

Abstract
β2-Microglobulin is a 99-residue protein with a propensity to form amyloid-like fibrils in vitro which exhibit distinct morphologies dependent on the solution conditions employed. Here we have used ion mobility spectrometry-mass spectrometry to characterize the oligomeric species detected during the formation of worm-like fibrils of β2-microglobulin at pH 3.6. Immediately upon sample dissolution, β2-microglobulin monomer and oligomers—the latter ranging in size from dimer to hexamer—are present as a pool of rapidly interconverting species. Increasing the ionic strength of the solution initiates fibril formation without a lag-phase whereupon these oligomers become more stable and higher-order species (7-mer to >14-mer) are observed. The oligomers detected have collision cross-sectional areas consistent with a linearly stacked assembly comprising subunits of native-like volume. The results provide insights into the identity and properties of the transient, oligomeric intermediates formed during assembly of worm-like fibrils and identify species that differ significantly from the oligomers previously characterized during the nucleated assembly of long, straight fibrils. The data presented demonstrate the interrelationship between different fibril-forming pathways and identify their points of divergence.
Introduction
The formation of amyloid-like fibrils from β2-microglobulin (β2m) in vitro has been studied extensively after the discovery of this protein as the primary causative agent of hemodialysis-related amyloidosis (reviewed in Heegaard (1)). In vitro, the morphology of fibrils formed depends on the solution conditions (2): incubation of β2m under acidic conditions (pH 1.0–5.0) and high ionic strength (>100 mM) gives rise to spontaneous, nonnucleated assembly of fibrils with a worm-like (WL) morphology. In contrast, at low pH (<3.0) and low ionic strength buffer (<100 mM), β2m assembles via a nucleation-dependent mechanism forming long, straight (LS) fibrils with an axial repeat periodicity of 50–100 nm which possess a structure akin to ex vivo amyloid fibrils (2,3). Significant progress has been made toward characterizing the structures of the different β2m fibrils formed in vitro using a plethora of techniques including limited proteolysis coupled with electrospray ionization-mass spectrometry (ESI-MS) (4,5), atomic force microscopy (AFM) (2,6), Fourier transform infrared (FT-IR) (7,8), cryo-electron microscopy (9), electron paramagnetic resonance (EPR) (10), and nuclear magnetic resonance (NMR) (11).
The conformation of both the LS and WL β2m fibrils has also been investigated at a residue-specific level by hydrogen deuterium exchange (12,13) and solid-state NMR (14,15) experiments. Protection factors indicate that the β-strands involved in the core of the native protein, and also their connecting loops, comprise the structured core of the LS fibrils, with the N- and C-terminal residues showing the least protection (12,13,16) in accord with proteolysis data (4,5). A different hydrogen deuterium exchange protection pattern was obtained for the WL fibrils with the data suggesting that the protected core of these fibrils is restricted to two regions involving residues Val37-Lys48 and Ser57-Asp69 (13). Continuous-wave EPR spectroscopy (10) and solid-state NMR (14) of LS fibrils reveal a core formed from β-strands arranged in stacks with a parallel, in-register structure that is highly nonnative. For the WL fibrils, FT-IR spectra indicate a more nativelike conformation within the fibril core (8,10) and EPR spectroscopy data demonstrate a lack of an ordered, parallel, in-register structure (10).
The differences in the architectures of LS and WL fibrils suggest that their pathways of formation may also be different. Other amyloid systems have also shown evidence for multiple assembly pathways: a truncated recombinant prion protein forms two structurally distinct isoforms, a β-oligomer and an α-helical conformer, although the former is not on-pathway to fibril formation (17), while a mutagenesis study on barstar, an amyloid protofibril-forming protein, led to the detection of alternative aggregation intermediates depending on the particular mutation (18). Techniques including AFM (3,19), NMR (20–22), single molecule fluorescence (23), EPR (24), and ESI-MS (recently including ESI-ion mobility spectrometry (IMS)-MS (3,17,25–31,33,34) have been used to observe protein oligomers created during amyloid formation.
The ability to characterize individual oligomers within a copopulated mixture is a key attribute of ESI-IMS-MS, as these species may be responsible for amyloid-related toxicity, rather than the fibrils themselves (35–37). As several oligomers may contribute to signals at a particular m/z ratio, individual oligomers cannot be quantified or characterized unless they are separated in situ. ESI-IMS-MS has been used to separate (and measure the collision cross-sectional area, Ω of) transient intermediates with ringlike assemblies of Aβ1-42 but a more open structure for Aβ1-40 (27). An alternative model involving stacked oligomers <16-mer detected during Aβ1-40 assembly has been reported separately (29). β2m oligomers (dimer, trimer, and tetramer) detected by ESI-IMS-MS during the lag time of LS fibril formation were also found to have collision cross-sectional areas consistent with stacked monomer units arranged in linear assemblies (31), and the subunit exchange dynamics of each oligomer, monitored directly within the ensemble by mixing 14N- and 15N- labeled oligomers, revealed a decrease in oligomer dynamics concomitant with increasing oligomer size (31).
In contrast with the plethora of information now available on β2m fibril formation at pH 2.5, our understanding of the mechanism of WL fibril formation at pH 3.6 is less advanced. ESI-MS has shown that monomer decline occurs in an exponential manner, in sharp contrast with the lag phase observed during LS fibril formation (3). Again in marked contrast with the formation of LS fibrils, higher order oligomers ≥14-mer in size are observed during WL fibril assembly (3). However, quantification of these copopulated oligomeric intermediates was not possible in the early studies due to the low intensity of the ions arising from the higher order species and the lack of any ion mobility separation to clarify the heterogeneity of the assembly reaction. Here we have extended these studies by using ESI-IMS-MS to separate different oligomers during WL fibril formation from β2m at pH 3.6, thus allowing the shape, dynamics, and population of each of the multitude of oligomers observed to be determined. We have identified a set of compact oligomeric species populated before WL fibril formation and have compared these directly with the limited number of transient oligomers detected during LS fibril assembly. We believe that, from these new data, the inter-relationship between the WL and LS fibril assembly pathways and their points of divergence has been ascertained.
