

Abstract
Lactophoricin (LPcin-I) is an antimicrobial, amphiphatic, cationic peptide with 23-amino acid residues isolated from bovine milk. Its analogous peptide, LPcin-II, lacks six N-terminal amino acids compared to LPcin-I. Interestingly, LPcin-II does not display any antimicrobial activity, whereas LPcin-I inhibits the growth of both Gram-negative and Gram-positive bacteria without exhibiting any hemolytic activity. Uniformly 15N-labeled LPcin peptides were prepared by the recombinant expression of fusion proteins in Escherichia coli, and their properties were characterized by electrospray ionization mass spectrometry, circular dichroism spectroscopy, and antimicrobial activity tests. To understand the structure-activity relationship of these two peptides, they were studied in model membrane environments by a combination of solution and solid-state NMR spectroscopy. We determined the tertiary structure of LPcin-I and LPcin-II in the presence of dodecylphosphorylcholine micelles by solution NMR spectroscopy. Magnetically aligned unflipped bicelle samples were used to investigate the structure and topology of LPcin-I and LPcin-II by solid-state NMR spectroscopy.
Introduction
Antimicrobial peptides are essential for the innate immune system of all life forms. Their broad spectrum antimicrobial activities make antimicrobial peptides potentially useful for pharmacological applications and commercial exploitation (1–3). These peptides are believed to kill the target cells by disrupting the ordered structures of cell membranes via barrel-stave, carpet, or toroidal mechanisms (4). Understanding the structure-activity relationship of these peptides is essential for developing them into novel therapeutics that can substitute for traditional antibiotics. Unfortunately, these membrane-targeting peptides are resistant to the growth of crystals in membranes or detergents, rendering x-ray diffraction inapplicable. The membrane-bound structures of antimicrobial peptides are primarily elucidated in membrane-mimetic models, such as organic solvents and lipid micelles, by solution NMR techniques (5). Organic solvents are very convenient to manipulate membrane proteins but differ greatly from environments based on biological membranes. Detergent micelles such as sodium dodecyl sulfate and dodecylphosphorylcholine (DPC) are able to better mimic membranes (6–9). Lipid bilayers or large bicelles (q >2.5) are regarded as the best membrane-mimetic model for the structural analysis of these peptides. However, they are not convenient for solution NMR, although transfer nuclear Overhauser effect (NOE) experiments have occasionally been performed to deduce valuable structural information (7,11). Solid-state NMR of aligned samples is well suited for determining the in-plane or transmembrane orientation of antimicrobial peptides. Therefore, a refined peptide structure from lipid micelles should be more transportable to lipid bilayers or large bicelles for orientational characterization by solid-state NMR spectroscopy (12,13).
LPcin-I is a cationic amphipathic peptide with 23-amino acid residues, and corresponds to the carboxy terminal 113–135 region of component-3 of proteose peptone from bovine milk. LPcin-I inhibits the growth of both Gram-negative and Gram-positive bacteria, but has no hemolytic activity at concentrations of <200 μM (14). In contrast to LPcin-I, it has been reported that LPcin-II, which corresponds to the 119–135 region of PP3, has no antibacterial activity, which is interesting because LPcin-I and LPcin-II have similar charge ratios and identical hydrophobic/hydrophilic sectors based on the helical wheel pattern (15–17). Furthermore, both peptides show cationic properties and interact with phospholipids. Nevertheless, only LPcin-I is incorporated into planar lipidic bilayers, in which it forms voltage-dependent channels (17).
To elucidate the structure-activity relationship of the peptides, we examined the tertiary structures of LPcin-I and LPcin-II in membrane-mimicking environments. Uniformly 15N-labeled peptides were prepared as described previously (18). Electrospray ionization mass spectrometry (ESI-MS, Applied Biosystems, Carlsbad, CA) was used to confirm the identity and purity of the recombinant LPcin-I and LPcin-II peptides prepared for NMR experiments. To examine the antimicrobial activities of the recombinant peptides, their antimicrobial activity was screened against five microorganisms using the agar hole diffusion method. The interactions between these peptides and the membrane were also studied by circular dichroism (CD, Jasco, Easton, MD), solution, and solid-state NMR spectroscopy. The tertiary structures of LPcin-I and LPcin II in the presence of DPC micelles were determined by solution NMR spectroscopy. The structure and topology of LPcin-I and LPcin-II in magnetically aligned unflipped bicelles were investigated by solid-state NMR spectroscopy. The structural differences between LPcin-I and LPcin-II peptides were compared and demonstrated in each case.
Materials and Methods
Expression and purification
The coding sequence for the polypeptides corresponding to LPcin-I and LPcin-II was synthesized and cloned into the pET31b(+) vector (Novagen, Darmstadt, Germany), as described previously (18). For the uniform 15N enrichment of the peptides, cells were grown in minimal medium, which included (15NH4)2SO4 (Sigma-Aldrich, St. Louis, MO) as the sole nitrogen source. The fusion proteins were purified by Ni-nitrilotriacetic acid affinity chromatography under denaturing conditions, and the target peptides were cleaved from their ketosteroid isomerase fusion protein with CNBr. The final purification of LPcin-I and LPcin-II was achieved by preparative reversed-phase high performance liquid chromatography.
Mass spectrometry
The mass spectrometry analysis of LPcin-I and LPcin-II was performed with a Q-TOF instrument (QSTAR Elite, Applied Biosystems) with an electrospray nano-ion source. The purified peptides were dissolved in 2% acetonitrile/98% water.
Antimicrobial activity test
The antimicrobial activity tests for the recombinant peptides, LPcin-I and LPcin-II, against the microorganisms were carried out using Mueller-Hinton agar (Difco) (BD, Sparks, MD). Five microorganisms belonging to two Gram-positive (Listeria innocua MC2 KCTC (Daejon, Korea) 3658 and Staphylococcus aureus ATCC 6538) and three Gram-negative (Pseudomonas aeruginosa ATCC27853, Salmonella ATCC (Manassas, VA) 19430, Escherichia coli KCTC 1682) bacterial species were tested for their susceptibility to the recombinant peptide based on the agar hole diffusion test. The inoculum was prepared from an overnight culture of bacteria in 5 mL of tryptic soy broth at 37°C. The turbidity of the suspension was adjusted with a spectrophotometer at 600 nm to obtain a final concentration matching that of a 0.5 McFarland standard. 20 mL of Mueller-Hinton agar was put in an Ø90 mm cell culture plate. The agar plates were inoculated with 40 μL of bacterial suspension using a sterile spreader. The inoculated agar plate was allowed to dry for 30 min at room temperature. The antimicrobial peptides were dissolved in sterilized water at a concentration of 6 μg/μL, and 30 μL of the solution was dropped onto the surface of the agar plate. After prediffusion at room temperature for 1 h, the plates were incubated at 37°C for 18 h. The susceptibility of the bacterial strains to the recombinant antimicrobial peptides was determined by examining the size and clearance of the inhibitory zone. All of the measurements were done in three replicates.
