

Abstract
Assembly of β-amyloid (Aβ) peptide into toxic oligomers is widely believed to initiate Alzheimer's disease pathogenesis. Under in vitro physiological conditions, zinc (Zn(II)) can bind to Aβ and redirect its assembly from amyloid fibrillar toward less toxic amorphous aggregation. Propensity of Aβ to go toward a specific form of aggregate state is determined by structural and dynamical properties of the initial monomeric as well as the aggregate state. Here we probe the structural and dynamical impact of binding of Zn(II) to monomeric Aβ40 using NMR spectroscopy. To obtain further support for the importance of intrinsic dynamics in the aggregation precursor, 15N relaxation measurements were also performed for Aβ42, the more fibrillar aggregation-prone variant of Aβ. The combined data suggest that, upon Zn(II)-binding to the N-terminus of Aβ40, a relatively rigid turnlike structure is induced at residues Val24-Lys28 whereas the residues flanking this region become more mobile on the picosecond-to-nanosecond timescale. This is in contrast to the increased rigidity of Aβ42 at the C-terminus, and proposed to be linked to the higher propensity of Zn(II)-bound peptide to form amorphous aggregates with less entropic penalties than their fibrillar counterparts.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder clinically characterized by a progression from an amnesic mild cognitive impairment to a slow global decline of cognitive function (1). Microscopically, AD is manifested by a widespread extracellular deposition of amyloid β-peptide (Aβ) as senile plaques and intracellular aggregation of hyperphosphorylated tau protein as neurofibrillary tangles (2). Genetic and pathological evidences support the so-called “amyloid cascade hypothesis,” according to which the main initial event in AD pathogenesis is aggregation of Aβ, and other elements of AD pathology, including tau dysfunction, prooxidative state, inflammation, synaptic impairment, and neuronal loss, are located downstream to Aβ aggregation (3,4). Inhibition of toxic Aβ aggregation is therefore seen as a major strategy to modify the disease pathology (1). In recent years, it has been revealed that small soluble Aβ oligomers that are formed early in the aggregation process are more toxic than the final amyloid fibrillar state present in the core of senile plaques (4). Accordingly, potential anti-aggregation drugs need to act in the very early stages of Aβ aggregation, through blockade of the toxic aggregation and/or redirection of the monomeric Aβ toward off-pathway less toxic aggregates. The rational design of antiaggregation compounds for AD treatment therefore requires a more detailed understanding of Aβ aggregation (toxic and nontoxic) in the earliest steps.
Aβ peptides are released into the extracellular medium after sequential proteolytic cleavages of a multidomain integral membrane type-I protein, the amyloid precursor protein, by β- and γ-secretases (5). The exact location of the trans-membrane cleavage site for γ-secretase results in a variability in the length of its Aβ product from 38 to 43 residues, with the dominance of Aβ40 and Aβ42 species (5). Aβ42 has an identical amino-acid sequence with Aβ40, except for the additional Ile41 and Ala42 at the C-terminus (Fig. 1). Solution NMR studies of full-length Aβ40 and Aβ42 have suggested that the average structures of the Aβ40 and Aβ42 monomers in solution are very similar (6).
Figure 1.
Amino-acid sequence of Aβ40 and Aβ42, with the main coordination sites of zinc (Asp1, His6, His13, and His14) highlighted.
Despite the nearly complete sequence similarity and structural resemblance, Aβ40 and Aβ42 show very different in vitro and in vivo properties. Aβ42 has a faster kinetics of amyloid fibrillar aggregation than Aβ40 in vitro (7,8), and apparently aggregates through a different mechanism involving a larger intermediate oligomeric species (9). Aβ42 is much more toxic to neurons in cell culture and animal models than Aβ40 (8,10,11). Aβ42 is the major variant of Aβ in the core of senile plaques whereas Aβ40 constitutes ∼90% of total Aβ in plasma (12), and in the most familial AD cases, the ratio of Aβ42/Aβ40 is shown to be elevated (13). The enhanced toxicity of Aβ42 most likely derives from its higher aggregation propensity, which in turn is determined by the structural and dynamical properties of the initial monomeric as well as the intermediate oligomeric and final aggregate state of the peptide.
Metal ion homeostasis is known to be affected in several neurodegenerative disorders (14). In AD, endogenous ions like Zn, Cu, and Fe are found in the amyloid plaques at concentrations as high as 1 mM (15). Zinc ions are coreleased along with glutamate in most of the glutamatergic synapses in the cerebral cortex, and reach high μM concentrations in the synaptic clefts (16). Under physiologic conditions, soluble monomeric Aβ has a high propensity to bind to zinc (17). Through a variety of spectroscopic methods and the use of different peptide fragments, it was suggested that the coordination site of Zn(Aβ) is most likely made of four or six ligands including the three histidines (His6, His13, and His14) and the N-terminus or the carboxylate group of Asp1 (Fig. 1) (17,18). A second site of lower affinity has been suggested for binding to zinc, which probably involves residues between Asp23 and Lys28, although considering the weak affinity of this site, it is quite unlikely that it is occupied by zinc in physiologically relevant conditions (18).
The effect of zinc on Aβ toxicity reveals a clear concentration variability, with promotion of toxicity at high (∼mM) and neuroprotection at low (<50 μM) zinc concentrations (19,20). In addition to the possible role of zinc in interfering with the copper-based generation of reactive oxygen species in Aβ aggregates (21), zinc could exert its protective role through differential interaction with Aβ aggregates (22). The effect of zinc on Aβ aggregation has been extensively studied in vitro, and it was shown that depending on the experimental conditions such as pH, ionic strength, and the concentration of zinc, Aβ aggregation and morphology of Aβ aggregates are variably influenced by the presence of zinc (23–26). The common notion is that, at very low zinc concentrations, this ion prevents Aβ aggregation, whether on-pathway fibrillar or off-pathway amorphous aggregate formation, but at higher concentrations when the metal/peptide ratio reaches ∼1, amorphous Aβ aggregation is promoted whereas fibrillar aggregation is blocked (18).
Here, we present a detailed study of the effect of Zn(II)-binding on the structure and backbone dynamics of Aβ40. Using 15N relaxation measurements we demonstrate that binding of zinc to Aβ introduces some rigidity in the N-terminus and in the Val24-Lys28 region, whereas the intervening region and the C-terminus are becoming more mobile. Through comparison with Aβ42, the variant of Aβ with increased rigidity in the C-terminus and higher propensity to form fibrillar aggregates, the association between Aβ dynamics in the monomeric state and its tendency to form aggregates of various morphologies is discussed.
Materials and Methods
Materials
The used chemicals were from Sigma-Aldrich (St. Louis, MO). 15N-Aβ40 and Aβ42 were purchased from rPeptide (Athens, GA). Uniformly labeled 15N,13C-Aβ40 was produced as described in the next paragraph.
