo transition structures. The s-trans conformer of 7 is more stable than s-cis isomer by 1.4 kcal/mol. All the calculated transition structures correspond to an asynchronous (carbon-carbon bond forming distance differences between 0.2 and 0.6 Å), but concerted, pathway. A 1:3.8 endo:exo ratio is predicted for the gas phase at 0 °C. Previous studies of the Diels-Alder reaction of cyclopentadiene with methyl vinyl ketone predicted an 1.2:1 endo:exo ratio in the gas phase, and 3.7:1 in nitromethane at 0 °C.23 According to preceding experimental results, in most of the cases an endo orientation of the carbonyl group is preferred, and this preference increases in polar solvents.23–25 In water the endo selectivity enhancement is accompanied by a considerable reaction acceleration due to enforced hydrophobic interactions between diene and dienophile.28–31 The introduction of an alkyl group at R2 slightly decreases the endo selectivity,26 while an inversion of the endo orientation is observed when the hydrogen atom at R3 is substituted by an alkyl group.26,27 The results were attributed to an attractive C-H···π interaction between the methyl group and the diene π-system.26,35
Figure 2.

S-cis- and s-trans-(E)-crotonaldehyde conformers and calculated transition structures for Diels-Alder reactions with cyclopentadiene. Relevant distances are in Å. Relative energies for the gas phase and activation energies (kcal/mol) are shown.
 |
(2) |
TS7 s-cis-exo is the most stable transition structure in the gas phase and benzene. A weak CH···O hydrogen bond involving the carbonyl oxygen of the dienophile and one of the hydrogen atoms of the CH2 group at the diene is likely a contribution (Figure 2). A partial negative charge of 0.48 u. a. is present at the carbonyl oxygen while there is a partial positive charge of 0.06 u.a. at Ha in the cyclopentadiene fragment (CHelpG charges).32 Both s-cis and s-trans-endo approaching are lower in energy than their corresponding exo transition structures, most likely due to the extisting C-H···π stabilizing interaction between the methyl group of the dienophile and the diene π-system in TS7 s-cis-exo and TS7 s-trans-exo. Increasing solvent polarity weakens these CH···O interactions.33,34 Single point calculations performed with the CPCM solvation model gave an endo:exo ratio of 1:2.4 in benzene for the reaction of 7 with cyclopentadiene at 0 °C, that is, the exo preference decreases with respect to the same reaction in the gas phase. In water, a selectivity reversal occurs, and the calculated endo:exo ratio is 1.2:1.
Figure 3 displays the most stable 4-hexen-3-one conformer (8) and transition structure for the Diels-Alder reaction (TS8 s-cis-exo) in the gas phase and in water. Conformer 8 s-cis is more stable than the s-trans isomer by 1.1 kcal/mol. The analysis of the calculated transition structures corresponding to the Diels-Alder reaction with cyclopentadiene in the gas phase gave similar results (see Supporting Information). The most stable transition state is TS8 s-cis-exo. The calculated endo:exo ratio at 0 °C in the gas phase was 1:4.5, slightly higher than predicted in the reaction of cyclopentadiene with (E)-crotonaldehyde. As a consequence of the steric hindrance by the ethyl group, TS8 s-cis-exo is also the most stable transition state in water, but the calculated endo:exo ratio decreases to 1:3.7. B3LYP/6-31G(d) overestimates the existing C-H···O and C-H···π stabilizing interactions in the s-cis-exo approaches.14,26,35
Figure 3.

Most stable 4-hexen-3-one conformer and transition structure for Diels-Alder reactions with cyclopentadiene. Relevant distances are in Å. Activation energy (kcal/mol) is shown.
Amine catalyzed Diels-Alder reactions with α,β-unsaturated aldehydes and ketones
The s-trans iminium formed from butenal and dimethylamine (9 s-trans) is more stable than 9 s-cis by 6.4 kcal/mol (Figure 4).16 Only the s-trans conformer was considered in the transition structure searching. The calculated endo- and exo- transition structures, shown in Figure 4, are concerted but very asynchronous. The carbon-carbon bond forming distances difference by ~ 1 Å. Electronic activation energies are lower than in the case of the uncatalyzed cycloaddition by about 13 kcal/mol (Figure 2). The predicted endo:exo ratio was 1:2 in the gas phase at 0 °C. In water the endo:exo ratio changes to 1:3.6.
Figure 4.

Calculated s-cis and s-trans-N-2-butenylidene-N-methylmethanaminium cation (9) and transition structures for Diels-Alder reactions with cyclopentadiene. Relevant distances are in Å. Relative energies for the gas phase (or water in brackets, full optimization in water in italic) and activation energies (kcal/mol) are shown.