Circular dichroism
The CD experiments were carried out using a Jasco J715 spectropolarimeter and 1 mm path-length quartz cuvettes. The spectra were recorded between 195 and 250 nm with a data pitch of 0.2 nm, a bandwidth of 1 nm, a scanning speed of 50 nm/min, and a response time of 0.25 s. 150 μM peptide samples were prepared in water and in water containing 20–100 mM DPC at pH 4.0. All of the CD spectra were collected at 25°C. The data shown were averaged from five individual spectra. The measurement of the buffer without the peptide was subtracted to correct the baseline of the final spectra. The CD signal Ψ (mdeg) was converted to the mean residue molar ellipticity [θ], in degrees cm2 dmol−1.
NMR sample preparation
The two kinds of samples used in the solution NMR experiments were prepared in the same manner as in the CD experiments. The recombinant peptides of the uniformly 15N-labeled LPcin-I and LPcin-II were dissolved at a concentration of 1 mM in 90% H2O and 10% D2O at pH 4.0. The micelle samples were then prepared by dissolving the uniformly 15N-labeled peptide at a concentration of 1 mM in 150 mM DPC-d38 containing 90% H2O and 10% D2O at pH 4.0. The bicelle samples used in the solid-state NMR experiments were prepared by dissolving 3 mg of the uniformly 15N-labeled peptide in a long-chain and short-chain lipid mixture. The dried peptide was mixed with 9.2 mg of 1,2-di-O-hexyl-sn-glycero-3-phosphocholine (6-O-PC) (Avanti) in 100 μL of water with vortexing. The dissolved protein/lipid mixture was added to 37.4 mg of 1,2-Di-Otetradecyl-sn-glycero-3-phosphocholine (14-O-PC) (Avanti) and 9.5 mg of dimyristoylphosphatidylglycerol (Avanti) in 100 μL of water to make a bicelle sample that was 28% lipid (w/v) and had a q-value (long chain/short chain) of 3.4. The bicelles were formed by putting the sample through several heating/cooling/vortexing cycles. The sample was transferred to a flat bottomed NMR tube (4 mmØ × 25 mm, Wilmad) and sealed with parafilm and Teflon tape.
Solution NMR spectroscopy
The solution NMR experiments were carried out at 308 K on a Varian VNMRS 900 MHz spectrometer (Agilent Technologies, Santa Clara, CA) equipped with a z-gradient cryogenic probe. Two-dimensional (2D) total correlation spectroscopy (TOCSY), nuclear Overhauser enhancement spectroscopy (NOESY), and double-quantum filtered correlation spectroscopy (DQF-COSY) were conducted to identify the spin systems and to assign all of the resonances from LPcin-I and LPcin-II. The mixing times for TOCSY were 30 ms and those for NOESY were 80 ms for both LPcins. The solvent suppression for these spectra was accomplished by the presaturation of the H2O resonance with continuous irradiation. All of the spectra were acquired with time domains of 1024 (F2) × 512 (F1) complex points and with spectral widths of 11,261 Hz for both dimensions. By using 15N-labeled peptides, 2D 1H-15N heteronuclear single quantum correlation (HSQC) spectra, and three-dimensional (3D) 15N-edited NOESY-HSQC spectra with a mixing time of 80 ms were also acquired to confirm the resonance assignments and to generate the distance restraints. All of the spectra were processed using NMRPipe/NMRDraw software (19) and analyzed by NMRView software (20).
Structural calculation
The NOE crosspeaks from the 2D NOESY spectra and 3D 15N-edited NOESY-HSQC spectra of LPcin-I and LPcin-II were assigned by a semiautomated method using the program ARIA 2.0 (21). The NOE-based distance restraints were derived based on the peak volume. The backbone dihedral angle restraints were obtained from the 3JHNCα coupling constants measured in the DQF-COSY and HMQC-J spectra of each peptide. The peptide structures were determined by using the programs, CNS 1.2 (22) and ARIA 2.0 (21). Hydrogen bond restraints were incorporated into the final calculation of the peptide structures based on the secondary structure of each peptide. In the final ARIA run, 100 structures each for LPcin-I and LPcin-II were calculated and the 20 lowest energy structures among them were selected for the statistical analysis using PROCHECK-NMR 3.4 (23).
Solid-state NMR spectroscopy
The solid-state NMR experiments were performed on a CMX Infinity plus-700 spectrometer (Jeol, Tokyo, Japan) with a three channel high-power amplifier. A Chemagnetics 4 mm T3 HXY CP/MAS Probe was double-tuned to the resonance frequencies of 1H (699.7 MHz) and 15N (70.9 MHz) in a wide-bore magnet. The bicelle samples were placed in a round coil and the static 15N NMR spectra of LPcin-I and LPcin-II in the magnetically aligned phospholipids bicelles were obtained at 35°C using a Chemagnetics variable-temperature stack and controller. The one-dimensional (1D) 15N chemical shift spectra were obtained using single-contact spin-lock cross-polarization (24) with a contact time of 1 ms and recycle delay of 5 s. The data were acquired for 5 ms with a TPPM 1H decoupling power of 50 kHz (25). 2D separated local field spectra were obtained using a SAMMY pulse sequence (26). The SAMMY spectra provide a measure of the 1H-15N heteronuclear dipolar coupling in one dimension and the 15N chemical shift in the other dimension. The chemical shift frequencies are referenced relative to that of external solid, powdered 15N ammonium sulfate, which is defined as 26.8 ppm.
