

Abstract
While the importance of viral fusion peptides (e.g., hemagglutinin (HA) and gp41) in virus-cell membrane fusion is established, it is unclear how these peptides enhance membrane fusion, especially at low peptide/lipid ratios for which the peptides are not lytic. We assayed wild-type HA fusion peptide and two mutants, G1E and G13L, for their effects on the bilayer structure of 1,2-dioleoyl-3-sn-phosphatidylcholine/1,2-dioleoyl-3-sn-phosphatidylethanolamine/Sphingomyelin/Cholesterol (35:30:15:20) membranes, their structures in the lipid bilayer, and their effects on membrane fusion. All peptides bound to highly curved vesicles, but fusion was triggered only in the presence of poly(ethylene glycol). At low (1:200) peptide/lipid ratios, wild-type peptide enhanced remarkably the extent of content mixing and leakage along with the rate constants for these processes, and significantly enhanced the bilayer interior packing and filled the membrane free volume. The mutants caused no change in contents mixing or interior packing. Circular dichroism, polarized-attenuated total-internal-reflection Fourier-transform infrared spectroscopy measurements, and membrane perturbation measurements all conform to the inverted-V model for the structure of wild-type HA peptide. Similar measurements suggest that the G13L mutant adopts a less helical conformation in which the N-terminus moves closer to the bilayer interface, thus disrupting the V-structure. The G1E peptide barely perturbs the bilayer and may locate slightly above the interface. Fusion measurements suggest that the wild-type peptide promotes conversion of the stalk to an expanded trans-membrane contact intermediate through its ability to occupy hydrophobic space in a trans-membrane contact structure. While wild-type peptide increases the rate of initial intermediate and final pore formation, our results do not speak to the mechanisms for these effects, but they do leave open the possibility that it stabilizes the transition states for these events.
Introduction
Membrane fusion occurs in all life processes in which cellular contents must be passed through a membrane barrier, such as viral infection, neurotransmission, fertilization, and intracellular protein trafficking. Infection by enveloped viruses involves fusion of viral and cellular membranes with subsequent transfer of viral genetic material into the cell. The virus components that mediate fusion are glycoproteins and the best-characterized such glycoprotein is influenza virus hemagglutinin (HA) (1,2). HA is a trimer of identical subunits, each of which is comprised of two glycopeptides linked by a single disulfide bond, with one glycopeptide accomplishing receptor binding (HA1, 328-residue) and the other working as a fusion machine (HA2, 221-residue) (3). A highly conserved 20-amino-acid amphipathic peptide at the N-terminus of HA2 is essential for fusion activity and is termed the “fusion peptide” (4). Fusion peptides of influenza virus strain X-31 (HA X-31) and HIV (gp41) cannot induce membrane fusion but do promote aggregation and rupture at high peptide/lipid ratios (P/Ls). However, fusion peptides promote fusion between vesicles brought into close apposition by poly(ethylene glycol) (PEG) (5).
Structural studies of the 20-residue HA fusion peptide have been of necessity low resolution, as the peptides have limited solubility and trigger membrane disruption at high P/Ls. One common model based on infrared spectroscopic experiments envisions a fully helical peptide with its N-terminus inserted into the bilayer at an angle of roughly 54° to the bilayer-normal (6). A detailed model derives from NMR studies of fusion peptide joined at its C-terminus to a charged carrier peptide that was designed to allow fusion peptide to be solubilized in dodecyl phosphocholine (DPC) micelles. By combining NMR data with electron spin resonance experiments on spin-labeled carrier-stabilized peptide associated with membranes, the peptide was proposed to adopt an inverted-V structure (7). Oriented Fourier transform infrared (FTIR) experiments along with membrane perturbation and fluorescence data obtained with the unmodified peptide at lower concentrations are consistent with the inverted-V model with both the C- and N-termini protruding somewhat into the bilayer but with polar residues near the middle of the peptide in the interfacial region of the bilayer (8).
NMR studies of a slightly longer (23 residues including the natural sequence, WTG at the C-terminus) peptide associated with DPC micelles suggest a helical hairpin structure with polar residues arranged on one face of the pair and the two tryptophans and nonpolar residues on the other (9). While this model would be consistent with FTIR data showing an orientation roughly parallel with a membrane surface (8), it should result in considerable perturbation of interfacial packing and little perturbation of core bilayer order—the opposite of what is observed with the 20-residue peptide (8). For this reason, we view the inverted-V model (an incompletely folded helical hairpin) as the best approximation for the structure of the 20-residue peptide examined here.
It remains to be seen how the HA fusion peptide (or any fusion peptide) functions to promote fusion in vivo. Based on studies of lamellar-to-nonlamellar phase transitions (particularly, lamellar to hexagonal) in lipid suspensions, some have proposed that fusion peptides might impose a negative curvature that is inconsistent with the intrinsic curvature of juxtaposed lamellar structures and thus promote formation of curved nonlamellar fusion intermediates (10,11). Of course, the stability of lamellar lipid phase relative to nonlamellar phases is influenced by factors other than intrinsic curvature, such as bending modulus (12), interstice or void energy (13,14), and Gaussian energy (15). Based on the influence of both the HA and HIV gp41 fusion peptides on bilayer structure (8) as well as on the mutual influence of hexadecane and fusion peptides on fusion, it has been suggested that fusion peptides might promote fusion by lowering void energy (5,16). One way to address these different possibilities is to compare the membrane-perturbing and fusion-promoting influences of wild-type fusion peptides with those of mutant peptides that fail to support fusion and/or lytic activity.