Cloning, expression, and purification of human Aβ40
A DNA duplex coding for Aβ40 was cloned into a modified pET28a vector coding for an N-terminal TEV-protease-cleavable His6-Z-tag fusion protein (27). 15N,13C-labeled Aβ40 fusion protein was expressed in Escherichia coli at 37°C in Toronto minimal medium. After purification on a 1 mL HisTrap HP nickel column (GE Healthcare, Waukesha, WI), the fusion protein was digested overnight on ice with recombinant TEV protease. The digestion mixture was loaded on a C4 reversed-phase HPLC column (Vydac, Hesperia, CA). The released Aβ40 peptide eluted in a linear (0–100%) acetonitrile gradient from this column as a single peak. According to electrospray ionization mass spectrometry, the isolated peptide was 100% pure. The purified peptide was lyophilized before use. The lyophilized peptide was then solubilized in 10 mM NaOH at a concentration of 2 mg/mL (∼460 μM), as recommended in Hou et al. (6) to remove the preformed aggregates, and stored at −80°C until use.
NMR spectroscopy
All two- and three-dimensional NMR spectra were recorded at 278 K and pH 7.2, buffered with 10 mM sodium phosphate. Chemical shift referencing at this temperature and pH was made with respect to the external 4,4-dimethyl-4-silapentane-1-sulfonic acid (0.0 ppm). The sample contained peptide at a concentration of 0.2 mg/mL (∼50 μM), with or without 50 μM ZnCl2. To eliminate the possible effect of pH variation on the NMR parameters (e.g., chemical shifts), pH of the sample before and after addition of zinc ion was carefully adjusted to the same value (7.2). The NMR experiments were performed on Avance 600- and 700-MHz spectrometers (Bruker, Karlsruhe, Germany), all equipped with cryogenic probeheads. 15N relaxation experiments were conducted on a 600 MHz spectrometer with a room temperature probe, using the singly labeled peptide. Temporal stability of the peptide solutions were checked with one-dimensional 1H-NMR spectra measured before and after each NMR experiment. All NMR spectra were processed and analyzed with NMRPipe (28) and Sparky 3 (T. D. Goddard and D. G. Kneller, http://www.cgl.ucsf.edu/home/sparky). To calculate secondary chemical shifts, random coil chemical shifts predicted by CamCoil were used (29).
1H-15N HSQC spectra of the backbone were obtained using an INEPT transfer delay of 5.4 ms. The experimental matrix had 2048 × 256 complex data points, which after zero filling was transformed to a final matrix of 8192 × 512 data points. The spectral width values were 10 and 26 ppm in 1H and 15N dimensions, respectively. HNCA spectra were recorded with 40 and 70 increments in the 15N and 13C dimension, respectively, with carrier frequencies set to 118 and 52 ppm. The 15N backbone relaxation rates, R1 and R2, were measured using relaxation delays of 10, 20, 80, 200, 400, 800, and 1200 ms for R1 and 10, 30, 50, 70, 110, 160, and 240 ms for R2 (30). The 15N longitudinal relaxation rate in the rotating frame (R1ρ) was measured in an on-resonance manner with a radio frequency field strength of 2.5 kHz and relaxation delays of 10, 40, 70, 110, 160, 240, and 320 ms (30).
Peak intensities were fitted to a single exponential decaying function. Heteronuclear nuclear Overhauser enhancements (NOEs) between 1H and 15N were measured with a 3.5 s irradiation of protons, which was shown to completely saturate amide proton resonances, and the NOE values were calculated through comparison of the peak intensities between saturated and reference spectra. The relaxation measurements were performed in an interleaved manner to ensure that the experimental conditions were the same for different relaxation delays, and in the case of R1, R2, and R1ρ measurements, the last interleaved experiment had the same relaxation delay as the first one to check the stability of the experiment. For R2 and R1ρ, a heat compensation element was implemented in the beginning of the pulse sequence before the recycle delay. The dipole-dipole (15N-1H)/chemical shift anisotropy (15N) cross-correlated relaxation rates (η) were measured using relaxation delays (2Δ) of 50, 100, 150, and 200 ms (31), after fitting to the equation
| (1) |
where IA and IB are the signal intensities obtained with pulse schemes A and B, which separately address relaxation of 15N downfield and upfield doublet components. The exchange-free R2 (R20) was calculated according to
| (2) |
where
| (3) |
with γN and γH as gyromagnetic ratios of 15N and 1H. The value κ was a constant value of 1.2589, calculated using theoretical expressions of 15N relaxation rates (32), for which the chemical shift anisotropy (CSA) of 15N was considered to be axially symmetric with a magnitude of −170 ppm and the angle between the symmetry axis of CSA tensor and N-H vector was assumed to be 15° (31). The exchange-mediated relaxation rates (Rex) were then calculated as Rex = R2 – R20 or Rex = R2 – R1ρ. The orientation spectral density function, J(ω), was calculated at three frequencies of 0, ωN and 0.87ωH as described in Palmer (33), with the exchange-free R20 calculated from cross-correlated relaxation rates taken as R2.
Results and Discussion
Identification of the Zn(II) binding site
The Aβ peptide tends to aggregate in a time- and concentration-dependent manner. To overcome this problem during the typically long NMR experiments, all the measurements were conducted at the low temperature of 5°C and peptide concentration of 50 μM, where the sample remains stable if it is devoid of any preformed aggregates. The latter condition was met by the initial treatment of Aβ with 10 mM sodium hydroxide (6). Comparison of one-dimensional 1H spectra before and after each NMR experiment verified that there was no significant peptide aggregation during the experiment. Because the three histidines of Aβ40 (His6, His13, and His14) constitute the main coordination sites of the zinc ion (17), the pH of the NMR sample was set to 7.2 to ensure that histidine side chains are mainly in a deprotonated state required for binding to zinc. At this pH, the apparent Kd of zinc binding to Aβ40 is ∼1 μM (18). To keep the temporal stability of Aβ solution upon addition of zinc, the zinc ion was added at a concentration of 50 μM, and the less aggregation-prone variant, Aβ40, was used. At the used metal/peptide ratio of 1:1, the reported apparent Kd of 1 μM implies that ∼90% of Aβ molecules are in the complex form with the zinc ion, and depending on the timescale of exchange between the free and metal-bound forms of Aβ in comparison with the NMR timescale, the NMR peaks are expected to be variably affected by addition of zinc.