Results and Discussion
ESI-MS spectrometry of LPcin-I and LPcin-II
Recombinant LPcin-I and LPcin-II peptides were prepared as described previously (18). The sequences of these two peptides are shown in Fig. 1, A and B, respectively. The underlined residues are from native bovine lactophoricin and six additional residues GGKKKK were added to enhance the solubility for the isolation, purification, and antibacterial activity test. The final residue Mhsl is homoserine lactone resulting from CNBr cleavage, which was determined from the theoretical molecular weight of the recombinant peptides compared with the experimental molecular weight obtained from the mass spectrum. The lyophilized peptides were dissolved in 2% acetonitrile and used for ESI-MS for further identification. The measured molecular masses of LPcin-I (Fig. 1 A) and LPcin-II (Fig. 1 B) obtained by HPLC purification were 3391 and 2719 Da, respectively, which matches well with their theoretical molecular weights.
Figure 1.
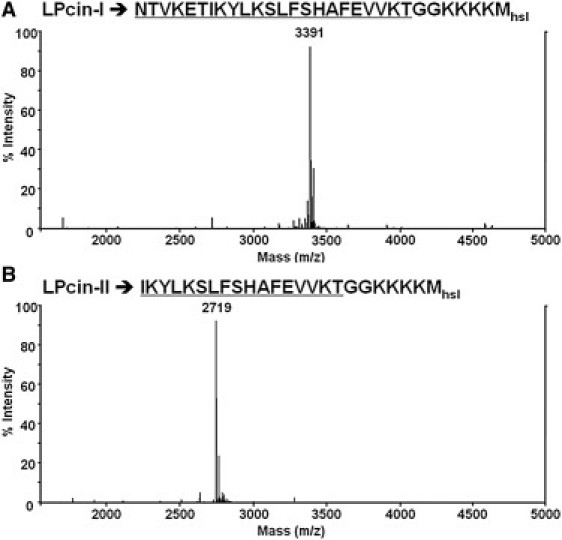
ESI-MS spectra of uniformly 15N-labeled LPcin-I (A) and LPcin-II (B) obtained by HPLC purification. The measured molecular masses of LPcin-I and LPcin-II were 3391 and 2719 Da, respectively, which matches well with their theoretical molecular weights.
Antimicrobial activity tests of recombinant LPcin-I and LPcin-II
We examined the antimicrobial activities of the recombinant peptides, LPcin-I and LPcin-II, against a representative set of bacterial strains including two Gram-positive (Listeria, S. aureus) and three Gram-negative (P. aeruginosa, Salmonella, E. coli) strains. The antimicrobial activity was screened using the agar hole diffusion test. The appearance of clear zones in the agar overlay was considered to represent strong inhibition, whereas a clearing but cloudy zone was considered to represent weak inhibition and no zone to no inhibition. LPcin-I peptide showed a strong inhibition zone against four of the tested bacteria (Listeria, P. aeruginosa, Salmonella, and E. coli) and a weak inhibition zone against one tested bacteria (S. aureus), as shown in Fig. 2 A. On the other hand, LPcin-II peptide showed a weak inhibition zone against only one of the tested bacteria (Salmonella) and no inhibition against the other tested bacteria, as shown in Fig. 2 B.
Figure 2.

Results of the antimicrobial activity tests for the recombinant LPcin-I (A) and LPcin-II (B) against five microorganisms belonging to two Gram-positive (Listeria innocua MC2 KCTC 3658 and Staphylococcus aureus ATCC 6538) and three Gram-negative (Pseudomonas aeruginosa ATCC27853, Salmonella ATCC 19430, Escherichia coli KCTC 1682) bacterial species.
CD measurements of LPcin-I and LPcin-II
We investigated the secondary structures of LPcin-I and LPcin-II in aqueous and membrane-like environments by analyzing their CD spectra. In Fig. 3, A and C, both peptides have random structures in aqueous solution, but exhibit conformational changes and form α-helical conformations in 100 mM DPC micelles, as is evident in the double minima absorption observed at 208 and 222 nm. These helical secondary structures are very stable and similar in 20, 40, 60, 80, and 100 mM DPC micelle environments (Fig. 3, B and D).
Figure 3.
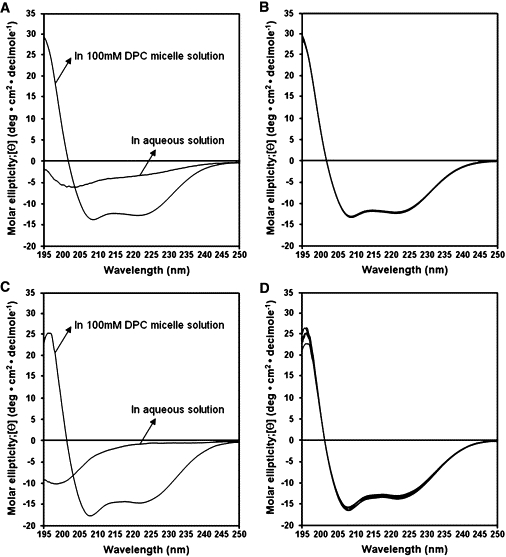
CD spectra of LPcin-I (A) and LPcin-II (C) in water solution and in 100 mM DPC micelles are shown on the left hand side. The CD spectra of LPcin-I (B) and LPcin-II (D) in 20, 40, 60, 80, and 100 mM DPC micelles are shown on the right hand side.
Solution NMR studies of LPcin-I and LPcin-II
The initial solution NMR studies of LPcin-I and LPcin-II provided valuable information about their structure and topology. To compare the structural differences between the two peptides, their 1H-15N HSQC spectra were recorded in aqueous and membrane-like environments. Each amide site in the uniformly 15N-labeled peptides contributes a single correlation resonance to the 2D 1H-15N HSQC spectra in Fig. 4. Each resonance is characterized by 1H and 15N chemical shift frequencies that reflect the local environment of the site in the peptides. As expected, there are fewer resonances in the spectrum obtained from LPcin-II than that obtained from LPcin-I. In Fig. 4 A, many of the resonances that are in the spectrum of LPcin-II (gray) can be matched to those found in the spectrum of LPcin-I (black). This suggests that the residues of LPcin-I and LPcin-II have essentially identical local structures in an aqueous environment. The backbone amide resonances from both peptides in an aqueous environment are observed with the narrow dispersion of the amide 1H chemical shifts in the 1H-15N HSQC and 1H-1H NOESY spectra, and they display no HN-HN NOE crosspeaks. This suggests that they are not structured in an aqueous environment. These results are consistent with the CD data in Fig. 3, A and C. Fig. 4 B shows a comparison of the 1H-15N HSQC spectra between LPcin-I (black) and LPcin-II (gray) in the 150 mM DPC micelles. The excellent dispersion of the amide 1H signals between 7.5 and 9.0 ppm indicates that the peptides are properly folded in a membrane-like environment. There are significant differences in their chemical shifts compared to those observed in an aqueous environment. To obtain a better understanding of the structure-antimicrobial activity relationship between the LPcin-I and LPcin-II peptides, their 3D structures were determined by solution NMR spectroscopy under micelle conditions, and the structure and topology of the helical peptides were also investigated by solid-state NMR spectroscopy under aligned large bicelle conditions, as discussed below.