Materials and Methods
Reagents
The Escherichia coli strain BL21 (DE3) was obtained from Promega UK (Southampton, Hampshire, UK). Dithiothreitol, Q-Sepharose, and des-Arg bradykinin were purchased from Sigma-Aldrich (Gillingham, Dorset, UK). Spectrapore membrane (molecular mass cutoff of 3500 Da) was obtained from Spectrum Laboratories (Breda, The Netherlands). Carbenicillin was obtained from Melford Laboratories (Ipswich, Suffolk, UK). 14N- and 15N-recombinant β2m were prepared as described previously (38). Solvents were purchased from Fisher Scientific (Loughborough, Leicestershire, UK).
Formation of amyloid fibrils from β2m and quantification of monomer concentration
β2m (38) (0.4 mg mL−1) was incubated in 100 mM ammonium acetate or formate (pH 3.6; 20°C) without agitation. Fibril growth kinetics were monitored using thioflavin-T fluorescence (10 μM) as described previously (39). Monomer quantification was achieved using ESI-MS; all observable monomer peaks visible in the mass spectra were summed and normalized to the (M+H)+ ions (m/z 904) of the internal standard, des-Arg bradykinin (1.4 μM, added immediately before analysis) (3).
14N/15N-β2m oligomer exchange
Samples of 14N- and 15N-β2m (31) (0.4 mg mL−1) in 100 mM ammonium acetate or formate were preincubated at 20°C for 1, 20, or 180 min. A quantity of 200 μL of each sample was then mixed and an aliquot removed and analyzed immediately by ESI-IMS-MS.
Electron microscopy
Collodion-coated copper electron microscopy (EM) grids were coated with protein samples as described (19). All images were taken using a CM10 electron microscope (Phillips Research, Eindoven, The Netherlands) operating at 80 keV.
ESI-IMS-MS
Electrospray ionization-mass spectrometry-mobility spectrometry (ESI-IMS-MS) experiments were carried out using a Synapt HDMS (Micromass UK, Manchester, UK) quadrupole-traveling wave IMS-orthogonal acceleration time-of-flight mass spectrometer, equipped with a Triversa NanoMate (Advion Biosciences, Ithaca, NY) automated nano-ESI interface. Positive ESI was used with a capillary voltage of 1.7 kV and a nitrogen nebulizing gas pressure of 0.7 psi. A sampling cone voltage of 175 V and a source backing pressure of 3.8 mbar were set on the mass spectrometer to optimize the observation of β2m oligomers. Samples were analyzed directly from the fibril-forming solutions without any further preparation.
Data were acquired over the range m/z 500–15,000 for 5 min per acquisition. Mass calibration was performed on a separate infusion of CsI cluster ions. Trap and transfer T-wave voltages were set at 4 and 40 V, respectively, ensuring that solvent clusters, but not the oligomers, are dissociated. The raw data were processed using MassLynx v.4.1 software (Micromass UK). For IMS-MS experiments, the wave height was ramped from 2 to 25 V at a speed of 300 ms−1. Drift times were corrected for both mass-dependent and mass-independent times (40). The drift time cross-section function was calibrated as described previously (34,41). Computer-based cross-sectional area estimations were calculated from in silico models generated using PyMOL Molecular Graphics System Version 1.2r3pre (Schrodinger, Camberley, UK). The β2m crystal structure (1LDS) (42) was used as a base for modeling to provide a comparison between gas-phase and solution-phase structures using an in-house projection approximation algorithm (34,40).
Results
Monitoring oligomers throughout the self-assembly pathway of β2m at pH 3.6
Here we report the profile of transiently populated oligomers detected during the self-aggregation of monomeric β2m (11,860.4 Da) into WL fibrils at pH 3.6. When incubated at pH 3.6 in low ionic strength solution (e.g., 10 mM ammonium acetate or formate, 20°C), β2m populates oligomers up to, and including, the hexamer. To induce WL fibril formation at this pH, the ionic strength of the solution was increased to a final buffer concentration of 100 mM. Under these conditions, WL fibril formation takes ∼1 h to reach completion at a protein concentration of 0.4 mg mL−1, as monitored by thioflavin-T fluorescence (Fig. 1). At time-points throughout fibril assembly, ESI-(IMS)-MS data were acquired and the decline in β2m monomer concentration was quantified using des-Arg bradykinin as an internal standard. As reported previously (3), monomer is lost in an exponential manner from the initiation of fibril assembly at a rate that mirrors the rate of WL fibril formation (Fig. 1).
Figure 1.

The formation of worm-like (WL) fibrils of β2m (0.4 mg mL−1 based on monomer; pH 3.6, 100 mM ammonium acetate; 20°C). β2m monomer (open circles) was quantified over time by ESI-IMS-MS. The m/z spectra were normalized to the (M+H)+ ions of des-Arg bradykinin (1.4 μM, added immediately before analysis) at m/z 904 for monomer quantification (3). (Error bars) Standard deviation of the mean over three replicate experiments. WL fibril formation was monitored simultaneously by thioflavin-T fluorescence (thick solid line). A control in which β2m (0.4 mg mL−1) was incubated in 10 mM ammonium acetate (pH 3.6; 20°C) was also analyzed; thioflavin-T fluorescence confirmed that fibrils do not form under these conditions (thick shaded line). (Inset) Negative stain EM image of fully formed WL fibrils (the scale bar = 100 nm).