Consistent with previous reported results, both s-trans-endo and exo transtition geometries for the reaction between 9 s-trans cyclopentadiene performing a full optimization using the CPCM solvation model at the B3LYP/6-31G(d) level of theory and employing the UAKS cavity model, are very close to that corresponding to the gas phase, and slightly more synchronous (Figure 4). The relative energy difference is calculated to be 0.6 kcal/mol very close to the 0.7 kcal/mol obtained using CPCM single point calculations.22,36
Structures TS9 are similar to the most stable closed transition states located for the alkylation reaction of pyrrole with (E)-crotonaldehyde organocatalyzed by dimethylamine.16 However, transition vector motion along the reaction coordinate of TS9 corresponds to a concerted asynchronous Diels-Alder cycloaddition reaction.37 In addition, no intermediate corresponding to a stepwise process could be found after perfoming intrinsic reaction coordinate (IRC) calculations in the gas phase and in water. The calculated transition structures TS9 optimize to the corresponding cycloadducts. Our results are in excellent agreement with previous results reported by Domingo related to the reaction between N,N-dimethyleneamonium cation and cyclopentadiene.36c The author concluded that the reaction takes place as a highly asynchronous concerted process with a large polar character and the process can be characterized by the nucleophilic attack of cyclopentadiene to the electron-poor cation. The transition states show a Michael-type addition character with a concomitant cyclization without formation of any zwitterionic intermediate.
One can explain the observed experimental data for the Diels-Alder cycloaddition of cyclopentadiene with α,β-unsaturated aldehydes organocatalyzed by imidazolidinones using our previous results on the theoretical study of alkylation reactions of pyrrole organocatalyzed by imidazolidinones 1 and 2.2,16 The small energy differences found between the endo and exo orientations of each conformer demonstrated that a distinct preference for one over the other does not occur. This fact is in agreement with the experimentally observed 1:1 endo:exo ratio (predicted endo:exo ratio is 1:1.5 for catalyst 1 and 1.3:1 for catalyst 2 in the gas phase at 25 °C in the case of the alkylation reaction of N-methyl pyrrole with (E)-crotonaldehyde). A theoretical ee value of 70% for the endo product, and an ee value of 46% for the exo cycloadduct were obtained. This results are also in agreement with the higher enantioselectivity experimentally observed for the endo product (endo 90% ee, exo 86% ee).2
In 2002 Zora reported the AM1 theoretical study of the cycloaddition reaction of cyclopentadiene with allylidenammonium cation.38 A stepwise pathway was found, and the activation barrier for the cycloaddition to the C=C bond was 4.2 kcal/mol lower than that to the C=N bond.
Figure 5 displays the calculated s-trans and s-cis N-2-butenylidene-N-1-ethyl-N-methylmethanaminium cations (10). The relative energy differences between the 10 s-trans iminium intermediate and the three located 10 s-cis1-3 conformers are lower than in the case of N-2-butenylidene-N-methylmethanaminium cations (9) (3.5, 2.9, and 3.8 kcal/mol respectively). This fact can be attributed to the presence of the ethyl group which generates steric hindrance in both s-cis and s-trans isomers. Only the s-trans conformer was considered in the transition structure searching.
Figure 5.

Calculated s-cis and s-trans N-2-butenylidene-N-1-ethyl-N-methylmethanaminium cation (10). Relative energy differences (kcal/mol) are shown.
Figure 6 shows the four calculated transition structures for the Diels-Alder reaction of 10 s-trans and cyclopentadiene. The carbon-carbon bond forming distances are very similar to the (E)-crotonaldehyde case. The calculated endo:exo ratio was 3.1:1 in the gas phase at 0 °C due to steric hindrance between the methylene group in the cyclopentadiene ring and the ethyl group in the ketone fragment. In water, the calculated endo:exo ratio increases to 4.1:1. The most stable transition state was TS10a s-trans-endo. Endo and exo transition structures TS10a, in which the methyl group of the ethyl moiety is pointing away from the cyclopentadiene fragment, are more stable than TS10b structures by 2.6 and 1.6 kcal/mol respectively. Only transition structures analogous to TS10a were considered in further transition structure searching involving a larger number of atoms. Activation energies for 10 are about 11 kcal/mol lower than for 8 (Figure 3).
Figure 6.

Calculated transition structures for the Diels-Alder reactions of 10 s-trans with cyclopentadiene. Relative distances are in Å. Relative energies for the gas phase (or water, in brackets) and activation energies (kcal/mol) are shown.