Figure 4.
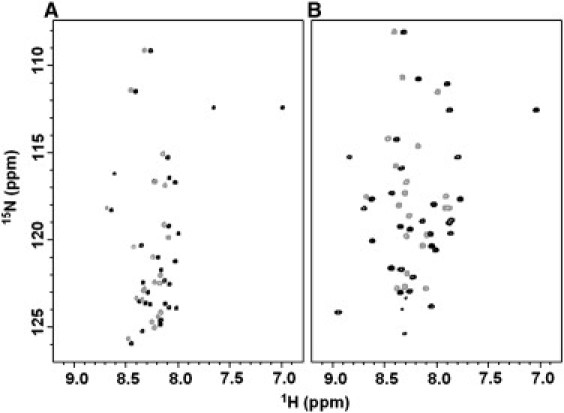
1H-15N HSQC spectra of uniformly 15N-labeled LPcin-I (black) and LPcin-II (gray) in water solution (A) and in 150 mM DPC micelles (B) are shown overlapped.
Resonance assignments of LPcin-I and LPcin-II
Most of the 1H and 15N resonances of LPcin-I and LPcin-II were unambiguously assigned in the analysis of the 2D NOESY, TOCSY, DQF-COSY, 1H-15N HSQC, and 3D 15N-edited NOESY-HSQC spectra of the peptides. The assigned resonances are summarized in Table 1 and the 1H-15N HSQC spectra of LPcin-I and LPcin-II are shown in Fig. 5. The C-terminal regions of LPcin-I (Ser-15-Met-30) and LPcin-II (Ser-9-Met-24) showed significant spectral overlap in the 1H-15N HSQC spectra, whereas the N-terminal 8 amino acids of LPcin-II revealed a significantly different chemical shift compared to the corresponding region of LPcin-I. Not only the backbone 1H and 15N resonances, but also the whole 1H resonances, showed the same spectral tendency. Because LPcin-II lacks 6 amino acids compared to LPcin-I, the chemical shifts differed only in the N-terminal part of the peptides. However, the overall structures of LPcin-I and LPcin-II resembled each other very well and will be discussed later. The resonances of the solubilization-tags (GGKKKK) from both peptides could not be assigned, because the signals from these residues were not separated in the 2D TOCSY and NOESY spectra, except for two glycines. At least 3 out of 4 lysines in the tag presented multiple 1H-15N correlation peaks in the lower center of the HSQC spectra, which suggests that these regions are very dynamic and under a slow exchange regime on the 900 MHz frequency timescale.
Table 1.
Summary of the resonance assignments of LPcin-I and LPcin-II
| LPcin-I |
LPcin-II |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HN | N | αH | βH | Others | HN | N | αH | βH | Others | ||||
| 1 | Asn | 3.04 | γNH2 7.87, 7.03 | ||||||||||
| 2 | Thr | 8.81 | 115.28 | 4.47 | 4.47 | γCH3 1.28 | |||||||
| 3 | Val | 8.94 | 124.15 | 3.67 | 2.22 | γCH3:1.12, 0.98 | |||||||
| 4 | Lys | 8.35 | 119.23 | 3.92 | 1.90, 1.84 | γCH2:1.57; δCH2:1.73; εCH2:2.98 | |||||||
| 5 | Glu | 8.01 | 117.99 | 4.09 | 2.14 | γCH2:2.42 | |||||||
| 6 | Thr | 8.24 | 119.21 | 4.00 | 4.38 | γCH3:1.25 | |||||||
| 7 | Ile | 8.41 | 121.53 | 3.65 | 2.03 | γCH2:1.81 1.19; γCH3:0.96; δCH3:0.86 | 1 | Ile | 3.85 | 1.96 | γCH2:1.63, 1.28; γCH3:1.01; δCH3:0.95 | ||
| 8 | Lys | 7.85 | 119.63 | 3.92 | 1.96 | γCH2:1.61, 1.43; δCH2:1.71; εCH2:2.96 | 2 | Lys | 4.16 | 1.63, 1.53 | γCH2:1.31, 1.15; δCH2:1.58; εCH2:2.90 | ||
| 9 | Tyr | 8.01 | 120.57 | 4.34 | 3.29, 3.23 | 2,6H:7.08; 3,5H:6.77 | 3 | Tyr | 8.24 | 117.08 | 4.41 | 3.18, 3.04 | 2,6H:7.08; 3,5H:6.83 |
| 10 | Leu | 8.61 | 120.03 | 3.88 | 2.02, 1.39 | γCH:2.09; δCH3:0.93 | 4 | Leu | 8.21 | 119.67 | 4.02 | 1.71, 1.59 | γCH:1.70; δCH3:0.98, 0.92 |
| 11 | Lys | 8.69 | 118.19 | 3.90 | 2.00, 1.94 | γCH2:1.35; δCH2:1.67; εCH2:2.88 | 5 | Lys | 8.29 | 117.94 | 3.98 | 1.91 | γCH2:1.52, 1.45; δCH2:1.76, 1.74; εCH2:2.96 |
| 12 | Ser | 7.79 | 115.26 | 4.01 | 4.28 | 6 | Ser | 8.10 | 114.67 | 4.33 | 3.98, 3.95 | ||
| 13 | Leu | 8.05 | 123.83 | 4.05 | 1.59, 1.45 | γCH:1.40; δCH3:0.78, 0.74 | 7 | Leu | 8.03 | 122.85 | 4.10 | 1.71, 1.60 | γCH:1.59; δCH3:0.88, 0.80 |