Fig. 2 A shows a superposition of the 1H-15N HSQC spectra of Aβ40, obtained in the absence or presence of an equimolar concentration of zinc. Clearly, the most remarkable effect on amide proton and nitrogen chemical shifts was observed in the N-terminal part of the peptide, namely, residues Glu3, Arg5, Asp7, Ser8, Gly9, Tyr10, Glu11, Val12, and Gln15 (Fig. 2 B). The intensity profile revealed that the same residues were prominently affected by signal broadening (Fig. 2 C). The broadening effect was extended further to nearby residues of Lys16 and Leu17, and the side-chain peaks of Arg5 and Gln15 underwent a similar effect. The general reduction in the signal intensity of the N-terminal residues upon addition of zinc may arise from several causes. For example, presence of chemical exchange between free and metal-bound forms of the peptide leads to a severe intensity loss if the exchange occurs in an intermediate regime with respect to the NMR timescale, or a lower mobility of the peptide backbone induced upon metal binding increases the relaxation rates and broadens the signals. In addition, the rate of exchange between amide protons and water might be enhanced in the zinc binding region.
Figure 2.
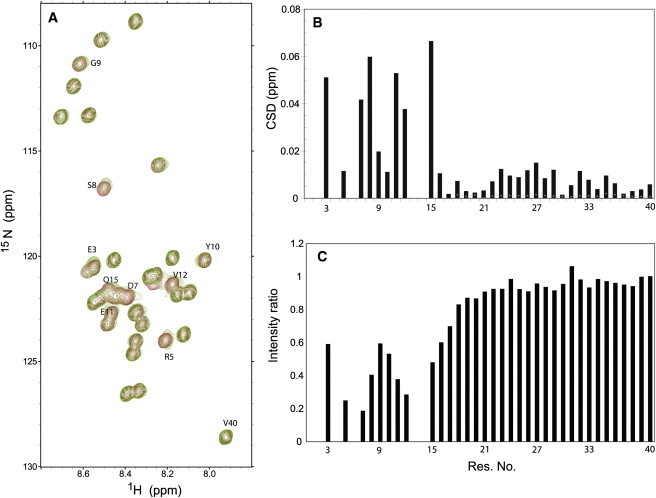
(A) 1H-15N HSQC spectra of Aβ40 in the absence (dark gray or red) or presence of equimolar zinc chloride (light gray or green). The more affected peaks, mainly in the N-terminus of Aβ40 sequence, are highlighted. (B) Average backbone N and HN chemical shift deviation in Aβ40 upon addition of an equimolar ratio of zinc. (Dotted line) Expected variability of chemical shift deviation for residues 18–40 if there is an 0.1 unit uncertainty in the measured pH. (C) Normalized intensity ratio, showing a striking loss of NMR signal intensity in the N-terminus of the peptide. Intensities were normalized based on the signal intensity of Val40.
The observed chemical shift perturbation and signal broadening effects are consistent with the known fact that the N-terminal part of Aβ40 constitutes the main coordination sites of the zinc ion, i.e., Asp1, His6, His13, and His14 (17). Because due to fast exchange with water the N-terminal Asp1 is principally missing from the 1H-15N HSQC spectra, and peaks of residues His6, His13, and His14 are severely affected by exchange broadening and/or overlap in Aβ40 spectra, we were not able to probe the effect of zinc ion on its directly binding residues. However, the observed intensity profile with its minima occurring at residues Arg5, Asp7, Val12, and Gln15, residues in the vicinity of His6, His13, and His14, support the direct role of these three histidines in zinc binding.
A second site of lower affinity for zinc binding has been recently suggested to be formed by residues Asp23 to Gly29 (18). In accord with this hypothesis, the stretch of residues between Asp23 and Gly29 showed a slightly higher than average perturbation in the backbone chemical shifts (Fig. 2 B). A significant chemical shift deviation was also observed for the side-chain peak of Asn27. These slight chemical shift deviations could not be caused by a pH change, as the pH was adjusted to 7.2 before and after addition of ZnCl2. To further ascertain that an uncertainty of ±0.1 unit in the measured pH would not cause the same level of chemical shift perturbation, the 1H-15N HSQC spectrum of metal-free Aβ40 was also measured at pH of 7.5 (0.3 unit above the pH of our study). To estimate the chemical shift changes that might be induced by a variation in pH by 0.1 unit, the observed pH-induced chemical shift deviations were divided by 3 and are shown in Fig. 2 B. Clearly, the chemical shift perturbations observed upon addition of zinc are mostly above this level, uniquely attributing the observed chemical shift changes to zinc binding.
Residues Asp23-Gly29 had intensities lower than the average in both the presence and absence of zinc ion. The change in intensity upon addition of zinc, however, showed no significant difference from nearby residues. The lack of significant signal broadening in this region upon zinc addition may be related to their smaller chemical shift difference in the free and metal-bound states, which takes their exchange between these states further away from the intermediate exchange regime and diminishes the exchange broadening effect. Although these data are consistent with the presence of a lower affinity zinc binding site in this region (18), a conformational change in residues 23–29—as a result of binding of zinc to the N-terminus—could present an alternative explanation.
Structural changes in the Aβ peptide induced by zinc binding
It is well known that the deviation of chemical shifts from their random coil values report on the local conformation of polypeptide chains (34,35). Cα chemical shifts are a very sensitive probe of local conformation, with its rise (drop) reflecting less (more) extended backbone conformation (34). To address how binding to zinc may affect Aβ40 conformation, Cα chemical shifts were measured through HNCA experiments. In the zinc-free form, two regions of the sequence showed negative secondary Cα chemical shifts: a continuous stretch of residues from Val8 to Glu22 and most of residues between Gly29 and Gly38 (see Fig. S1 in the Supporting Material). This reveals a tendency of these residues to adopt an extended conformation, whereas the intervening region remains in a less extended conformation.
Upon addition of zinc, residues Ala2, Glu3, Ser8, Val12, Gln15, and Lys16 of the N-terminal part were significantly affected, and except for Glu3, all decreased in Cα chemical shifts (Fig. 3 A). Because these residues are very close to the bound zinc ion, the changes of Cα chemical shift are not purely caused by conformational alterations, but the direct (de)shielding effect of the positively charged zinc ion results in a prominent contribution. Therefore, it is not possible to distinguish between the direct effects of the zinc ion on Cα chemical shift from those caused by conformational changes. However, the increased Cα chemical shift of Glu3, whereas the other nearby residues showed a decrease in Cα chemical shift, might support a model according to which binding to zinc induces a turn centered on Glu3 (18).
Figure 3.