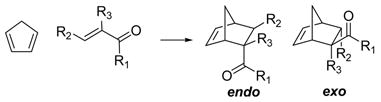
MacMillan performed the reaction in aqueous media and in the presence of HClO4 (one equivalent with respect to the amine catalyst, 20 mol% with respect to ketone 8).10 The computations were performed for the reaction involving protonated 4-hexen-3-one, and the results for the lowest energy path are shown in Figure 7. The isomeric reactants complexes, and transition states are given in the supporting information. The s-cis protonated species forms with cyclopentadiene an ion-molecule complex in the gas phase, and the activation barrier from this complex is only 3 kcal/mol, even lower than the activation barrier for the iminium case. However, the formation of the protonated ketone from the protonated amine catalyst will be highly endothermic and little protonated ketone will be formed. The energies of formation of compound 11 s-cis from 8 s-cis and protonated form of amines 1 and 5 were calculated to be 17.4 and 18.1 kcal/mol respectively in the gas phase. The calculated transition structures are very asynchronous (carbon bond forming distance differences ~ 1 Å), similarly to dimethylamine catalyzed cycloadditions. These results predicted an endo:exo ratio of 2.8:1 in the gas phase at 0 °C. In water the calculated endo:exo ratio was 7.5:1 the same value found in the experiment for the reaction catalyzed by imidazolidinone 1.10
Figure 7.
Most stable protonated s-cis 4-hexen-3-one conformer, Diels-Alder transition structure, and ion-molecule complex. Relevant distances are in Å. Activation and formation energies (kcal/mol) are shown.
Imidazolidinone catalyzed Diels-Alder cycloadditions
Iminium ion intermediates
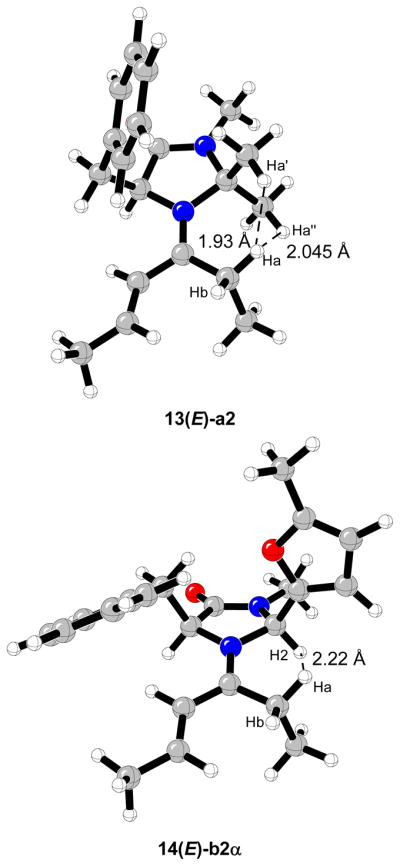
A complete conformational study of iminium complexes formed from (E)-crotonaldehyde and the chiral imidazolidinone 1 has been recently reported.15f,16 Contrary to previous force field energy minimizations, conformer (E)-12a,2,39 which includes a stabilizing C-H···π interaction between one of the methyl groups at position C2 of the imidazolidinone ring, and the phenyl ring of the benzyl group at position C5, was found to be the global minimum.40 A number of conformers of similar energy were located; attack from the bottom, away from the benzyl group is favored as noted by MacMillan.2,4,6,16
Figures 9 and 10 show the twelve different conformers found in the case of imidazolidinone 1 and 4-hexen-3-one. These correspond to the (E) and (Z) configurations about the N+=C bond, the three staggered conformations involving the bond to the benzyl group, and two conformations of the ethyl group attached to the carbon-nitrogen double bond.
Figure 9.

Optimized structures, relative energy differences (kcal/mol), calculated respect to the lowest energy isomer 13(E)-a2, and percentage of each structure in the gas phase at 0 °C for the 13(E)-iminium complexes formed from chiral imidazolidinone 1 and 4-hexen-3-one.
Figure 10.

Optimized structures, relative energy differences (kcal/mol), calculated respect to the lowest energy isomer 13(E)-a2 (Figure 9), and percentage of each structure in the gas phase at 0 °C for the 13(Z)-iminium complexes formed from chiral imidazolidinone 1 and 4-hexen-3-one.