The oligomers populated under the low ionic strength conditions at pH 3.6 from which fibrillation of β2m was initiated are readily separated by ESI-IMS-MS. Ion separation by IMS is achieved as the speed with which an ion travels through the IMS drift cell depends on its shape and charge: more compact ions traverse the cell faster than extended ions of the same charge and mass, while ions of the same m/z ratio but different charge and mass separate with the more highly charged ions having the shorter drift times. The drift times measured can be calibrated using proteins of known collision cross-sectional area and thus the cross-sectional areas of the analytes measured (34,43), as well as their mass, in a single experiment. Fig. 2 A presents these results in the form of a driftscope plot which shows drift time (ms) versus m/z versus intensity. Upon increasing the ionic strength of the solution to initiate fibril formation, oligomers ≤9-mer are populated within the first minute (Fig. 2, C and D). After 20 min, all oligomers ≤14-mer (166,045.6 Da) are observed (Fig. 2, E and F). Higher-order species than this may well be populated, but their signal intensities were too weak for confident characterization. As the separative feature of ESI-IMS-MS allows the individual charge-state ions of each oligomer to be identified, the relative population of any individual oligomer can be estimated at a given time-point by summing the peak areas of each of its charge-state ions and expressing this as a fraction of the total ion intensity.
Figure 2.

ESI-IMS-MS driftscope plots (drift time versus m/z versus intensity; left) and m/z spectra (right) showing β2m (0.4 mg mL−1 based on monomer; 20°C; pH 3.6) oligomers detected. (A and B) 10 mM ammonium acetate; (C and D) 100 mM ammonium acetate, after 1 min; (E and F) 100 mM ammonium acetate, after 20 min. The numbers above the peaks indicate the size of each oligomer (e.g., 3 = trimer). The oligomer region (>m/z 6000) in the m/z spectra has been magnified by a factor of 10.
Although this approach does not allow absolute quantification of oligomer concentration because the ionization potential of each species may differ depending on the size and conformation of the ions, it provides a robust method to quantify the relative population of each oligomer along the assembly pathway of fibril formation. The relative populations of β2m monomer, trimer, hexamer, and 9-mer during the time-course of fibril assembly (t = 0–180 min) are shown in Fig. 3. The first time-point (−10 min) indicates the relative populations of the trimer and hexamer before assembly initiation, both of which increase by a factor of ∼2 when the ionic strength of the solution is increased.
Figure 3.
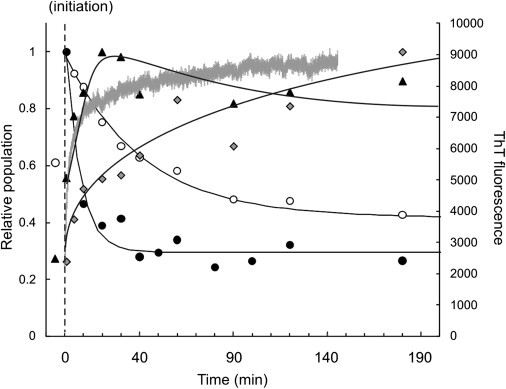
β2m oligomer population (0.4 mg mL−1 based on monomer) before (t = −10–0 min; 10 mM ammonium acetate; pH 3.6) and during WL fibril formation (t = 0–180 min; 100 mM ammonium acetate, pH 3.6). (Solid circles) β2m monomer; (open circles) trimer; (solid triangles) hexamer; and (shaded diamonds) 9-mer. For each oligomer, signals were normalized to a value of 1 for the most intense signal detected during the time course. The data are fitted to exponential functions (solid lines). (Thick shaded line) Thioflavin-T fluorescence signal.
As self-assembly progresses, populations of the monomer and lower-order oligomers (≤ trimer) decrease exponentially, with the loss of monomer preceding the reduction in the population of the trimer. The hexamer population initially increases during assembly, peaking at 20 min before slowly decreasing. The increases in the populations of higher-order oligomers (>9-mer) are much slower in comparison to the lower-order oligomers and do not reach a maximum in intensity by the end of the experimental time-course (180 min). The successive formation of oligomers of increasing molecular mass as a function of assembly time, combined with the apparent exponential decrease in the population of monomer and lower-order oligomers, suggests that WL fibril formation occurs by a monomer addition mechanism, beginning with lower-order oligomers followed by the sequential population of larger oligomers as self-assembly proceeds.
Determining the shape and stability of the β2m oligomers
The kinetic stabilities of the different β2m oligomers were assessed by investigating their propensity to undergo subunit exchange using 14N- and 15N-labeled protein mixing experiments. Uniform 15N-labeling of β2m results in a mass increase of 140 Da per protein monomer compared with 14N-labeled protein. WL fibril formation from both 14N- and 15N-labeled β2m was initiated separately and aliquots from the two solutions were subsequently mixed in an equimolar ratio at different times during self-assembly. If the subunits of the oligomers are in rapid exchange, the oligomers detected by ESI-MS will be formed stochastically from the mixed protein pool. In the case of the dimer this would manifest as three peaks in the mass spectrum in the ratio 1:2:1 corresponding to 14N/14N-, 14N/15N-, and 15N/15N-labeled dimers. For the trimer, tetramer, and pentamer, random subunit exchange would result in four, five, or six peaks, respectively, with ratios of 1:3:3:1, 1:4:6:4:1, and 1:5:10:10:5:1 for the different resolvable isomeric assemblies.
Fully exchanged oligomers larger than the pentamer cannot be resolved into their individually labeled components completely due to the broad nature of the peaks resulting from multiple salt adducts; nevertheless, resolution of the all-14N- from the all-15N-labeled entities can be achieved for all oligomers under these conditions. Fig. 4 summarizes the subunit exchange data for β2m dimer, trimer, tetramer, and pentamer. When the two protein samples are mixed just 1 min after the initiation of fibril assembly, all the β2m oligomers detected had undergone rapid subunit exchange and a stochastic mix of 14N/15N-labeled oligomers was observed for each oligomer. If the two protein samples are mixed 20 min into fibril growth, all the charge-state ions associated with the dimer, together with the more highly charged trimer (+10 and +11 charge states) and tetramer (+12 and +13) ions, remain in rapid subunit exchange. However, the lower charge-state ions of the trimer (+6, +7, +8, and +9) and the tetramer (+9, +10, and +11), plus all the pentamer ions (+11, +12, and +13), are significantly less amenable to subunit exchange. The latter observation is manifest by domination of the 14N- and 15N-only oligomeric states, with the 14N/15N-mixed oligomers displaying only weak intensity after mixing the 14N- and 15N-labeled samples at 20 min. Although the +12 and +13 charge-state ions of the tetramer and the +10 and +11 charge-state ions of the trimer still display dynamic behavior at these later time points, their intensity has decreased.