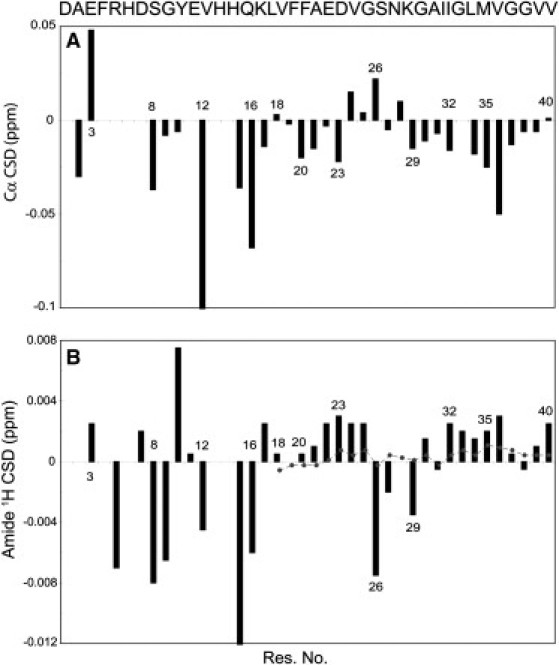
Carbon alpha (Cα) (A) and amide proton (B) chemical shift deviation of Aβ40, after addition of an equimolar ratio of zinc chloride. (B, Dotted line) Expected variability of amide proton chemical shift for residues 18–40 if there is an 0.1 unit uncertainty in the measured pH. The observed deviations suggest that residues 24–28 adopt a turnlike structure, whereas the flanking residues in nearby N- and C-terminal sides tend to form more extended structures.
On the other hand, for residues that are relatively far from the bound metal ion and their Cα chemical shifts are expected to more purely represent the backbone conformation, an interesting pattern was observed. Although residues Val24, Ser26, and Lys28 showed a considerable rise in Cα chemical shifts, most of the flanking residues on both N- and C-terminal sides had diminished shifts. It is interesting that residues 24–28, hypothesized to form a second binding site of lower affinity, changed their Cα chemical shifts oppositely to residues in the main N-terminal binding site. This makes it more probable that the chemical shift alterations we observe in this region are not due to direct binding of zinc, but instead caused by a secondary conformational change. Indeed, several experimental and theoretical studies point to the inherent tendency of the decapeptide Aβ(21–30) toward turn formation at residues Val24-Lys28 (36–38). This turn is stabilized by the intrinsic propensity of Val-Gly-Ser-Asn and Gly-Ser-Asn-Lys sequences to form a β-turn, a long-range Coulombic interaction between Lys28 and either Glu22 or Asp23, and a hydrophobic interaction between the isopropyl and butyl side chains of Val24 and Lys28 (37).
Accordingly, we hypothesize that after a zinc ion binds to the N-terminal site of the peptide, the region Ala21-Ala30 is released from some long-range contacts with the N-terminus and gains the possibility to reveal its intrinsic tendency to form a turn at residues Val24-Lys28, whereas the flanking sides will be slightly more extended. This hypothesis is further supported by the observed drop in amide proton chemical shifts of Ser26, Asn27, and Gly29, concomitant with a rise in the shifts of the flanking residues (Fig. 3 B) (35).
Solid-state NMR spectroscopy of Aβ40 fibrils showed that the region between residues 23 and 29 forms an exposed turnlike structure between two β-strands (39), and a salt bridge between Asp23 and Lys28 might contribute in stabilizing this structure (39). A similar structure also exists in Aβ oligomers (40), suggesting that formation of this turn and its resultant proximity of the N- and C-terminal parts of the Aβ sequence may occur relatively early in the aggregation process. In fact, occurrence of conformational exchange with transient adoption of a turnlike structure at residues 24–28 seems to be an inherent feature of the monomeric Aβ, as evidenced by the lower intensity of 1H-15N HSQC peaks in this region described above and secondary Cα chemical shifts.
Binding to zinc appears to enhance this feature, and considering the detailed structural models of Aβ40 oligomers (40) and fibrils (39), this alteration in conformational tendency of Aβ40 is in line with the observed promoting effect of zinc on Aβ aggregation. However, the higher aggregation propensity of the zinc-bound Aβ monomer does not result in formation of ordered fibrillar aggregates, as it is the case with metal-free Aβ40 and the more aggregation-prone variant of Aβ, Aβ42, but instead mainly produces amorphous aggregates. This difference in aggregation propensity may have its origin in the dynamical properties of the Aβ monomer; so we addressed in the next step how the Aβ40 backbone dynamics is altered by binding to zinc.
Binding of zinc increases the mobility on the picosecond-to-nanosecond timescale in Aβ regions involved in formation of fibrillar β-structure
To investigate the effects of zinc binding on the picosecond-to-nanosecond dynamics of the Aβ40 backbone, we measured 15N R1, R2, and steady-state 1H-15N NOE (Fig. 4). R1 values (Fig. 4 A) showed small fluctuations along the sequence of Aβ, with the values decreasing toward the N- and C-termini in agreement with increased internal dynamics on the picosecond-to-nanosecond timescale. The average R1 was 1.67 ± 0.17 s−1 for Aβ40 in the absence of zinc, and it remained nearly unchanged upon binding to zinc (with an average of 1.73 ± 0.23 s−1). The corresponding value for Aβ42 was 1.70 ± 0.19 s−1. Because 15N R1 is dominated by the value of the spectral density function J(ω) at ωN, motions with frequencies near ωN = 60 MHz (with a correlation time of ∼2.7 ns, in the same order as the expected global correlation time for Aβ monomer) are nearly identical for these peptides. This indicates that the assembly state of the NMR-observable Aβ molecules does not differ among these three conditions, i.e., all three are in the monomeric state. The similarity of global tumbling motions then allows a proper comparison of the internal motions of Aβ on the picosecond-to-nanosecond timescale.
Figure 4.
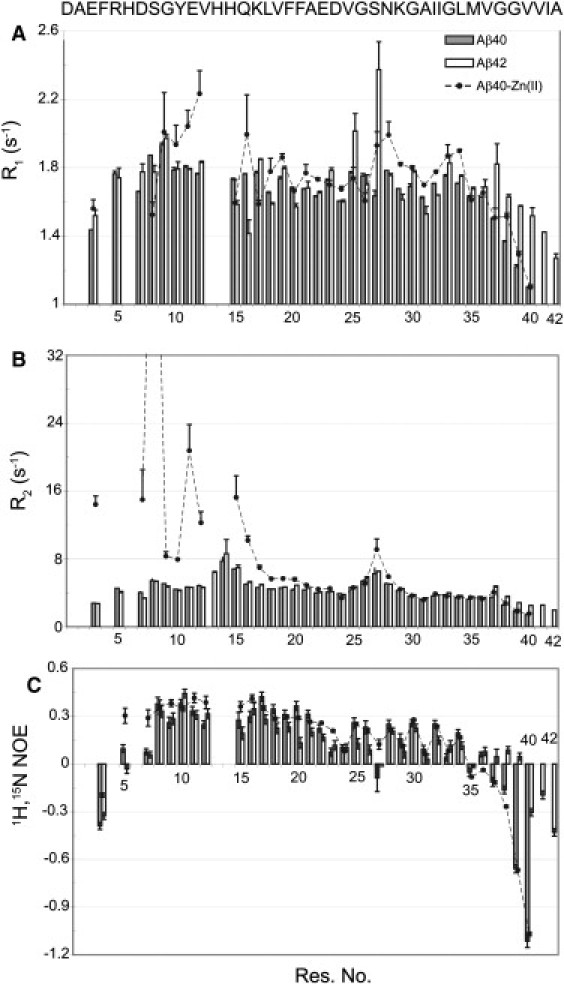
15N longitudinal (A) and transverse (B) relaxation rates and steady-state 1H-15N heteronuclear NOE values (C), measured for Aβ40 in the absence or presence of equimolar zinc, and Aβ42. R1 values showed little variability among the samples and indicated the same monomeric state of the three samples. The most remarkable change of R2 occurred at the N-terminus, reflecting the exchange-mediated relaxation contribution.