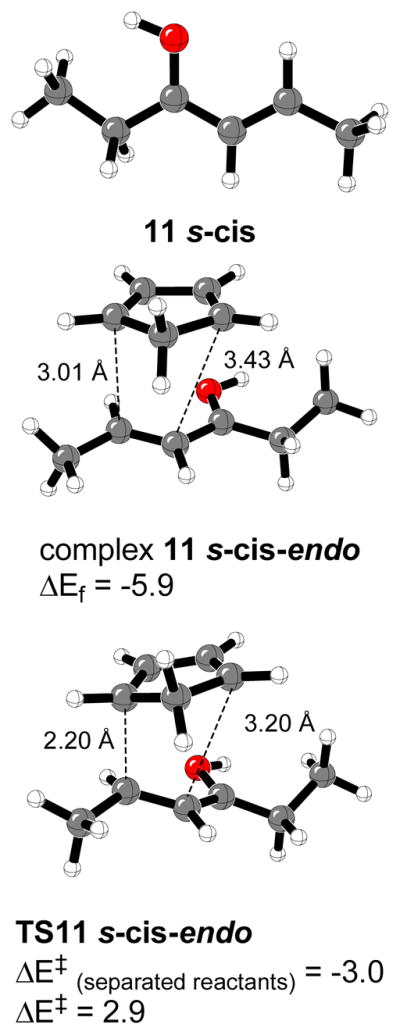
As in previous studies on the iminium complexes formed from imidazolidinone 1 and (E)-crotonaldehyde,16 the most stable conformer (13(E)-a2) includes a stabilizing C-H···π interaction between one of the methyl groups at the phenyl ring of the benzyl group at position C5.38 The ethyl conformer 13(E)-a1 and Z-isomer 13(Z)-a2 are only 0.1 and 0.2 kcal/mol higher in energy, respectively these three conformers constitute the 70% of all the species in the gas phase at 0 °C (Figures 9 and 10). The conformers commonly drawn by MacMillan et al. (b1 and b2) are 0.6–3.3 kcal/mol higher in energy and constitute 12% of the equilibrium mixture. The least stable structures correspond to 13(Z)-b1 and 13(Z)-c1 iminium intermediates due to strong steric repulsions between the ethyl group attached to the iminium carbon and the benzyl group at position C5 of the imidazolidinone ring.
In the case of imidazolidinone 5, the derived iminium ions have even more isomers because of rotation and isomerism around C2-furyl bond. A total number of 26 different conformers were located. The 4 most stable calculated iminium intermediates are shown in Figure 11. The “α” means that H2-2-C1′-O′ dihedral angle is close to 180°.
Figure 11.

Most stable calculated structures, relative energy differences (kcal/mol), calculated respect to the lowest energy isomer 14(E)-b2α, and percentage of each structure in the gas phase at 0 °C for the 14-iminium complexes formed from 5 and 4-hexen-3-one.
The most stable 14(E)-b2α structure is like that reported by MacMillan and coworkers based on a MM3 conformational search.12 Surprisingly, the 14(Z)-b2α isomer is only 0.2 kcal/mol higher in energy. A large number of species will exist in the gas phase at 0 °C, and a total of 26 different conformers could be located (see Supporting Information). Conformers 14(E)-b2α, 14(E)-c2α and 14(Z)-b2α are predicted to constitute 64% of all of them (Figure 11). This percentage is slightly lower than in the case of iminium intermediates 13.
Transition structure searching
For transition structure searching involving catalyzed reactions of 4-hexen-3-one with cyclopentadiene, the most stable iminium intermediates, 13 and 14, were considered. The lowest energy calculated transition states involving iminium ions 13 and 14 are depicted in Figures 12 and 13, respectively (“δ” means a H2-C2-C1′-O′ dihedral angle of about -30°). Tables 1 and 2 list the relative energy differences, activation energies (calculated as the difference between the electronic energies of the transition structures and the sum of the electronic energies of cyclopentadiene and the corresponding iminium intermediate) and percentage of transition structures, TS13 and TS14, in the gas phase at 0 °C.
Figure 12.

Most stable calculated transition structures for the Diels-Alder reaction of cyclopentadiene with 4-hexen-3-one catalyzed by chiral imidazolidinone 1. Relevant distances are in Å. Relative energies for the gas phase (or water, in brackets) and activation energies (kcal/mol) are shown.
Figure 13.

Most stable calculated transition structures for the Diels-Alder reaction of cyclopentadiene with 4-hexen-3-one catalyzed by chiral imidazolidinone 5. Relevant distances are in Å. Relative energies for the gas phase (or water, in brackets) and activation energies (kcal/mol) are shown.
Table 1.
Calculated relative energy difference and percentage of transition structures TS13 in the gas phase and water at 0 °C.
| Transition Structure | Gas phase | Water | ||
|---|---|---|---|---|
| Energy Difference (kcal/mol) | Percent | Energy Difference (kcal/mol) | Percent | |
| TS13 (E)-a1-endo | 0.0 | 35 | 1.8 | 4 |
| TS13 (E)-a1-exo | 0.9 | 6 | 3.9 | 0 |
| TS13 (E)-a2-endo | 3.9 | 0 | 5.5 | 0 |
| TS13 (E)-a2-exo | 4.6 | 0 | 6.2 | 0 |
| TS13 (E)-c1-endo | 0.4 | 16 | 5.4 | 0 |
| TS13 (E)-c1-exo | 1.2 | 4 | 6.4 | 0 |
| TS13 (E)-c2-endo | 3.1 | 0 | 6.6 | 0 |
| TS13 (E)-c2-exo | 5.5 | 0 | 7.8 | 5 |
| TS13 (Z)-a1-endo | 3.9 | 0 | 4.4 | 0 |
| TS13 (Z)-a1-exo | 5.4 | 0 | 5.6 | 0 |
| TS13 (Z)-a2-endo | 0.4 | 18 | 0 | 96 |
| TS13 (Z)-a2-exo | 1.4 | 3 | 5.6 | 0 |
| TS13 (Z)-b2-endo | 1.4 | 3 | 6.3 | 0 |
| TS13 (Z)-b2-exo | 2.2 | 1 | 6.9 | 0 |
| TS13 (Z)-c2-endo | 0.5 | 13 | 4.5 | 0 |
| TS13 (Z)-c2-exo | 1.7 | 2 | 5.7 | 0 |
Table 2.