Figure 4.
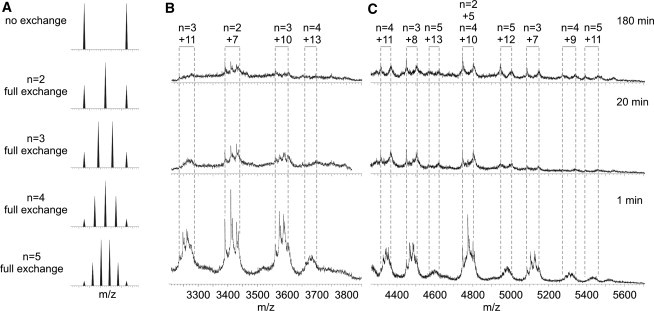
β2m oligomer dynamics change as a function of assembly time. 14N- and 15N-labeled β2m were incubated separately under WL fibril-forming conditions (0.4 mg mL−1 based on monomer; 100 mM ammonium acetate, pH 3.6). The two samples were mixed in an equimolar ratio at various time points and ESI-MS spectra acquired within 1 min of mixing. (A) Predicted m/z spectra for dimer, trimer, tetramer, and pentamer, assuming full 14N/15N-labeled subunit exchange has occurred. If subunit exchange with the bulk protein pool is rapid then, in the case of the dimer (n = 2), a 1:2:1 ratio of 14N/14N:14N/15N:15N/15N will be observed. If the exchange is slow or does not occur, then a 1:1 ratio of 14N/14N:15N/15N will be observed. (B) ESI-MS spectra (m/z 3200–3800) at t = 1, 20, and 180 min. The dimer and the more highly charged trimer (+10, +11) and tetramer (+13) ions exchange rapidly throughout the time course, but are barely populated by 180 min. (C) ESI-MS spectra (m/z 4300–5600) at t = 1, 20, and 180 min. The lower charge-state ions of the trimer (+7, +8), the tetramer (+9, +10, +11) and all the pentamer ions exchange rapidly at t = 1 min but display slow exchange at 180 min, indicating less-dynamic quaternary structures.
This suggests that the trimers and tetramers shift to a more compact structural conformation during aggregation, consistent with the increase in their stability. Although we cannot rule out the possibility that the more stable oligomers may have a greater propensity to ionize, the data presented here are consistent with the observation by the area under the curve that the oligomer population in solution increases during the time-course of fibril assembly (3). These stable oligomeric species remain during the time-course of fibril growth (≤180 min after initiation), decreasing in population as they assemble into larger species and eventually fibrils. These data indicate a WL fibril assembly pathway whereby β2m first populates a range of highly dynamic species ≤ hexamer. Under fibril growth conditions, these oligomers become less dynamic and higher-order oligomers are formed from these now stable lower-order oligomers. Once WL fibril formation has reached steady state, the concentration of monomer is ∼30% of its initial ion count (i.e., ∼0.12 mg mL−1) (Fig. 1), and population of trimer and tetramer is barely detectable, presumably because the concentration of the protein monomer has decreased to a concentration below which further assembly does not occur noticeably on this timescale. Indeed, when WL fibril formation under these conditions is carried out at 0.8 mg mL−1 initial β2m monomer concentration, the residual monomer at the steady state is again ∼0.12 mg mL−1.
To determine how changes in oligomer stability may relate to structural properties, the collision cross-sectional area of each oligomer populated 20 min into the assembly time-course was measured ((31,34,43) and see Fig. 5). The average cross-sectional area of a given globular protein can be estimated from its molecular mass if a spherical structure and average density (0.44 Da Å−3) are assumed (31,44). This analysis shows that the β2m oligomers populated en route to WL fibrils are significantly larger than predicted for globular proteins of the same molecular mass (Fig. 5). FT-IR spectra of WL fibrils suggest that monomers retain native-like structure within the fibrils (8,10), while x-ray fiber diffraction studies indicate a cross-β structure in which β-sheets run parallel to the fibril axis (39). Therefore, in silico models were constructed from different arrangements of native-like monomers organized with native-like β-strands oriented perpendicular to the long axis of the fibril, and the cross-sectional areas of these models were calculated (31,34).
Figure 5.
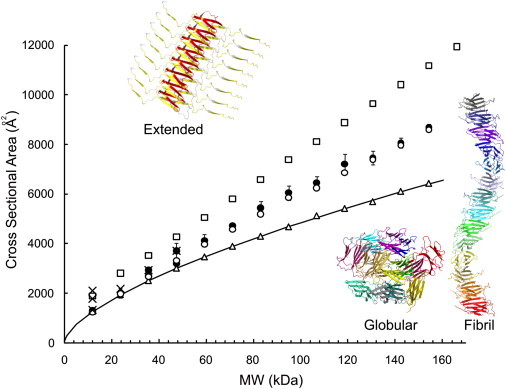
ESI-IMS-MS collision cross-sectional areas (Ω; Å2) for β2m oligomers detected during WL fibril assembly versus molecular mass (kDa). (Solid circles) β2m oligomers (dimer to 14-mer) populated during WL fibril assembly (100 mM ammonium acetate, pH 3.6). (Solid crosses) β2m oligomers (dimer to tetramer) populated during LS fibril assembly (100 mM ammonium formate, pH 2.5). (Error bars) One standard deviation in Ω between charge states of a protein. In silico models were constructed using the PyMOL Molecular Graphics System and their Ω-values calculated (34,40). In silico predicted Ω-values are shown for a linear assembly of nativelike β2m monomers stacked edge-to-edge (open circles), spherical oligomeric structures (open triangles), and an extended model with the N-terminal strand A and the C-terminal strand B of each protein subunit unstructured (open squares). (Solid line) The Ω expected for a protein of a given mass assuming a spherical assembly with a density of 0.44 Da Å−3 (31).