| 14 | Phe | 8.43 | 117.32 | 4.22 | 3.13 | 2,6H:7.20; 3,5H:7.11; 4H:7.03 | 8 | Phe | 8.23 | 116.75 | 4.27 | 3.18, 3.15 | 2,6H:7.24; 3,5H:7.17; 4H:7.08 |
| 15 | Ser | 8.36 | 114.23 | 4.19 | 4.07, 4.03 | 9 | Ser | 8.40 | 114.20 | 4.21 | 4.04, 4.01 | ||
| 16 | His | 8.13 | 118.86 | 4.58 | 3.37 | 2H:7.26 | 10 | His | 8.17 | 118.65 | 4.57 | 3.32 | 2H:7.26 |
| 17 | Ala | 8.34 | 121.65 | 3.95 | 1.42 | 11 | Ala | 8.34 | 121.64 | 3.97 | 1.43 | ||
| 18 | Phe | 8.32 | 115.87 | 4.18 | 3.25, 3.03 | 2,6H:7.23; 3,5H:7.27 | 12 | Phe | 8.32 | 115.88 | 4.20 | 3.24, 3.04 | 2,6H:7.24; 3,5H:7.08, 4H:7.28 |
| 19 | Glu | 7.77 | 117.42 | 4.03 | 2.21, 2.19 | γCH2:2.49 | 13 | Glu | 7.78 | 117.31 | 4.03 | 2.21, 2.17 | γCH2:2.46, 2.44 |
| 20 | Val | 7.85 | 118.86 | 3.77 | 2.16 | γCH3:0.90 | 14 | Val | 7.82 | 118.40 | 3.80 | 2.19 | γCH3:0.97, 0.92 |
| 21 | Val | 7.86 | 119.10 | 3.80 | 2.11 | γCH3:1.01, 0.92 | 15 | Val | 7.79 | 118.36 | 3.81 | 2.13 | γCH3:1.01, 0.92 |
| 22 | Lys | 8.05 | 119.80 | 4.08 | 1.70, 1.64 | γCH2:1.29; δCH2:1.55 | 16 | Lys | 8.01 | 119.73 | 4.09 | 1.70, 1.94 | γCH2:1.29; δCH2:1.56 |
| 23 | Thr | 7.87 | 111.15 | 4.30 | 4.34 | γCH3:1.25 | 17 | Thr | 7.88 | 111.44 | 4.33 | 4.33 | γCH3:1.25 |
| 24 | Gly | 8.17 | 110.73 | 3.98 | 18 | Gly | 8.22 | 110.76 | 3.97 | ||||
| 25 | Gly | 8.30 | 108.11 | 3.94 | 19 | Gly | 8.32 | 108.05 | 3.94 | ||||
| 26 | Lys | 8.06 | 120.38 | 4.29 | 1.82, 1.75 | γCH2:1.45 | 20 | Lys | 8.04 | 120.30 | |||
| 27 | Lys | 21 | Lys | ||||||||||
| 28 | Lys | 22 | Lys | ||||||||||
| 29 | Lys | 23 | Lys | ||||||||||
| 30 | Met | 8.62 | 117.68 | 4.64 | 24 | Met | 8.61 | 117.61 | |||||
Figure 5.
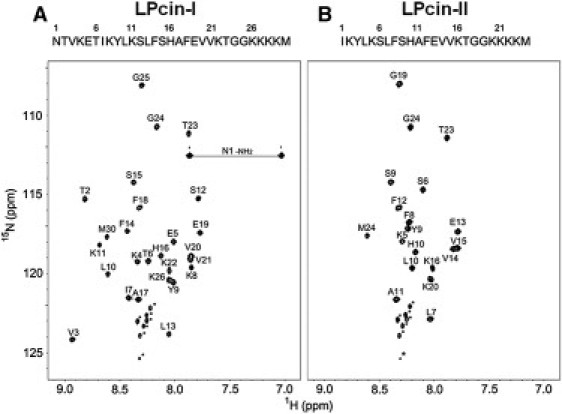
1H-15N HSQC spectra of LPcin-I (A) and LPcin-II (B). On top of each spectrum, the primary sequences of LPcin-I and LPcin-II were indicated by the one-letter codes of the amino acids. The C-terminal sequence of GGKKKK was added to express and solubilize each peptide and the last methionine was introduced for the cleavage by CNBr. The assignments of the backbone 1H and 15N resonances were also indicated on the HSQC spectra of LPcin-I and LPcin-II. The asterisked crosspeaks in the lower center of each spectrum could not be assigned, but were attributed to the C-terminal lysine residues, because of the slow conformational exchange of these residues (see text).
Overall structures of LPcin-I and LPcin-II
The 3D structures of LPcin-I and LPcin-II were determined based on the NOE-derived distance restraints, dihedral angle restraints, and hydrogen bond restraints. The numbers of experimental restraints and statistics for the calculated LPcin-I and LPcin-II are summarized in Table 2. Each peptide consists of a single α-helix spanning from Thr-2 to Thr-23 for LPcin-I and from Tyr-3 to Thr-17 for LPcin-II (Fig. 6, A and B). The C-terminal additional residues are disordered in each peptide. The backbone root mean-square deviations of LPcin-I and LPcin-II were 0.44 and 0.30 Å, respectively. The Ramachandran plots showed disallowed spaces of 1.3 and 1.2% in LPcin-I and LPcin-II, respectively, however, these disallowances occurred only on the C-terminal lysine residues. The α-helix of LPcin-I was slightly curved, whereas that of LPcin-II was straight.
Table 2.