The 15N transverse relaxation rates (R2) are shown in Fig. 4 B. In the zinc-free Aβ40, R2 values gradually rose from the N- and C-termini, and the largest values were observed for residues His14, Gln15, and Gly25-Gly29. There was no significant difference in R2 values between Aβ40 and Aβ42, except for residues close to the C-terminus, and the same maxima of R2 were observed at His13-Gln15 and Gly25-Gly29. The larger R2 values for His13-Gln15 likely originate from chemical exchange due to protonation and deprotonation of the histidine side chains, and residues Gly25-Gly29 may also undergo a conformational exchange (see below for exchange-mediated relaxation rates, Rex). At the C-terminus, Aβ42 had significantly larger R2 values than Aβ40, even if the sequences were aligned from their C-termini, showing that the observed difference is not simply caused by the longer sequence of Aβ42. Upon addition of zinc to Aβ40, a general rise of R2 was observed in the N-terminal half up to Glu22, with residues close to the zinc binding site, namely Glu3, Arg5, Ser8, Glu11, Val12, and Gln15, mostly affected. The enhanced R2 is due to a chemical exchange between free and bound forms and/or a higher rigidity of the Aβ backbone induced upon zinc binding. A significant increase of R2 was detected for Asn27 and Lys28, whereas the C-terminal part was relatively unaffected.
The low steady-state 1H-15N heteronuclear NOE values, shown in Fig. 4 C, indicate that the Aβ backbone is remarkably dynamic on the picosecond-to-nanosecond timescale. In the free Aβ40, residues at the N- and C-terminus had negative NOE values, as expected from their higher mobility. The NOE values of Ser8-Ala20 were larger than in the C-terminal half. The data suggest that the backbone of Aβ40 is relatively less mobile in the N-terminal half, in agreement with the existence of some structured regions in the N-terminus of Aβ40 as suggested by molecular dynamics simulations (41). The C-terminal part showed an alternating NOE pattern between residues Lys28 and Gly37. This interesting finding may reflect a transient adoption of β-stranded conformation within residues 28–37. In the middle part of the sequence, residues Asp23 and Val24 had lower NOEs than the nearby residues and a negative NOE was observed for Asn27. This is likely due to a rapid interconversion between different conformational states on the picosecond-to-nanosecond timescale.
Compared to Aβ40, Aβ42 had slightly diminished NOE values in the N-terminal half (up to Ile32), except for Val12, Lys16, and Asn27. On the other hand, larger NOE values were observed at the C-terminus of Aβ42. This is in agreement with molecular dynamics simulations suggesting the formation of a β-hairpin structure at the C-terminus of Aβ42 (41). Addition of zinc to Aβ40 introduced some rigidity in the peptide backbone in close proximity to its binding site, as reflected by a considerable rise in the NOE of residues Glu3, Arg5, Asp7, Gly9, Glu11, Val12, Gln15, and Lys16. A similar change was also observed for Glu22, Asp23, and Asn27.
To further quantify the influence of Zn(II)-binding on the backbone dynamics of Aβ, reduced spectral density analysis of 15N relaxation parameters was carried out. Because the measured R2 were significantly affected by exchange-mediated relaxation (Rex), it was not possible to use R2 for this analysis. Instead, we measured the 15N longitudinal relaxation rate in the rotating frame (R1ρ) (see Fig. S2) in which the Rex contribution is scaled down. The R1ρ values of Aβ40 and Aβ42 were very similar, except at the C-terminus where Aβ42 had larger values. Upon addition of zinc, residues close to the zinc binding site in the N-terminus and Ser26-Gly29 increased in R1ρ. Because elimination of the exchange contribution is not complete in the R1ρ experiment, we further measured the cross-correlated relaxation rate (η) between 15N-1H dipolar and 15N CSA interactions. This parameter is dominated by J(0) and largely unaffected by the exchange process. Fig. S3 shows how η varies along the Aβ sequence.
In the free Aβ40, the largest η-values were observed for Tyr10-Ala21 and η gradually decreased toward the N- and C-termini. Upon addition of zinc Glu3, Tyr10, Gln15, and Lys16 showed an increased η (Fig. 5 A). There was also a rise of η for Ser26 and Lys28, whereas their flanking residues showed a slight drop in η. This interesting finding suggests that in the zinc-bound Aβ40, residues 26–28 lose some of their internal mobility on the picosecond-to-nanosecond timescale whereas the flanking residues at both N- and C-terminal sides become more mobile. On the other hand, in Aβ42, the rise of η at residues 25, 26, and 28 continued toward the C-terminus, indicating that the C-terminal half of the sequence is more rigid in Aβ42 than Aβ40.
Figure 5.
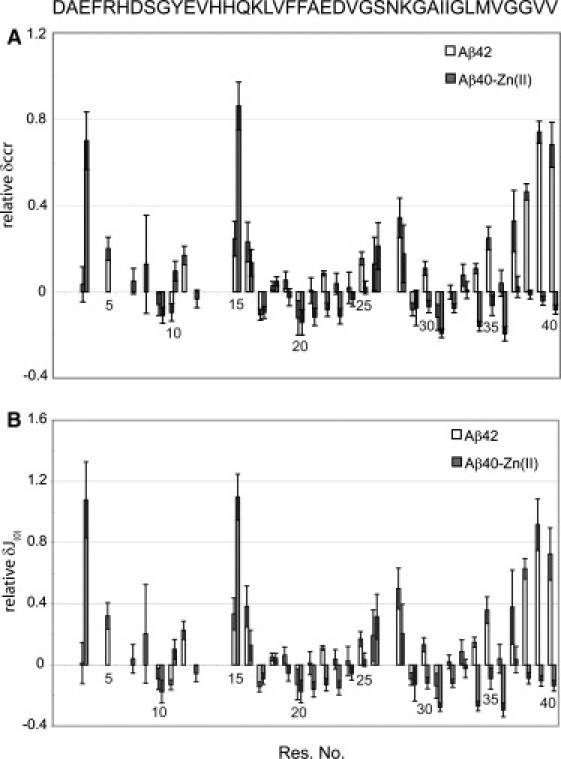
Differences in cross-correlated relaxation rates (CCR) (A) and spectral density function at frequency 0, J(0) (B), with respect to free Aβ40. Note that both zinc-bound Aβ40 and Aβ42 show larger values in the region Val24-Lys28, but change oppositely in the C-terminus. For calculation of J(0),CCR-based exchange-free R2 values were used. The changes were calculated relative to the value of free Aβ40 (i.e., a value of 1 means that it has increased to twice its value in the free Aβ40).