An elongated stacked assembly of β2m monomers based on edge-to-edge interactions of native monomers closely matches the observed data (Fig. 5). Molecular models of a more closely packed globular assembly give rise to cross-sectional areas smaller than the experimentally measured values (Fig. 5). Alternative models in which the N-terminal strand A (residues 1–16) and C-terminal strand G (residues 86–99) were unstructured and exposed to solvent, consistent with their weak hydrogen exchange protection and exposure to proteases (4,5,12), greatly overestimated the observed cross-sectional areas. Hence, the β2m oligomers detected during WL fibril formation are consistent with assembly via elongated polymers composed of subunits with native-like volume. This linear expansion of cross-sectional area with an increasing number of subunits strongly supports the notion that these oligomers are growing in two dimensions as observed in the case of Aβ1-40 oligomers (29), rather than being packed in a three-dimensional, spherical assembly.
Comparing oligomers detected during different β2m self-assembly pathways
Dimers, trimers, and tetramers are populated during both WL fibril assembly at pH 3.6 and the formation of LS fibrils at pH 2.5 (31). To determine whether these oligomers are similar in conformation, and if the two fibril assembly pathways have common, low-molecular mass oligomeric precursors, 14N-labeled β2m under LS-fibril forming solution conditions at pH 2.5 and 15N-labeled β2m under WL fibril-forming conditions at pH 3.6 were incubated separately for 20 min, allowing their respective oligomer populations to establish, before being mixed together in an equimolar ratio and analyzed directly by ESI-IMS-MS. After 20 min of assembly, the lower charge-state trimer and tetramer ions formed on the WL pathway (similar to the behavior shown by the trimer and tetramer ions formed on the LS pathway (31)) as well as the pentamer ions, are stable to subunit exchange. Thus, a direct comparison of the conformeric properties of the individual oligomers originating from each pathway, now analyzed under the same, final solution conditions, can be made (Fig. 6).
Figure 6.

ESI-IMS-MS collision cross-sectional areas (Ω; Å2) of individual charge-state ions of β2m trimer (triangles) and tetramer (circles) arising under long, straight (LS) fibril-forming conditions at pH 2.5 (open symbols) and WL fibril-forming conditions at pH 3.6 (solid symbols). 14N-labeled β2m (0.4 mg mL−1 based on monomer; pH 3.6) and 15N-labeled β2m (0.4 mg mL−1 based on monomer; pH 2.5) were incubated separately for 20 min before being mixed in an equimolar ratio and analyzed immediately. The trimer ions and the +10 to +13 tetramer ions are common to the oligomers detected on-pathway to both LS and WL fibrils. Only the tetramers detected on-pathway to WL fibrils populate the more compact +7 to +9 charge states.
The ESI-MS charge-state distribution of a protein is sensitive to its tertiary or quaternary structure, with higher charge-state ions representing more extended conformations and lower charge-state ions indicative of more compact states (45). The charge-state distributions for the tetramers formed at pH 3.6 and pH 2.5 were found to exhibit significant differences. The tetramer detected during WL fibril assembly populates the +7 to +13 charge states, while the tetramer detected during LS fibril assembly populates only the +10 to +13 charge-state ions, suggesting distinct conformers of protein assemblies of the same molecular mass (Fig. 6). This is substantiated by ESI-IMS-MS measurement of the cross-sectional area of each species: the cross-sectional area of the tetramer populated during WL fibril formation at pH 3.6 is 3424 Å2 (averaged over all charge states) while the tetramer observed during LS fibril formation at pH 2.5 is larger (3658 Å2) (31).
On closer data interrogation, it can be seen that the cross-sectional areas of the commonly populated charge states, i.e., the +10 to +13 ions, are indistinguishable under both sets of fibril assembly conditions (Fig. 6). By contrast, the average cross-sectional area of the +7 to +9 charge-state ions populated uniquely under WL growth conditions is 3112 Å2, implying these ions are ∼15% more compact than the higher charged ion series under these conditions. It is clear from Fig. 6 that the +10 to +13 and +7 to +9 charge-state ions are representative of two distinct tetrameric species, the latter of which is uniquely populated under WL fibril-forming conditions. By contrast, the trimer is also populated on both the WL and the LS fibril pathways; in both cases the +6 to +11 charge-state ions are detected and their measured cross-sectional areas are indistinguishable. Control experiments in which oligomers formed at pH 2.5 and pH 3.6 were diluted with buffer lacking protein at pH 3.6 and pH 2.5, respectively, showed no change in cross-sectional area or population over the time-course of the experiment. It was noted previously (31) that two forms of trimer are populated during the lag phase of LS fibril formation—one displaying rapid subunit exchange and the other displaying much slower exchange. The data presented here show that the +10 and +11 trimer charge-state ions are more dynamic than the +6 to +9 charge-state ions on both fibril pathways and thus indicate that the solution conditions not only determine the pathway of fibril formation, but also influence the conformational properties of the oligomeric intermediates in terms of their subunit dynamics, charge-state distribution, and cross-sectional areas.
Previous data have shown that self-assembly of β2m into WL or LS fibrils occurs via alternative, parallel pathways that depend on the initial solution conditions (2). The data presented in Fig. 6 suggest that population of compact tetramer ions (+7, +8, and +9 charge states) may provide unique access to the WL fibrillation pathway in which this tetramer self-assembles rapidly into higher-order oligomers. To determine whether population of compact β2m tetramers opens the route toward WL fibril formation, LS fibril assembly was initiated at pH 2.5, and then the ionic strength of this solution was increased further while maintaining the pH at 2.5: conditions that have been shown previously to result in WL fibril formation (2). This resulted in an immediate increase in thioflavin-T fluorescence intensity (Fig. 7), consistent with an instantaneous switch to the formation of WL fibrils that occurs without a lag-phase. The effect caused by this alteration in buffer concentration on the early oligomer population is significant (Fig. 7). As the salt concentration increases, ESI-IMS-MS experiments indicate that the intensities of the more compact, lower charge-state tetramer ions (+7 to +9) increase and higher-order oligomers become populated rapidly, as would be expected by a switch to the WL fibril assembly pathway.