Calculated relative energy difference, electronic activation energies, and percentage of transition structures TS14 in the gas phase at 0 °C.
| Transition Structure | Gas phase | Water | ||
|---|---|---|---|---|
| Energy Difference (kcal/mol) | Percent | Energy Difference (kcal/mol) | Percent | |
| TS14 (E)-b1α-endo | 0.6 | 11 | 0.0 | 79 |
| TS14 (E)-b1α-exo | 1.4 | 3 | 2.1 | 2 |
| TS14 (E)-b2α-endo | 1.4 | 3 | 5.0 | 0 |
| TS14 (E)-b2α-exo | 4.3 | 0 | 7.7 | 0 |
| TS14 (E)-b2δ-endo | 2.2 | 1 | 5.9 | 0 |
| TS14 (E)-b2δ-exo | 4.8 | 0 | 11.1 | 0 |
| TS14 (E)-c1α-endo | 0.3 | 22 | 0.9 | 16 |
| TS14 (E)-c1α-exo | 0.9 | 6 | 2.8 | 0 |
| TS14 (E)-c2α-endo | 0.8 | 8 | 2.8 | 0 |
| TS14 (E)-c2α-exo | 2.7 | 0 | 4.6 | 0 |
| TS14 (E)-c2δ-endo | 1.8 | 1 | 4.2 | 0 |
| TS14 (E)-c2δ-exo | 3.4 | 0 | 5.4 | 0 |
| TS14 (Z)-b2α-endo | 0.0 | 35 | 1.9 | 2 |
| TS14 (Z)-b2α-exo | 1.1 | 5 | 2.6 | 1 |
| TS14 (E)-b2δ-endo | 1.0 | 6 | 3.4 | 0 |
| TS14 (E)-b2δ-exo | 2.8 | 0 | 4.2 | 0 |
All the transition structures are concerted but very asynchronous, and very similar to that previously calculated for the dimethylamine organocatalyzed reaction shown in Figure 5. In the gas phase, the lowest energy transition state related to imidazolidinone 1 catalyzed reaction (TS13 (E)-a1-endo) corresponds to the endo approach of the cyclopentadiene ring to the Si,Re face (bottom face) of the most stable iminium ion conformer 13 (E)-a1; this approach accounts for 35% of all the transition structures. In water, the most stable transition structure is TS13 (Z)-a2-endo, corresponding to cyclopentadiene attack on the Si,Re face (top face) of 13 (Z)-a2 conformer. TS13 (Z)-a2-endo accounts for more than 96% of all the transition structures in water at 0 °C. In both cases, gas phase and water, the most stable calculated transition structures lead to the endo-enantiomer that is isolated as the major product of reaction. In the case of imidazolidinone 5, the lowest energy transition structure in the gas phase corresponds to the endo approach of the diene on the Si,Re face (top face) of 14 (Z)-b2α isomer. TS14 (Z)-b2α-endo accounts for only 35% of all the transition structures while the most stable calculated transition state in water, TS14 (E)-b1α-endo, accounts for 80%. As in the case of the calculations on amine 1 catalyzed reactions the lowest energy transition structures in both gas phase and water predict the product isolated as the major compound. The average calculated electronic activation energies are very similar for both 1 and 5 organocatalysis (13.4 and 13.1 kcal/mol, respectively in the gas phase).