Cross-correlated relaxation rates (η) were then converted through Eq. 2 to R20, the exchange-free R2, and compared with R1ρ values. As displayed in Fig. S4, R20 was strongly correlated with R1ρ, with correlation coefficients of 0.78, 0.87, and 0.87 observed for Aβ40, Aβ42, and Aβ40-Zn(II), and corresponding slopes of best-fitted lines were 1.01, 0.97, and 0.92, respectively. The slopes of <1 indicate that, in the case of Aβ42 and Aβ40-Zn(II), R1ρ still has contributions from chemical exchange. Therefore, we used R20 to perform spectral density analysis. J(0), derived from the reduced spectral density analysis and shown in Fig. S5, is a measure of backbone dynamics and directly varies with the backbone rigidity. As expected from the other relaxation parameters of Aβ40, J(0) was higher for residues 8–21 and gradually decreased toward the N- and C-termini. Upon addition of zinc, residues 26–28 showed a raised J(0), but nearly all the nearby residues slightly dropped in J(0) (Fig. 5 B). On the other hand, the rise of J(0) at residues 26–28 of Aβ42 was accompanied with similar changes in the C-terminal half, especially after Gly33.
The 15N relaxation parameters discussed so far probe the picosecond-to-nanosecond dynamics of the peptide backbone. In contrast, the exchange-mediated relaxation rate, Rex, arises from processes that usually occur at longer timescales. Fig. 6 manifests how Rex varies along the Aβ sequence. In the free Aβ40, the largest Rex values were observed in two regions: first, close to the histidines 6, 13, and 14, probably reflecting an exchange due to protonation-deprotonation of histidine side chains; and second, for residues 26–28 that most likely undergo a conformational exchange. A similar but less pronounced pattern was found for Aβ42. On the other hand, addition of zinc to Aβ40 caused a striking rise in the Rex of the N-terminal part, most likely due to the exchange between free and metal-bound forms of the peptide. Interestingly, whereas several residues in the N- and C-terminal halves showed an enhanced Rex after zinc binding, residues 23–29 had either unchanged or diminished Rex. This supports our above suggestion that this region is not involved in direct binding to zinc but undergoes a secondary conformational change. The elevation of Rex in the C-terminus, which is far from the zinc-binding site, is not likely to be caused by binding, instead it may represent a higher mobility of this segment on the microsecond-to-millisecond timescale.
Figure 6.
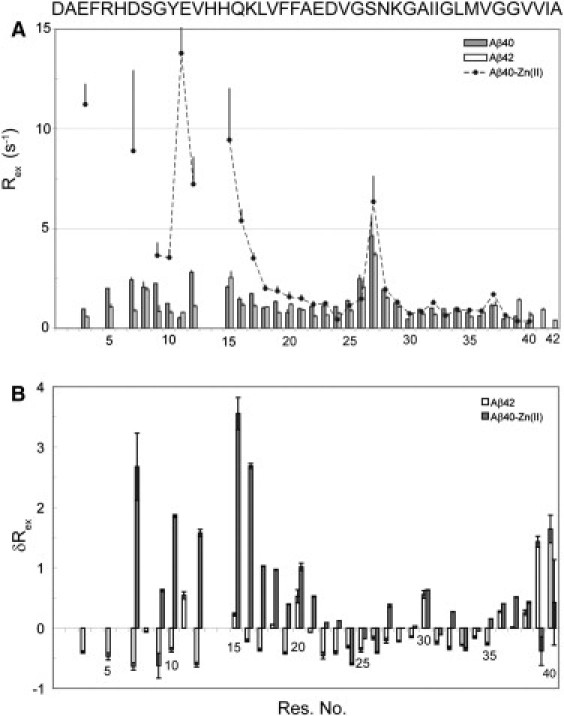
(A) Exchange-mediated relaxation rates (Rex), calculated as the difference between apparent R2 and R1ρ values, for Aβ40 in the absence or presence of equimolar zinc and Aβ42. A similar pattern was observed using CCR-based R2 instead of R1ρ (data not shown). (B) Differences in Rex with respect to the free Aβ40. Note the rise of Rex in the N-terminus and its drop at residues 24–26. The changes were calculated relative to the value of free Aβ40.
The importance of intrinsic dynamics in the Aβ peptide on its modes of aggregation
The effect of zinc binding on the dynamics of Aβ40 was investigated earlier by Danielsson et al. (18). Our data are in general agreement with their conclusion that binding of zinc induces an increased order in the N-terminus of Aβ40. The picosecond-to-nanosecond timescale dynamics of Aβ42 was also studied before (42,43), and a higher rigidity of the C-terminal half was demonstrated. Here, we have extended the earlier studies in the following areas.
First, whereas the conclusions by Danielsson et al. (18) were mainly based on the measurement of R1 and (apparent) R2, we have additionally measured R1ρ and cross-correlated relaxation (η) rates and 1H-15N heteronuclear NOEs. Through these relaxation parameters, we were able to distinguish the exchange-mediated contribution in transverse relaxation (Rex) and use an exchange-free R2 (R20) for reduced spectral density analysis. Through calculation of the spectral density function, the dynamics of the peptide backbone in the picosecond-to-nanosecond timescale is more accurately probed than the simple use of differences in apparent R2. The separation of Rex also enabled us to qualitatively address the effects of zinc binding on dynamics occurring on longer timescales.
Second, the dynamics measurements on zinc-bound Aβ40 were accompanied with the similar measurements on Aβ42. This made it more reliable to compare the relaxation properties of zinc-bound Aβ40 and Aβ42, which both have a higher aggregation propensity than free Aβ40 but form aggregates of different morphologies.
The combined 15N relaxation data, Cα chemical shifts, and 1H-15N HSQC intensities suggest the following model for the zinc-induced alterations in the structure and dynamics of Aβ40. Binding to zinc introduces conformational changes in the N-terminal part of Aβ40, including a turn formation centered at Glu3, and causes a higher rigidity of the backbone in this part. This induces a change in the conformations sampled by residues Val24 to Lys28 toward turnlike structures that can bring the N- and C-terminal parts of Aβ40 closer together. Although flexibility of this region is partially lost upon zinc-induced turn formation, the nearby residues on both N- and C-terminal sides gain in flexibility.