Figure 7.
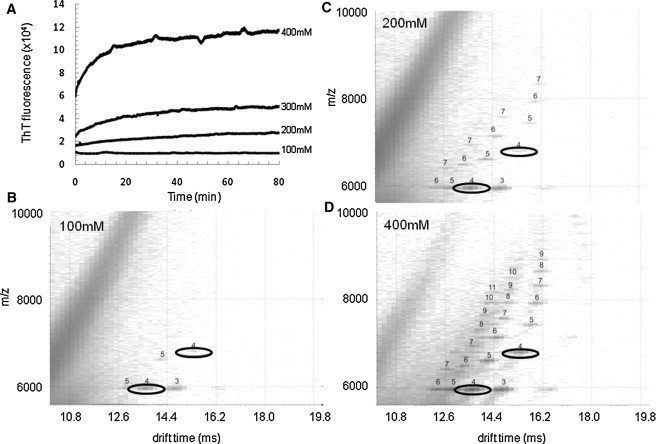
Compact tetramers divert assembly to WL fibrils at pH 2.5. The LS fibril-assembly of β2m was initiated (β2m 0.4 mg mL−1 based on monomer; pH 2.5) and then subjected to increasing buffer concentrations. (A) Thioflavin-T fluorescence analyses showing the spontaneous formation of WL fibrils with >300 mM ammonium formate buffer; (B–D) ESI-IMS-MS driftscope plots showing β2m oligomers at t = 1 min during fibril assembly with increasing ammonium formate buffer (B, 100 mM; C, 200 mM, and D, 400 mM). The numbers above each peak indicate the oligomer size. The +7 and +8 charge-state ions of the tetramer are highlighted to show their increase in their population with increasing ionic strength.
Discussion
Understanding the sequence of events leading to the formation of amyloid fibrils is key to the development of therapies against amyloidosis, especially as the oligomers populated en route to fibrils are thought to be responsible for the toxicity associated with several amyloid disorders (37). In vitro, β2m forms amyloid-like fibrils with different morphologies dependent on the buffer conditions (2). Here, ESI-IMS-MS has been used to probe the details of these assembly pathways, focusing on the properties of the oligomers formed during WL fibril assembly in terms of their population, conformation, and kinetic stability. It has been demonstrated previously that the species populated during the lag phase of LS fibril assembly at pH 2.5 range from monomer to tetramer, with no evidence for the significant population of larger species (3,31).
Furthermore, these oligomers have been shown to display a decrease in subunit exchange dynamics as the time of assembly increases, and to have cross-sectional areas consistent with elongated oligomers (31). How LS fibrils assemble from the tetramer remains unclear, although the nucleus has been modeled as a hexamer in size, rationalizing the lack of higher-order species detected during assembly (46). In sharp contrast, WL fibril formation from β2m at pH 3.6 occurs spontaneously without a lag phase (3). At this pH, β2m initially populates primarily a partially folded monomer (3,39,47) together with oligomers ranging from dimer to hexamer in size. Here we have shown that immediately after initiation of fibril assembly by increasing the ionic strength of the solution, higher order oligomers ≤9-mer are detected, all of which initially undergo rapid subunit exchange. Twenty minutes into fibril assembly, the rates of subunit exchange for the trimer, tetramer, and pentamer decrease, as indicated by 14N- and 15N-β2m exchange experiments, likely by conformational rearrangements into more stable architectures. This increase in stability is accompanied by the formation of oligomers >14-mer in size. Quantification of the relative populations of oligomers during fibril formation infers that the higher-order species arise by stepwise monomer addition to the lower-order oligomers, suggestive of an assembly mechanism involving sequential monomer addition.
Comparing the molecular mass of the oligomers versus their ESI-IMS-MS-derived cross-sectional areas indicates a linear relationship in the increase in size as the number of monomeric subunits increases (Fig. 5), suggesting a two-dimensional assembly as observed for Aβ1-40 assembly (29). Consistent with this, molecular modeling of linearly-stacked assemblies of oligomers provided estimated cross-sectional areas which agree closely with the measured values. The models were constructed from nativelike β2m monomers assuming that the monomeric subunits within WL fibrils retain a native-like β-sheet structure, as suggested by FT-IR (8).
Protein tetramers are populated on both the WL and LS pathways, although there are distinct differences in the conformational properties of these species (Fig. 6). Under WL fibril-forming conditions at early time points a wider charge-state distribution (+7 to +13) is detected by ESI-IMS-MS compared with that seen under the LS fibril-forming conditions (+10 to +13). The lower charge-state ions of the tetramer on the WL fibril pathway are associated with a significantly more compact conformer (cross-sectional area, Ω = 3112 Å2) than that associated with the +10 to +13 charge-state ions common to both pathways (Ω = 3658 Å2). We have noted previously that two forms of the trimer are present during LS fibril formation (31) and the same behavior is observed here for the trimer detected under WL fibril conditions. Under both assembly conditions, the 10+ and 11+ charge-state trimer ions display highly dynamic subunit exchange while the lower charge-state ions (+6 to +9) have a more compact structure and are more stable to subunit exchange, becoming more highly populated in comparison to the lower charge states during the course of aggregation into WL fibrils.
The results here suggest a relationship between the conformeric state of the protein monomer and the structure of the assembled fibrils. The β2m monomer populates three distinct conformeric families at pH <5: the acid unfolded state, the partially folded state, and a native-like state (33,47). Specifically, under LS fibril-forming conditions at pH 2.5, the acid unfolded state is highly populated, while under WL fibril-forming conditions at pH 3.6, the partially folded conformer predominates (33,47). Increasing the ionic strength of the protein solution at pH 2.5 to the higher ionic strength required for WL fibril assembly not only results in an increased population of the partially folded compared with the acid unfolded monomer, but also leads to the population of tetrameric +7 to +9 charge-state ions, indicating the population of a more compact, stable tetramer when the conditions are switched to those favoring the formation of WL fibrils. Together, the ESI-IMS-MS data presented here suggest that the population of the lower charged, more compact tetramers appears to govern the switch between the two fibril pathways.