The Boltzmann distribution analysis predicted an endo:exo ratio of 6:1 and a theoretical ee value > 99% for the cycloaddition reaction catalyzed by 1 in the gas phase at 0 °C. MacMillan and coworkers reported an endo:exo product ratio of 7:1 and 0% ee for the same reaction performed in water at 0 °C. Surprisingly, catalyst 5 which showed a remarkably improved endo- and enantioselectivities (25:1 endo:exo ratio and 99% ee) gave an endo:exo ratio of 6:1 and a theoretical 70% ee value.12 The energies of all the transition structures in water with the CPCM solvation model at the HF/6-31G(d)//B3LYP/6-31G(d) level of theory and, with the UAKS cavity model have been also computed (see computational methods section).22 Solvation energies increase both endo selectivity and enantioselectivity. For catalyst 1, an almost 100% endo selectivity was predicted, and in the case of amine 5 a 35:1 endo:exo ratio was calculated, versus the 25:1 experimental product ratio. Both catalysts are predicted to give > 99% ee, whereas experiments give 0% and 90% for catalysts 1 and 5 respectively.12
The possibility that one or both of these reactions could be reversible was also considered. The relative stabilities of the Diels-Alder cycloadducts obtained from the most stable calculated transition structures, TS13 (E)-a1-endo and TS14 (Z)-b2α-endo (Figure 14). The potential-energy profiles show that the amine 5 catalyzed reaction is only 1.0 kcal/mol more exothermic than the amine 1 catalyzed reaction. According to the relative cycloadducts formation energy differences, thermodynamic control does not account for the differences experimentally observed between catalyst 1 and 5.12
Figure 14.

Potential-energy profile for cycloaddition reactions involving the most stable transition structures in the gas phase TS13 (E)-a1-endo and TS14 (Z)-b2α-endo.
However, catalyst 1 is sterically more hindered than 5, and the possibility that the intermediate iminium is formed very slowly from 1, allowing the uncatalyzed background reaction to occur, was considered. The net result would be the negligible stereocontrol observed experimentally.
Figure 15 displays the potential-energy profile for the formation of the most stable iminium ions 13(E)-a2 and 14(E)-b2α (see Figures 9, 10 and 11) from protonated amines 1 and 5, and 8 s-cis. These data indicate that formation of 14(E)-b2α iminium ion is thermodynamically more favored than formation of 13(E)-a2 by 8.5 kcal/mol in the gas phase. These results can explain the different behavior observed between catalyst 1 and 5 in the cycloaddition reaction of cyclopentadiene with α,β-unsaturated ketones. Iminium ions derived from chiral amine 1 and 4-hexen-3-one are formed slowly or not at all and, only the uncatalyzed background reaction is observed (48 hours, 20% yield, 7:1 endo:exo ratio, 0% ee).12 This conclusion is also in agreement with the experimental study reported by Jørgensen et al. on asymmetric addition of nitroalkanes to α,β-unsaturated enones organocatalyzed by several imidazolines.39 Organocatalyst 1 gave no conversion after 60 hours.
Figure 15.

Computed energies of formation of 13(E)-a2 and 14(E)-b2α iminium ions from protonated amines 1 and 5 in the gas phase.
The different observed stabilities are due to strong steric hindrance produced between the ethyl fragment and the dimethyl groups at position C2 of the imidazolidinone ring of 1. Figure 16 depicts the closest distances between Ha at the methylene group of the ethyl fragment and substituents at C2 of the imidazolidinone moiety. Iminium intermediate 13(E)-a2 presents a Ha-Ha′ distance of 1.93 Å and a Ha-Ha″ distance of 2.05 Å. Ha-H2 = 2.22 Å was the closest distance found in compound 14(E)-b2α between the methylene group and the imidazolininone ring substituents.
Figure 16.
Most stable calculated 13(E)-a2 and 14(E)-b2α iminium intermediates. Closest distances between methylene group and C2 imidazolidinone ring substituents are shown (Å).
Conclusions
DFT calculations have demonstrated that secondary amines decrease the activation energies of the Diels-Alder reactions of cyclopentadiene with α,β-usaturated aldehydes and ketones by 13 and 11 kcal/mol, respectively. The formation of the iminium complex produces a much more reactive dienophile. Although a number of different conformers of iminium intermediates and transition states are located, there is a preference for attack in a sterically unencumbered fashion that leads to a family of preferred transition structures and high stereoselectivity.
The different reactivities observed for imidazolidinone catalysts 1–5, in the [4+2] cycloaddition reactions of 4-hexen-3-one and cyclopentadiene have been explored. Chiral amines 1 and 2 form the corresponding iminium intermediates reluctantly with ketone 8 due to strong steric hindrance between the ethyl group and substituents at position C2 of the imidazolidinone ring.41 When reactions are performed in the presence of 1 and 2, only the uncatalyzed background reaction is observed (48 hours, 20% yield, 7:1 endo:exo ratio, 0% ee).12
Supplementary Material
Figure 8.
Most stable iminium ion conformer formed from catalyst 1 and (E)-crotonaldehyde.16
Acknowledgments
We are grateful to the National Institute of General Medical Sciences, National Institutes of Health, for financial support. This research was facilitated through the Partnerships for Advanced Computational Infrastructure (PACI) through the support of the National Science Foundation. The computations were performed on the National Science Foundation Terascale Computing System at the Pittsburgh Supercomputing Center (PSC) and on the UCLA Academic Technology Services (ATS) Hoffman Beowulf cluster. Ruth Gordillo thanks the Ministerio de Educación, Cultura y Deporte-Spain for a postdoctoral fellowship. We thank Professor David MacMillan and his research group, and Dr. Yu Takano for helpful discussions and suggestions.