Structural statistics for the final 20 structures of LPcin-I and LPcin-II
| LPcin-I | LPcin-II | |
|---|---|---|
| Number of experimental restraints | ||
| NOE distance restraints | 336 | 234 |
| Hydrogen bond restraints | 32 | 22 |
| Torsion angle restraints | 42 | 26 |
| Number of violations | ||
| NOE > 0.3 (Å) | 0 | 0 |
| Dihedral angle > 0.5 (°) | 0 | 0 |
| Energies (kcal/mol) | ||
| Etotal | −1013.70 ± 48.72 | −774.97 ± 40.67 |
| Ebond | 0.67 ± 0.02 | 0.45 ± 0.07 |
| Eangle | 12.42 ± 0.37 | 9.92 ± 0.27 |
| Eimproper | 0.77 ± 0.12 | 0.52 ± 0.05 |
| Edihed | 140.24 ± 2.08 | 109.39 ± 1.57 |
| Evdw | −237.96 ± 3.46 | −186.13 ± 3.60 |
| Eelec | −929.83 ± 49.48 | −679.11 ± 41.36 |
| ENOE | 2.91 ± 1.53 | 0.98 ± 0.27 |
| Root mean-square deviations (RMSD) from experimental restraints | ||
| NOE (Å) | 0.012 ± 0.003 | 0.09 ± 0.001 |
| Torsion angle (°) | 0.025 ± 0.038 | 0.131 ± 0.092 |
| RMSD from the idealized geometry | ||
| Bonds (Å) | 0.001 ± 0.000 | 0.001 ± 0.000 |
| Bond angles (°) | 0.294 ± 0.004 | 0.291 ± 0.004 |
| Improper angles (°) | 0.142 ± 0.011 | 0.127 ± 0.007 |
| Ramachandran plot (%)∗ | ||
| Most favored | 88.7 | 81.0 |
| Additionally allowed | 8.5 | 13.8 |
| Generously allowed | 1.5 | 4.0 |
| Disallowed† | 1.3 | 1.2 |
| RMSD of well-ordered region (Å)‡ | ||
| Backbone | 0.44 | 0.30 |
| Heavy atoms | 1.28 | 1.16 |
As determined y PROCHECK-NMR.
Disallowed regions were from the seven lysine residues in the disordered region in LPcin-I and from five lysines in LPcin-II.
RMSD was calculated by using the software MOLMOL (27) for the well-ordered residues of LPcin-I (Thr-2–Thr-23) and LPcin-II (Tyr-3–Thr-17).
Figure 6.
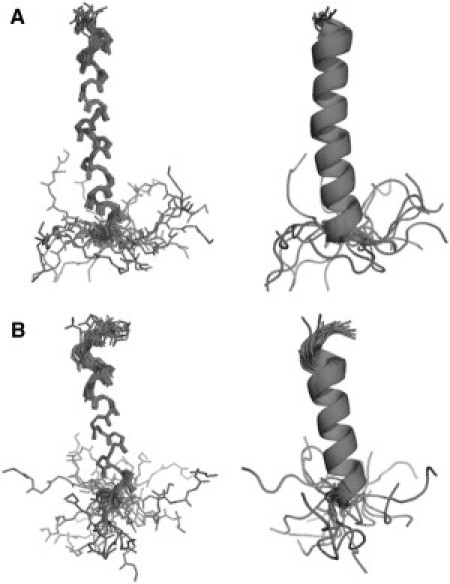
Overall structures of LPcin-I and LPcin-II. The ensembles of the 20 lowest energy structures of LPcin-I (A) and LPcin-II (B). Each peptide consists of a single α-helix spanning from Thr-2 to Thr-23 for LPcin-I and from Tyr-3 to Thr-17 for LPcin-II, and the C-terminal additional residues are disordered. LPcin-I was slightly curved, whereas LPcin-II was straight.
Distribution of hydrophobic residues in LPcin peptides
Because each peptide was dissolved in DPC micelles, the binding interface of the peptides to the micelles was of great interest to investigate the interaction of the LPcins with the cell membrane. The 3D structures determined in this work provided clues to the interaction and orientation of the peptides with respect to the membrane using solid-state NMR spectroscopy. The hydrophilic residues of LPcin-I were distributed on one side of a single α-helix and the hydrophobic and hydrophilic residues were situated on the other side of the helix. Fig. 7 A shows the distribution of the hydrophobic, polar, and charged residues on the surface of LPcin-I. One more interesting point was the slight curvature of the helix of LPcin-I. The concave surface of LPcin-I consisted of the hydrophobic residues, although the convex surface of the peptide consisted of the polar and charged residues. This curvature might be induced on the surface of the micelle sphere where the binding between LPcin-I and micelles occurs. On the other hand, the structure of LPcin-II was straight, because the length of the peptide is insufficient. The pattern of side-chain distribution was quite similar to that shown in LPcin-I and is indicated in Fig. 7 B.
Figure 7.
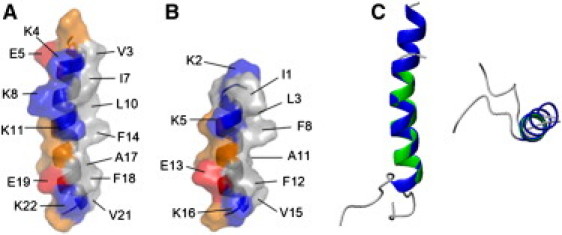
Distribution of side chains in LPcin-I (A), LPcin-II (B), overlaid structures (C). For each peptide, the hydrophobic residues are situated on one side of the helix and the charged and polar residues on the other side. The concave surface of the slightly curved LPcin-I consisted of the hydrophobic residues, which might be the micelle-binding site. The hydrophobic, positively and negatively charged, and polar amino acids were indicated in gray, blue, red, and orange, respectively. Each residue was labeled with one-letter amino acid codes and the sequence.
Solid-state NMR studies of LPcin-I and LPcin-II
The initial experimental results consist of the 1D and 2D solid-state NMR spectra of the uniformly 15N-labeled samples of LPcin-I and LPcin-II containing a single amphipathic in-plane helix in magnetically aligned unflipped bicelles that have their normals perpendicular to the direction of the applied magnetic field. Solid-state NMR spectra are strongly affected by the molecular motion and orientation. In the spectrum of an unoriented sample with multiple labeled sites without additional sample manipulations, no resolution among the resonances is feasible. This is because most of the backbone sites are structured and immobile on the timescale of the 15N chemical shift interaction (10 kHz) and contribute to the characteristic amide powder pattern between ∼220 and 60 ppm (28). In contrast, sample orientation gives well-resolved spectra that retain the orientational information used for structure determination. Each 15N-labeled site on the aligned samples contributes a single-line resonance, whose frequency is determined by the orientation of the peptide plane containing the amide group relative to the direction of the applied magnetic field. There is no evidence of residual powder pattern line shapes in the 1D 15N NMR spectra of LPcin-I (Fig. 8 A) and LPcin-II (see Fig. 8 C), suggesting that both peptides are well aligned with the phospholipids in the bicelle samples. Many of the resonances overlap in the spectra of the uniformly 15N-labeled samples, because the amide sites in an α-helix have similar orientations. The 15N resonance linewidth of LPcin-I is ∼10 ppm broader than that of LPcin-II, as shown in Fig. 8, A and C. This broadening is to be expected, based on the additional six N-terminal residues of LPcin-I and the presence of a kinked or curved helical structure in the membrane environment.