The data presented highlight the power of ESI-IMS-MS to probe the details of two fibril assembly pathways from the same protein, indicating that the conformational structures of intermediates populated during self-aggregation play an important role in determining the final fibril morphology. Specifically in this case, the capability of characterizing transient individual oligomers and conformers from within a heterogeneous mixture indicates there is a fine balance between different conformeric arrangements of the tetramer dependent on the precise solution conditions employed. A compact tetrameric species unique to the WL fibril-forming pathway has been detected. Whether the presence of this species is related to the formation of higher-order oligomers (5-mer to >14-mer) unique to the WL fibril pathway remains to be discerned. However, the ability to control the morphology of fibrils and relate this to the assembly pathway in terms of the oligomer population is a key step forward to understanding fibril formation in vitro, and the role of individual oligomeric species in defining the assembly route.
Acknowledgments
We thank members of the Ashcroft and Radford groups for helpful discussions.
D.P.S. and L.A.W. were supported by the Biotechnology and Biological Sciences Research Council (grants No. BB/D010284/1 and No. BB/526502/1). The Synapt HDMS mass spectrometer was purchased with funds from the Biotechnology and Biological Sciences Research Council Research Equipment Initiative (grant No. BB/E012558/1), Micromass UK Ltd., and the University of Leeds.
Footnotes
David P. Smith's present address is Faculty of Health and Wellbeing, Sheffield Hallam University, Sheffield, UK.
Contributor Information
Sheena E. Radford, Email: s.e.radford@leeds.ac.uk.
Alison E. Ashcroft, Email: a.e.ashcroft@leeds.ac.uk.
References
- 1.Heegaard N.H. β2-microglobulin: from physiology to amyloidosis. Amyloid. 2009;16:151–173. doi: 10.1080/13506120903151775. [DOI] [PubMed] [Google Scholar]
- 2.Gosal W.S., Morten I.J., Radford S.E. Competing pathways determine fibril morphology in the self-assembly of β2-microglobulin into amyloid. J. Mol. Biol. 2005;351:850–864. doi: 10.1016/j.jmb.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 3.Smith A.M., Jahn T.R., Radford S.E. Direct observation of oligomeric species formed in the early stages of amyloid fibril formation using electrospray ionization mass spectrometry. J. Mol. Biol. 2006;364:9–19. doi: 10.1016/j.jmb.2006.08.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monti M., Principe S., Pucci P. Topological investigation of amyloid fibrils obtained from β2-microglobulin. Protein Sci. 2002;11:2362–2369. doi: 10.1110/ps.0206902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myers S.L., Thomson N.H., Ashcroft A.E. Investigating the structural properties of amyloid-like fibrils formed in vitro from β2-microglobulin using limited proteolysis and electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2006;20:1628–1636. doi: 10.1002/rcm.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gosal W.S., Myers S.L., Thomson N.H. Amyloid under the atomic force microscope. Protein Pept. Lett. 2006;13:261–270. doi: 10.2174/092986606775338498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jahn T.R., Tennent G.A., Radford S.E. A common β-sheet architecture underlies in vitro and in vivo β2-microglobulin amyloid fibrils. J. Biol. Chem. 2008;283:17279–17286. doi: 10.1074/jbc.M710351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fabian H., Gast K., Naumann D. Early stages of misfolding and association of β2-microglobulin: insights from infrared spectroscopy and dynamic light scattering. Biochemistry. 2008;47:6895–6906. doi: 10.1021/bi800279y. [DOI] [PubMed] [Google Scholar]
- 9.White H.E., Hodgkinson J.L., Saibil H.R. Globular tetramers of β2-microglobulin assemble into elaborate amyloid fibrils. J. Mol. Biol. 2009;389:48–57. doi: 10.1016/j.jmb.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ladner C.L., Chen M., Langen R. Stacked sets of parallel, in-register β-strands of β2-microglobulin in amyloid fibrils revealed by site-directed spin labeling and chemical labeling. J. Biol. Chem. 2010;285:17137–17147. doi: 10.1074/jbc.M110.117234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Platt G.W., Xue W.F., Radford S.E. Probing dynamics within amyloid fibrils using a novel capping method. Angew. Chem. Int. Ed. Engl. 2009;48:5705–5707. doi: 10.1002/anie.200901343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoshino M., Katou H., Goto Y. Mapping the core of the β2-microglobulin core by H/D exchange. Nat. Struct. Biol. 2002;9:332–336. doi: 10.1038/nsb792. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi K., Katou H., Goto Y. Core and heterogeneity of β2-microglobulin amyloid fibrils as revealed by H/D exchange. J. Mol. Biol. 2004;338:559–571. doi: 10.1016/j.jmb.2004.02.067. [DOI] [PubMed] [Google Scholar]
- 14.Debelouchina G.T., Platt G.W., Griffin R.G. Intermolecular alignment in β2-microglobulin amyloid fibrils. J. Am. Chem. Soc. 2010;132:17077–17079. doi: 10.1021/ja107987f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skora L., Becker S., Zweckstetter M. Molten globule precursor states are conformationally correlated to amyloid fibrils of human β2-microglobulin. J. Am. Chem. Soc. 2010;132:9223–9225. doi: 10.1021/ja100453e. [DOI] [PubMed] [Google Scholar]
- 16.Skora L., Becker S., Zweckstetter M. Characterization of amyloid fibrils of human β2-microglobulin by high-resolution magic-angle spinning NMR. ChemBioChem. 2010;11:1829–1832. doi: 10.1002/cbic.201000336. [DOI] [PubMed] [Google Scholar]
- 17.Baskakov I.V., Legname G., Cohen F.E. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem. 2002;277:21140–21148. doi: 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