Footnotes
Supporting Information Available: Complete references 18 and 21, Cartesian coordinates of all reported structures, as well as the total electronic and zero-point vibrational energies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews on enantioselective organocatalysis: Dalko PL, Moisan L. Angew Chem Int Ed. 2001;40:3726–3748. doi: 10.1002/1521-3773(20011015)40:20<3726::aid-anie3726>3.0.co;2-d.Dalko PI, Moisan L. Angew Chem Int Ed. 2004;43:5138–5175. doi: 10.1002/anie.200400650.Houk KN, List B. Asymmetric Organocatalysis. Acc Chem Res. 2004;37:487. doi: 10.1021/ar0300524.
- 2.Ahrendt KA, Borths CJ, MacMillan DWC. J Am Chem Soc. 2000;122:4243–4244. [Google Scholar]
- 3.Jen WS, Wiener JJM, MacMillan DWC. J Am Chem Soc. 2000;122:9874–9875. [Google Scholar]
- 4.Paras NA, MacMillan DWC. J Am Chem Soc. 2001;123:4370–4371. doi: 10.1021/ja015717g. [DOI] [PubMed] [Google Scholar]
- 5.Brochu MP, Brown SP, MacMillan DWC. J Am Chem Soc. 2004;126:4108–4109. doi: 10.1021/ja049562z. [DOI] [PubMed] [Google Scholar]
- 6.Austin JF, MacMillan DWC. J Am Chem Soc. 2002;124:1172–1173. doi: 10.1021/ja017255c. [DOI] [PubMed] [Google Scholar]
- 7.Paras NA, MacMillan DWC. J Am Chem Soc. 2002;124:7894–7895. doi: 10.1021/ja025981p. [DOI] [PubMed] [Google Scholar]
- 8.Brown SP, Goodwin NC, MacMillan DWC. J Am Chem Soc. 2003;125:1192–1194. doi: 10.1021/ja029095q. [DOI] [PubMed] [Google Scholar]
- 9.Austin JF, Kim SG, Sinz CJ, Xiao WG, MacMillan DWC. Proc Natl Acad Sci USA. 2004;101:5482–5487. doi: 10.1073/pnas.0308177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mangion IK, Northrup AB, MacMillan DWC. Angew Chem Int Ed. 2004;43:6722–6724. doi: 10.1002/anie.200461851. [DOI] [PubMed] [Google Scholar]
- 11.(a) Yang JW, Hechavarría-Fonseca MT, List B. Angew Chem Int Ed. 2004;43:6660–6662. doi: 10.1002/anie.200461816. [DOI] [PubMed] [Google Scholar]; (b) Yang JW, Hechavarría-Fonseca MT, Vignola N, List B. Angew Chem Int Ed. 2004;44:108–110. doi: 10.1002/anie.200462432. [DOI] [PubMed] [Google Scholar]
- 12.Northrup AB, MacMillan DWC. J Am Chem Soc. 2002;124:2458–2460. doi: 10.1021/ja017641u. [DOI] [PubMed] [Google Scholar]
- 13.Molecular mechanics Monte Carlo conformational search with MM3 force field; Macromodel V6.5.
- 14.(a) Becke AD. J Chem Phys. 1993;98:1372–1377. [Google Scholar]; (b) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (c) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (d) Ditchfield R, Hehre WJ, Pople JA. J Chem Phys. 1971;54:724–728. [Google Scholar]; (e) Hehre WJ, Ditchfield R, Pople JA. J Chem Phys. 1972;56:2257–2261. [Google Scholar]; (f) Hariharan PC, Pople JA. Theor Chim Acta. 1973;28:213–222. [Google Scholar]
- 15.(a) Bahmanyar S, Houk KN. J Am Chem Soc. 2001;123:11273–11283. doi: 10.1021/ja011403h. [DOI] [PubMed] [Google Scholar]; (b) Hoang L, Bahmanyar S, Houk KN, List B. J Am Chem Soc. 2003;125:16–17. doi: 10.1021/ja028634o. [DOI] [PubMed] [Google Scholar]; (c) Bahmanyar S, Houk KN, Martin HJ, List B. J Am Chem Soc. 2003;125:2475–2479. doi: 10.1021/ja028812d. [DOI] [PubMed] [Google Scholar]; (d) Bahmanyar S, Houk KN. Org Lett. 2003;5:1249–1251. doi: 10.1021/ol034198e. [DOI] [PubMed] [Google Scholar]; (e) Wayner G, Houk KN, Sun YK. J Am Chem Soc. 2004;126:199–203. doi: 10.1021/ja035147f. [DOI] [PubMed] [Google Scholar]; (f) Alleman C, Gordillo R, Clemente FR, Cheong PHY, Houk KN. Acc Chem Res. 2004;37:558–569. doi: 10.1021/ar0300524. [DOI] [PubMed] [Google Scholar]