This may be due to loss of long-range contacts between the central hydrophobic cluster (Leu17-Ala21) and Val39, shown in the free form of Aβ40 (41). The higher mobility results in a more frequent sampling of the favorable extended conformation by those residues, reflected in their Cα and amide 1H chemical shift deviations. Because a similar turn exists in Aβ40 oligomeric aggregates (40), the higher propensity of Aβ40-Zn(II) to adopt this conformation in the monomeric state is expected to decrease the entropic cost of aggregation, and thermodynamically favor the aggregation process (see Fig. 7). This mechanism of aggregation promotion may act in addition to the simple electrostatic role of the zinc ion, according to which a decrease in the net negative charge of Aβ40 after zinc binding weakens the electrostatic repulsion among them (18).
Figure 7.
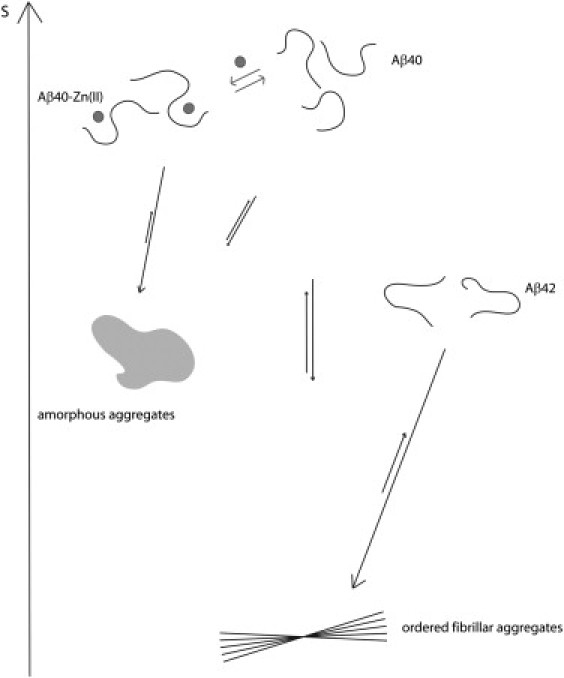
Cartoon representation of Aβ dynamics and implications for its aggregation. Formation of ordered fibrillar aggregates from the initial monomeric Aβ implies a huge entropic price. The monomeric state of Aβ42 is relatively restricted at the C-terminus, so it needs to pay less entropic cost than Aβ40 and therefore shows a higher tendency to form fibrils. On the other hand, zinc binding to Aβ40 introduces rigidity around its binding site but mobilizes the C-terminus. As a result, formation of amorphous aggregates with less ordered C-termini is favored compared to fibril formation. The entropic scale is only used for illustration and should not be taken rigorously.
The higher mobility of the C-terminal part of Aβ40 in complex with a zinc ion is in contrast with the lower mobility of the corresponding part of Aβ42. This feature of Aβ42 has been used to explain its higher (than Aβ40) propensity to form fibrillar aggregates (42,43). Because dynamics of Aβ42 fibrils in the C-terminus is not expected to be different from those in Aβ40 fibrils, the lower mobility of the C-terminus of Aβ42 monomers should decrease the entropic penalty of fibril formation. Similarly, the enhanced dynamics of the C-terminus of Aβ40 in complex with zinc should increase the configurational entropic cost of fibril formation and thermodynamically disfavor fibrillar aggregation.
However, amorphous aggregates that are likely less ordered in the C-terminal part are expected to be favored. Induction of a specific conformation in the N-terminus to accommodate the zinc ion may present an additional mechanism to prevent the N-terminal β-strand formation necessary for fibrillar aggregation. However, it was shown for copper that binding to Aβ was independent of aggregation state, i.e., the same coordination sphere was observed for Aβ monomers, oligomers, and fibrils (17,44,45). Binding of Cu(II) ion to monomeric Aβ did not interfere with fibrillar aggregation, nor did it affect the fibrillar structure when it was added to preformed fibrils (44,45). Given that similar residues are involved in zinc coordination, the new conformation induced in the N-terminus upon metal binding is unlikely to explain its amorphous aggregation propensity.
Conclusion
Our study confirmed the presence of a zinc binding site at the N-terminus of Aβ40 and suggested that a turnlike conformation is formed by residues Val24-Lys28, probably as a result of zinc binding to the N-terminus. Furthermore, relaxation measurements revealed that zinc binding leads to a reduction in mobility on the picosecond-to-nanosecond timescale in the N-terminus of Aβ40 and for Val24-Lys28. On the other hand, residues in the intervening region and at the C-terminus became more mobile on the picosecond-to-nanosecond timescale, as well as on the microsecond-to-millisecond timescale. At the same time, Aβ42 revealed a higher rigidity than Aβ40 at the C-terminus.
We propose that the increased rigidity of the N-terminus and residues Val24-Lys28 causes a net decrease of configurational entropy in the monomeric peptide, leading to a thermodynamic destabilization of the monomer and promotion of aggregation. In contrast, the higher mobility of the C-terminus may favor amorphous aggregation resulting in less ordered C-termini and therefore requires less entropic cost than fibrillar aggregation. Taken together, our study provides detailed mechanistic insights on how the Aβ peptide is directed toward different aggregation pathways.
Acknowledgments
We thank Christian Griesinger for useful discussions.
This work was supported by the Max Planck Society and the Deutsche Forschungsgemeinschaft through a Heisenberg scholarship (ZW 71/2-2 and 3-2) to M.Z.
Contributor Information
Nasrollah Rezaei-Ghaleh, Email: nare@nmr.mpibpc.mpg.de.
Markus Zweckstetter, Email: mzwecks@gwdg.de.