Figure 8.
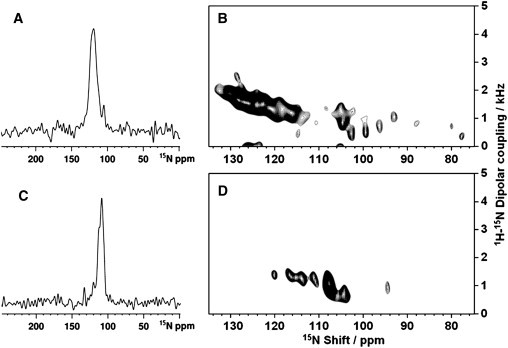
1D solid-state NMR spectra of uniformly 15N-labeled LPcin-I (A) and LPcin-II (C) in magnetically aligned bicelles and 2D SAMMY spectra of uniformly 15N-labeled LPcin-I (B) and LPcin-II (D) samples. The spectra were obtained at 700 MHz on 160 μL samples with 3 mg of 15N-labeled peptides. The mix time was 1 ms; the acquisition time was 5 ms during which TPPM 1H decoupling (25) was applied, and the recycle delay was 5 s. The data were processed with 100 Hz of exponential line broadening applied in t2 followed by Fourier transformation.
2D SAMMY spectra contain much more structural information than 1D spectra. Each resonance in the SAMMY spectra can be characterized by two orientationally dependent frequencies, 15N chemical shift and 1H-15N heteronuclear dipolar coupling. The observed 15N chemical shift is a function of the angle between the 1H-15N heteronuclear bond axis and the magnetic field. Orientational information can also be obtained from the N-H dipolar interaction (29). Furthermore, high-resolution SAMMY spectra of uniformly 15N-labeled helical peptides have resonances in the form of characteristic PISA wheel patterns that provide a direct measurement of the helix tilt and rotation (30–32). Fig. 8, B and D, show the initial experimental SAMMY spectra of the uniformly 15N-labeled samples of LPcin-I and LPcin-II in the unflipped bicelles, respectively. Significantly, there are signal intensities that are discernible at the isotropic frequency (∼120 ppm), which indicates that nearly all of the residues are structured and immobilized along with the rest of the protein in the phospholipid bilayers. The partially resolved resonance intensity between 70 and 120 ppm is consistent with transmembrane helices that are well aligned in unflipped bicelles (33). As indicated in Fig. 8, B and D, most of the residues in the LPcin-I and LPcin-II peptides are integrated into the membrane, respectively. However, the observed resonances in the SAMMY spectrum of LPcin-I (Fig. 8 B) are distributed over a wide range of 76–134 ppm in the 15N chemical shift dimension. On the other hand, the resonances from LPcin-II are present in a narrow range of 94–121 ppm. These results suggest that the structure of LPcin-II is a nearly straight helix, whereas that of LPcin-I has some deviation from ideality, such as kinks or curvatures.
Conclusion
Lactophorin is a minor phophoglycoprotein found in bovine milk and its homologous proteins are also found in milk of other species such as camel, llama, ewe, and goat, but not in human milk (14). LPcin-I is a cationic amphipathic peptide with 23-amino acid residues, and corresponds to the carboxy terminal 113–135 region of component-3 of proteose peptone and inhibits the growth of both Gram-negative and Gram-positive bacteria. In contrast to LPcin-I, it has been reported that LPcin-II, which corresponds to the 119–135 region of PP3, has no antibacterial activity, which is interesting because LPcin-I and LPcin-II have similar charge ratios and identical hydrophobic/hydrophilic sectors and show cationic properties and interact with phospholipids. Nevertheless, only LPcin-I is incorporated into planar lipidic bilayers, in which it forms voltage-dependent channels (17). We examined the tertiary structures of LPcin-I and LPcin-II in membrane-mimicking environments to elucidate the structure-activity relationship of these peptides.
Based on the antibacterial activity test, recombinant LPcin-I showed much stronger antimicrobial activity than recombinant LPcin-II in the agar hole diffusion test against five microorganisms. The high-resolution solution and solid-state NMR spectra demonstrate that the polypeptides are well folded in micelle and bilayer membrane environments. The 3D solution structure of LPcin-I determined under DPC micelle conditions was a slightly curved α-helix, whereas that of LPcin-II was a straight α-helix. And the solid-state NMR studies indicate that most of the residues in both the LPcin-I and LPcin-II peptides are integrated into the membrane. However, the larger chemical shift dispersion of LPcin-I in 2D SAMMY spectra than that of LPcin-II suggests that the structure of LPcin-I exhibits some deviation from ideal straight α-helix, such as kinks or curvatures in the membrane environments. This structural specificity of curved α-helix of LPcin-I help to make a hole in the bacterial membrane than straight α-helix of LPcin-II. Therefore, the difference in the antimicrobial activities between LPcin-I and LPcin-II peptides might result from these different structural characteristics. The solid-state NMR spectra in Fig. 8 are an early result and could be improved by further optimizing the sample conditions.
Acknowledgments
This work was supported by the Gyeonggi Research Center Program of Gyeonggi Province (GRRC HUFS 2011-B01) and the HUFS Research Fund of 2011. This study made use of the NMR facility, which is supported by the NMR Research Program of the Korea Basic Science Institute. H.-C.A. was supported by a grant from the Korea Healthcare technology R&D Project, Ministry for Health & Welfare, Republic of Korea (A092006), and wishes to thank the staff at the Korea Institute of Science and Technology 900 MHz NMR facility.
Contributor Information
Hee-Chul Ahn, Email: hcahn@dongguk.edu.
Yongae Kim, Email: yakim@hufs.ac.kr.