- 16.Gordillo R, Carter J, Houk KN. Adv Synth Catal. 2004;346:1175–1185. [Google Scholar]
- 17.Dudding T, Houk KN. Proc Natl Acad Sci USA. 2004;101:5770–5775. doi: 10.1073/pnas.0307256101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frisch MJ, et al. Gaussian 98, revision A.6. Gaussian, Inc; Pittsburgh, PA: 1998. [Google Scholar]
- 19.Scott AP, Radom L. J Phys Chem. 1996;100:16502–16513. [Google Scholar]
- 20.(a) Barone V, Cossi M. J Phys Chem A. 1998;102:1995–2001. [Google Scholar]; (b) Barone B, Cossi M, Tomasi J. J Comput Chem. 1998;19:404–417. [Google Scholar]
- 21.Frisch MJ, et al. Gaussian 03, revision B. 04. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 22.Takano Y, Houk KN. J Chem Theory Comput. 2005;1:70–77. doi: 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- 23.Fu YS, Tsai SC, Huang CH, Yen SY, Hu WP, Yu SJ. J Org Chem. 2003;68:3068–3077. doi: 10.1021/jo026596l. [DOI] [PubMed] [Google Scholar]
- 24.Breslow R, Guo T. J Am Chem Soc. 1998;110:5613–5617. [Google Scholar]
- 25.Breslow R, Maitra U, Rideout D. Tetrahedron Letters. 1983;24:1901–1904. [Google Scholar]
- 26.Kobuke Y, Fueno T, Furukawa J. J Am Chem Soc. 1970;92:6548–6553. [Google Scholar]
- 27.Mellor JM, Webb CF. J Chem Soc, Perkin Trans 2. 1974:17–22. [Google Scholar]
- 28.Blokzijil W, Blandamer MJ, Engberts JBFN. J Am Chem Soc. 1991;113:4241–4246. [Google Scholar]
- 29.Kumar A, Phalgune U, Pawar SS. J Phys Org Chem. 2000;13:555–557. [Google Scholar]
- 30.Baldwin JE, Herchen SR, Schulz G. J Am Chem Soc. 1980;102:7816–7817. [Google Scholar]
- 31.Meijer A, Otto S, Engberts JBFN. J Org Chem. 1998;63:8989–8994. [Google Scholar]
- 32.Breneman CM, Wiberg KB. J Comp Chem. 1990;11:361–373. [Google Scholar]
- 33.Washington I, Houk KN. Angew Chem Int Ed. 2001;40:4485–4487. doi: 10.1002/1521-3773(20011203)40:23<4485::aid-anie4485>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 34.García Ruano JL, Fraile A, González G, Martín MR, Clemente FR, Gordillo R. J Org Chem. 2003;68:6522–6534. doi: 10.1021/jo0345603. [DOI] [PubMed] [Google Scholar]
- 35.Nishio M. Tetrahedron. 2005;61:6923–6950. [Google Scholar]
- 36.a) Domingo LR, Aurell MJ, Pérez P, Contreras R. J Org Chem. 2003;68:3884–3890. doi: 10.1021/jo020714n. [DOI] [PubMed] [Google Scholar]; b) Domingo LR. Tetrahedron. 2002;58:3765–3774. [Google Scholar]; c) Domingo LR. J Org Chem. 2001;66:3211–3214. doi: 10.1021/jo001332p. [DOI] [PubMed] [Google Scholar]; d) Evanseck JD, Kong S. J Am Chem Soc. 2000;122:10418–10427. [Google Scholar]
- 37.Schaftenaar G, Noordik JH. Vibrational mode visualization was performed using the MOLDEN program: Molden: a pre- and post-processing program for molecular and electronic structures. J Comput-Aided Mol Design. 2000;14:123–124. doi: 10.1023/a:1008193805436. [DOI] [PubMed] [Google Scholar]
- 38.Zora M. J Mol Struct (THEOCHEM) 2002;619:121–133. [Google Scholar]
- 39.Kozlowisky MC, Panda M. J Org Chem. 2003;68:2061–2076. doi: 10.1021/jo020401s. [DOI] [PubMed] [Google Scholar]
- 40.Distances (3.7 and 3.8 Å) are in good agreement with previous theoretical studies on benzene-methane complexes: Tsuzuki S, Honda K, Uchimaru T, Mikami K, Tanabe K. J Am Chem Soc. 2000;122:3746–3753.
- 41.Halland N, Hazell RG, Jørgensen KA. J Org Chem. 2002;67:8331–8338. doi: 10.1021/jo0261449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.