- 18.Kumar S., Udgaonkar J.B. Conformational conversion may precede or follow aggregate elongation on alternative pathways of amyloid protofibril formation. J. Mol. Biol. 2009;385:1266–1276. doi: 10.1016/j.jmb.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 19.Kad N.M., Myers S.L., Thomson N.H. Hierarchical assembly of β2-microglobulin amyloid in vitro revealed by atomic force microscopy. J. Mol. Biol. 2003;330:785–797. doi: 10.1016/s0022-2836(03)00583-7. [DOI] [PubMed] [Google Scholar]
- 20.Carulla N., Zhou M., Dobson C.M. Experimental characterization of disordered and ordered aggregates populated during the process of amyloid fibril formation. Proc. Natl. Acad. Sci. USA. 2009;106:7828–7833. doi: 10.1073/pnas.0812227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed M., Davis J., Smith S.O. Structural conversion of neurotoxic amyloid-β (1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chimon S., Shaibat M.A., Ishii Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid. Nat. Struct. Mol. Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 23.Orte A., Birkett N.R., Klenerman D. Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2008;105:14424–14429. doi: 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J.W., Breydo L., Glabe C. Fibrillar oligomers nucleate the oligomerization of monomeric amyloid β but do not seed fibril formation. J. Biol. Chem. 2010;285:6071–6079. doi: 10.1074/jbc.M109.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clemmer D.E., Hudgins R.R., Jarrold M.F. Naked protein conformations—cytochrome-c in the gas-phase. J. Am. Chem. Soc. 1995;117:10141–10142. [Google Scholar]
- 26.Giles K., Pringle S.D., Bateman R.H. Applications of a traveling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun. Mass Spectrom. 2004;18:2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein S.L., Dupuis N.F., Bowers M.T. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the etiology of Alzheimer's disease. Nat. Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jablonowska A., Bakun M., Dadlez M. Alzheimer's disease Aβ peptide fragment 10–30 forms a spectrum of metastable oligomers with marked preference for N to N and C to C monomer termini proximity. J. Mol. Biol. 2004;344:1037–1049. doi: 10.1016/j.jmb.2004.09.083. [DOI] [PubMed] [Google Scholar]
- 29.Kłoniecki M., Jabłonowska A., Dadlez M. Ion mobility separation coupled with MS detects two structural states of Alzheimer's disease Aβ1–40 peptide oligomers. J. Mol. Biol. 2011;407:110–124. doi: 10.1016/j.jmb.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Nettleton E.J., Tito P., Robinson C.V. Characterization of the oligomeric states of insulin in self-assembly and amyloid fibril formation by mass spectrometry. Biophys. J. 2000;79:1053–1065. doi: 10.1016/S0006-3495(00)76359-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith D.P., Radford S.E., Ashcroft A.E. Elongated oligomers in β2-microglobulin amyloid assembly revealed by ion mobility spectrometry-mass spectrometry. Proc. Natl. Acad. Sci. USA. 2010;107:6794–6798. doi: 10.1073/pnas.0913046107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reference deleted in proof.
- 33.Smith D.P., Giles K., Ashcroft A.E. Monitoring copopulated conformational states during protein folding events using electrospray ionization-ion mobility spectrometry-mass spectrometry. J. Am. Soc. Mass Spectrom. 2007;18:2180–2190. doi: 10.1016/j.jasms.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith D.P., Knapman T.W., Ashcroft A.E. Deciphering drift time measurements from travelling wave ion mobility spectrometry-mass spectrometry studies. Eur. J. Mass Spectrom. (Chichester, Eng.) 2009;15:113–130. doi: 10.1255/ejms.947. [DOI] [PubMed] [Google Scholar]
- 35.Glabe C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh D.M., Klyubin I., Selkoe D.J. Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 37.Aguzzi A., O'Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 38.McParland V.J., Kalverda A.P., Radford S.E. Structural properties of an amyloid precursor of β2-microglobulin. Nat. Struct. Biol. 2002;9:326–331. doi: 10.1038/nsb791. [DOI] [PubMed] [Google Scholar]
- 39.McParland V.J., Kad N.M., Radford S.E. Partially unfolded states of β2-microglobulin and amyloid formation in vitro. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- 40.Knapman T.W., Berryman J.T., Ashcroft A.E. Considerations in experimental and theoretical collision cross-section measurements of small molecules using traveling wave ion mobility spectrometry-mass spectrometry. Int. J. Mass Spectrom. 2009;298:17–23. [Google Scholar]
- 41.Knapman T.W., Morton V.L., Ashcroft A.E. Determining the topology of virus assembly intermediates using ion mobility spectrometry-mass spectrometry. Rapid Commun. Mass Spectrom. 2010;24:3033–3042. doi: 10.1002/rcm.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trinh C.H., Smith D.P., Radford S.E. Crystal structure of monomeric human β2-microglobulin reveals clues to its amyloidogenic properties. Proc. Natl. Acad. Sci. USA. 2002;99:9771–9776. doi: 10.1073/pnas.152337399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruotolo B.T., Benesch J.L., Robinson C.V. Ion mobility-mass spectrometry analysis of large protein complexes. Nat. Protoc. 2008;3:1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 44.Benesch J.L., Ruotolo B.T., Robinson C.V. Protein complexes in the gas phase: technology for structural genomics and proteomics. Chem. Rev. 2007;107:3544–3567. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- 45.Heck A.J. Native mass spectrometry: a bridge between interatomic and structural biology. Nat. Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 46.Xue W.F., Homans S.W., Radford S.E. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. USA. 2008;105:8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borysik A.J., Radford S.E., Ashcroft A.E. Co-populated conformational ensembles of β2-microglobulin uncovered quantitatively by electrospray ionization mass spectrometry. J. Biol. Chem. 2004;279:27069–27077. doi: 10.1074/jbc.M401472200. [DOI] [PubMed] [Google Scholar]