References
- 1.Saido-Sakanaka H., Ishibashi J., Yamakawa M. In vitro and in vivo activity of antimicrobial peptides synthesized based on the insect defensin. Peptides. 2004;25:19–27. doi: 10.1016/j.peptides.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Chinchar V.G., Bryan L., Rollins-Smith L. Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology. 2004;323:268–275. doi: 10.1016/j.virol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 3.Hancock R.E.W. Peptide antibiotics. Lancet. 1997;349:418–422. doi: 10.1016/S0140-6736(97)80051-7. [DOI] [PubMed] [Google Scholar]
- 4.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z., Wang G. APD: the Antimicrobial Peptide Database. Nucleic Acids Res. 2004;32(Database issue):D590–D592. doi: 10.1093/nar/gkh025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauterwein J., Bösch C., Wüthrich K. Physicochemical studies of the protein-lipid interactions in melittin-containing micelles. Biochim. Biophys. Acta. 1979;556:244–264. doi: 10.1016/0005-2736(79)90046-4. [DOI] [PubMed] [Google Scholar]
- 7.Henry G.D., Sykes B.D. Methods to study membrane protein structure in solution. Methods Enzymol. 1994;239:515–535. doi: 10.1016/s0076-6879(94)39020-7. [DOI] [PubMed] [Google Scholar]
- 8.Gesell J., Zasloff M., Opella S.J. Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an alpha-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. J. Biomol. NMR. 1997;9:127–135. doi: 10.1023/a:1018698002314. [DOI] [PubMed] [Google Scholar]
- 9.Rozek A., Friedrich C.L., Hancock R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry. 2000;39:15765–15774. [PubMed] [Google Scholar]
- 10.Reference deleted in proof.
- 11.Wakamatsu K., Takeda A., Matsuzaki K. Dimer structure of magainin 2 bound to phospholipid vesicles. Biopolymers. 2002;64:314–327. doi: 10.1002/bip.10198. [DOI] [PubMed] [Google Scholar]
- 12.Bechinger B., Zasloff M., Opella S.J. Structure and dynamics of the antibiotic peptide PGLa in membranes by solution and solid-state nuclear magnetic resonance spectroscopy. Biophys. J. 1998;74:981–987. doi: 10.1016/S0006-3495(98)74021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hori Y., Demura M., Asakura T. Interaction of mastoparan with membranes studied by 1H-NMR spectroscopy in detergent micelles and by solid-state 2H-NMR and 15N-NMR spectroscopy in oriented lipid bilayers. Eur. J. Biochem. 2001;268:302–309. doi: 10.1046/j.1432-1033.2001.01880.x. [DOI] [PubMed] [Google Scholar]
- 14.Campagna S., Mathot A.G., Gaillard J.L. Antibacterial activity of lactophoricin, a synthetic 23-residues peptide derived from the sequence of bovine milk component-3 of proteose peptone. J. Dairy Sci. 2004;87:1621–1626. doi: 10.3168/jds.S0022-0302(04)73316-0. [DOI] [PubMed] [Google Scholar]
- 15.Campagna S., Van Mau N., Gaillard J.L. Specific interaction between anionic phospholipids and milk bovine component PP3 and its 119–135 C-terminal fragment. Colloids Surf. B Biointerfaces. 1999;13:299–309. [Google Scholar]
- 16.Campagna S., Vitoux B., Gaillard J.L. Conformational studies of a synthetic peptide from the putative lipid-binding domain of bovine milk component PP3. J. Dairy Sci. 1998;81:3139–3148. doi: 10.3168/jds.S0022-0302(98)75879-5. [DOI] [PubMed] [Google Scholar]
- 17.Campagna S., Cosette P., Gaillard J.L. Evidence for membrane affinity of the C-terminal domain of bovine milk PP3 component. Biochim. Biophys. Acta. 2001;1513:217–222. doi: 10.1016/s0005-2736(01)00360-1. [DOI] [PubMed] [Google Scholar]
- 18.Park T.J., Kim J.S., Kim Y. Cloning, expression, isotope labeling, purification, and characterization of bovine antimicrobial peptide, lactophoricin in Escherichia coli. Protein Expr. Purif. 2009;65:23–29. doi: 10.1016/j.pep.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 20.Johnson B.A. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 21.Linge J.P., O'Donoghue S.I., Nilges M. Automated assignment of ambiguous nuclear overhauser effects with ARIA. Methods Enzymol. 2001;339:71–90. doi: 10.1016/s0076-6879(01)39310-2. [DOI] [PubMed] [Google Scholar]
- 22.Brünger A.T., Adams P.D., Warren G.L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Laskowski R.A., Rullmannn J.A., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 24.Pines A., Gibby M.G., Waugh J.S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973;59:569–571. [Google Scholar]
- 25.Khitrin A.K., Fujiwara T., Akutsu H.J. Phase-modulated heteronuclear decoupling in NMR of solids. J. Magn. Reson. 2003;162:46–53. doi: 10.1016/s1090-7807(02)00173-8. [DOI] [PubMed] [Google Scholar]
- 26.Nevzorov A.A., Opella S.J. A “magic sandwich” pulse sequence with reduced offset dependence for high-resolution separated local field spectroscopy. J. Magn. Reson. 2003;164:182–186. doi: 10.1016/s1090-7807(03)00240-4. [DOI] [PubMed] [Google Scholar]
- 27.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- 28.Marassi F.M., Ramamoorthy A., Opella S.J. Complete resolution of the solid-state NMR spectrum of a uniformly 15N-labeled membrane protein in phospholipid bilayers. Proc. Natl. Acad. Sci. USA. 1997;94:8551–8556. doi: 10.1073/pnas.94.16.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.North C.L., Barranger-Mathys M., Cafiso D.S. Membrane orientation of the N-terminal segment of alamethicin determined by solid-state 15N NMR. Biophys. J. 1995;69:2392–2397. doi: 10.1016/S0006-3495(95)80108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marassi F.M., Opella S.J. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J., Denny J., Cross T.A. Imaging membrane protein helical wheels. J. Magn. Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 32.Park S.H., Opella S.J. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 2005;350:310–318. doi: 10.1016/j.jmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Park S.H., Prytulla S., Opella S.J. High-resolution NMR spectroscopy of a GPCR in aligned bicelles. J. Am. Chem. Soc. 2006;128:7402–7403. doi: 10.1021/ja0606632. [DOI] [PMC free article] [PubMed] [Google Scholar]