Supporting Material
References
- 1.Citron M. Alzheimer's disease: strategies for disease modification. Nat. Rev. Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 2.Trojanowski J.Q., Lee V.M. “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer's disease and other neurodegenerative disorders. Ann. N. Y. Acad. Sci. 2000;924:62–67. doi: 10.1111/j.1749-6632.2000.tb05561.x. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Walsh D.M., Selkoe D.J. A β oligomers —a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 5.Reinhard C., Hébert S.S., De Strooper B. The amyloid-β precursor protein: integrating structure with biological function. EMBO J. 2005;24:3996–4006. doi: 10.1038/sj.emboj.7600860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hou L., Shao H., Zagorski M.G. Solution NMR studies of the Aβ (1–40) and A β1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 7.Jarrett J.T., Berger E.P., Lansbury P.T., Jr. The carboxy terminus of the β-amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 8.El-Agnaf O.M., Mahil D.S., Austen B.M. Oligomerization and toxicity of β-amyloid-42 implicated in Alzheimer's disease. Biochem. Biophys. Res. Commun. 2000;273:1003–1007. doi: 10.1006/bbrc.2000.3051. [DOI] [PubMed] [Google Scholar]
- 9.Bitan G., Kirkitadze M.D., Teplow D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., McLaughlin R., LeBlanc A. Selective cytotoxicity of intracellular amyloid β peptide 1–42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mucke L., Masliah E., McConlogue L. High-level neuronal expression of Aβ 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gravina S.A., Ho L., Younkin S.G. Amyloid β protein (Aβ) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ 40 or Aβ 42(43) J. Biol. Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 13.De Jonghe C., Esselens C., De Strooper B. Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 2001;10:1665–1671. doi: 10.1093/hmg/10.16.1665. [DOI] [PubMed] [Google Scholar]
- 14.Zatta P., Lucchini R., Taylor A. The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res. Bull. 2003;62:15–28. doi: 10.1016/s0361-9230(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 15.Miller L.M., Wang Q., Miklossy J. Synchrotron-based infrared and x-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer's disease. J. Struct. Biol. 2006;155:30–37. doi: 10.1016/j.jsb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Frederickson C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989;31:145–238. doi: 10.1016/s0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- 17.Faller P., Hureau C. Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-β peptide. Dalton Trans. 2009;7:1080–1094. doi: 10.1039/b813398k. [DOI] [PubMed] [Google Scholar]
- 18.Danielsson J., Pierattelli R., Gräslund A. High-resolution NMR studies of the zinc-binding site of the Alzheimer's amyloid β-peptide. FEBS J. 2007;274:46–59. doi: 10.1111/j.1742-4658.2006.05563.x. [DOI] [PubMed] [Google Scholar]
- 19.Lovell M.A., Xie C., Markesbery W.R. Protection against amyloid β peptide toxicity by zinc. Brain Res. 1999;823:88–95. doi: 10.1016/s0006-8993(99)01114-2. [DOI] [PubMed] [Google Scholar]
- 20.Cardoso S.M., Rego A.C., Oliveira C.R. Protective effect of zinc on amyloid-β 25–35 and 1–40 mediated toxicity. Neurotox. Res. 2005;7:273–281. doi: 10.1007/BF03033885. [DOI] [PubMed] [Google Scholar]
- 21.Cuajungco M.P., Goldstein L.E., Bush A.I. Evidence that the β-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of Aβ by zinc. J. Biol. Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 22.Garai K., Sahoo B., Maiti S. Zinc lowers amyloid-β toxicity by selectively precipitating aggregation intermediates. Biochemistry. 2007;46:10655–10663. doi: 10.1021/bi700798b. [DOI] [PubMed] [Google Scholar]
- 23.Huang X., Atwood C.S., Bush A.I. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer's Aβ peptides. J. Biol. Inorg. Chem. 2004;9:954–960. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 24.House E., Collingwood J., Exley C. Aluminum, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Aβ42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease. J. Alzheimers Dis. 2004;6:291–301. doi: 10.3233/jad-2004-6310. [DOI] [PubMed] [Google Scholar]
- 25.Yoshiike Y., Tanemura K., Takashima A. New insights on how metals disrupt amyloid β-aggregation and their effects on amyloid-β cytotoxicity. J. Biol. Chem. 2001;276:32293–32299. doi: 10.1074/jbc.M010706200. [DOI] [PubMed] [Google Scholar]
- 26.Innocenti M., Salvietti E., Messori L. Trace copper(II) or zinc(II) ions drastically modify the aggregation behavior of amyloid-β1-42: an AFM study. J. Alzheimers Dis. 2010;19:1323–1329. doi: 10.3233/JAD-2010-1338. [DOI] [PubMed] [Google Scholar]
- 27.Edlich C., Stier G., Muhle-Goll C. Structure and phosphatidylinositol-(3,4)-bisphosphate binding of the C-terminal PH domain of human pleckstrin. Structure. 2005;13:277–286. doi: 10.1016/j.str.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 29.De Simone A., Cavalli A., Vendruscolo M. Accurate random coil chemical shifts from an analysis of loop regions in native states of proteins. J. Am. Chem. Soc. 2009;131:16332–16333. doi: 10.1021/ja904937a. [DOI] [PubMed] [Google Scholar]
- 30.Palmer A.G., 3rd NMR probes of molecular dynamics: overview and comparison with other techniques. Annu. Rev. Biophys. Biomol. Struct. 2001;30:129–155. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- 31.Tjandra N., Szabo A., Bax A. Protein backbone dynamics and 15N chemical shift anisotropy from quantitative measurement of relaxation interference effects. J. Am. Chem. Soc. 1996;118:6986–6991. [Google Scholar]
- 32.Cavanagh J.F.W., Palmer A.G., 3rd, Skelton N.J. Academic Press; San Diego, CA: 2007. Protein NMR Spectroscopy, Principles and Practice. [Google Scholar]
- 33.Palmer A.G., 3rd NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 34.Wishart D.S., Sykes B.D. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 35.Wishart D.S., Sykes B.D., Richards F.M. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 36.Fawzi N.L., Phillips A.H., Head-Gordon T. Structure and dynamics of the Aβ (21–30) peptide from the interplay of NMR experiments and molecular simulations. J. Am. Chem. Soc. 2008;130:6145–6158. doi: 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazo N.D., Grant M.A., Teplow D.B. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14:1581–1596. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grant M.A., Lazo N.D., Teplow D.B. Familial Alzheimer's disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc. Natl. Acad. Sci. USA. 2007;104:16522–16527. doi: 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petkova A.T., Ishii Y., Tycko R. A structural model for Alzheimer's β -amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L., Edalji R., Olejniczak E.T. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 41.Sgourakis N.G., Yan Y., Garcia A.E. The Alzheimer's peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan Y., Wang C. Aβ42 is more rigid than Aβ40 at the C terminus: implications for Aβ aggregation and toxicity. J. Mol. Biol. 2006;364:853–862. doi: 10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 43.Lim K.H., Henderson G.L., Louhivuori M. Structural, dynamic properties of key residues in Aβ amyloidogenesis: implications of an important role of nanosecond timescale dynamics. ChemBioChem. 2007;8:1251–1254. doi: 10.1002/cbic.200700194. [DOI] [PubMed] [Google Scholar]
- 44.Karr J.W., Akintoye H., Szalai V.A. N-Terminal deletions modify the Cu2+ binding site in amyloid-β. Biochemistry. 2005;44:5478–5487. doi: 10.1021/bi047611e. [DOI] [PubMed] [Google Scholar]
- 45.Karr J.W., Szalai V.A. Cu(II) binding to monomeric, oligomeric, and fibrillar forms of the Alzheimer's disease amyloid-β peptide. Biochemistry. 2008;47:5006–5016. doi: 10.1021/bi702423h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.