Factors that affect lamellar-to-nonlamellar phase equilibria may influence the free energies of semistable intermediates on the path to fusion (e.g., stalk) but may also influence the rates at which transitions occur between these states. To examine this possibility, one needs a mechanistic model within which to interpret fusion kinetics. We have previously proposed such a model and have shown it to account for multiple observations (17). Here we report the kinetics of PEG-mediated fusion of vesicles composed of an optimal fusogenic mix of lipids in the presence and absence of wild-type and two mutant forms, G1E and G13L, of HA fusion peptide. A G1E mutant peptide from influenza virus A/Japan/305/57 has reduced efficacy for fusion in a cell-based assay (4), while a G13L mutant fusion peptide from the A/PR/8/34 virus strain was impaired in lysis of model membranes (18). We also compare to wild-type peptide the structures of these membrane-bound mutant peptides as well as their influence on bilayer structure. From these results, we identify at what stage(s) of the fusion process, and in what ways, the wild-type influenza X-31 HA fusion peptide exerts its unique fusion-promoting influence.
Finally, by comparing these results with the effects of these peptides on lipid phase behavior (B. G. Tenchov, D. P. Siegel, R. C. MacDonald, and B. R. Lentz, unpublished), we hope to gauge the following:
Does the HA fusion peptide influence fusion by affecting the curvature energy, the void energy, or some combination of both? Or does it influence fusion by some other contribution to the free energy of lamellar or nonlamellar lipid structures, or perhaps by the transitions between these structures?
Experimental Procedures
Materials
Chloroform stock solutions of 1,2-dioleoyl-3-sn-phosphatidylcholine (DOPC), 1,2-dioleoyl-3-sn-phosphatidylethanolamine (DOPE), bovine sphingomyelin (SM), and 1-palmitoyl-2-n-(4-nitrobenzo-2-oxa-1,3-diazole) aminohexanoyl phosphatidylcholine (C6NBDPC) were purchased from Avanti Polar Lipids (Birmingham, AL) and used without further purification. The concentration of the stock lipids was determined by phosphate assay. Cholesterol was purchased from Avanti Polar Lipids and was further purified by published procedures (20). 1,6-diphenyl-1,3,5-hexatriene (DPH) and 1-(4-trimethylammonium)-6-diphenyl-1,3,5-hexatriene (TMA-DPH) were purchased from Molecular Probes (Eugene, OR). 2-(n-Morpholino)ethanesulfonic acid sodium salt was purchased from Sigma Chemical (St. Louis, MO). All other reagents were of the highest purity available. Poly(ethylene glycol) of molecular weight 7000–9000 (PEG 8000) was purchased from Thermo Fisher Scientific (Waltham, MA) and purified as previously reported (21).
Fusion model and data analysis
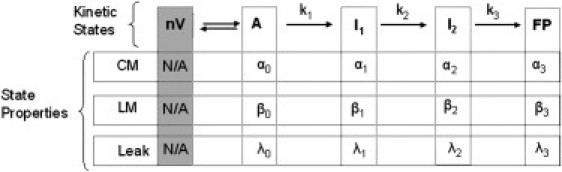
Fusion is widely (but not universally) viewed in terms of a multistep process that proceeds through semistable transient intermediates (14). We have treated the kinetics of the process in terms of a sequential, two-intermediate (four-state) model that relates observable events associated with PEG-mediated fusion (lipid mixing, contents mixing, leakage, light scattering, and pyrene cholesterol fluorescence) to rate constants for evolution of fusion intermediates and probabilities of events occurring in each state. For all the systems considered here, lipid mixing, content mixing, and content leakage data were sufficient to determine the three rate constants and other parameters (17).
This model is summarized in the following Scheme:Here nV represents n separate vesicles, A is a state with vesicles in contact within aggregates, I1 and I2 are the intermediate states (stalk and extended trans-membrane contact (TMC) (22)), and FP is the final fusion pore state. Each state is characterized by the probability of observing fusion events (i.e., content mixing {αi}, lipid mixing {βi}, and leakage rate (λi}) and by rate constants (k1, k2, and k3) for interconversion of states. The A-state is achieved using PEG to aggregate vesicles into molecular contact (23). The aggregation rate was approximately fivefold greater than k1 and independent of the presence of peptide. Step 1 can be reversed if aggregation is reversed by removing PEG, but is irreversible when aggregation is maintained (24), which it is in our experiments.
The sequential, two-intermediate model has 15 parameters: 3 rate constants and 12 values that describe the probabilities of lipid mixing, content mixing, and rates of leakage in each state. Four of these probabilities can be set to zero by observation (α0 = 0, β0 = 0, β3 ≈ 0, λ3 = 0), and two normalization conditions (α1 + α2 + α3 = 1 and β1 + β2 =1) constrain another two, leaving nine parameters (three rate constants, three leakage rate constants, two αi and βi) to be set by comparison to experiment. Each data set (lipid mixing, content leakage, content leakage) is normally a double exponential described by four parameters. Global or simultaneous fitting of all three data sets can thus define 9–12 parameters so that the parameters should, in theory, not be underdetermined. In practice, we have found this to be true in that
-
1.
Fitting with different sets of starting parameters always returns the same final set, and
-
2.
Separate analyses of triplicate experiments yields the same parameters within parameter uncertainties determined by fitting the individual data sets or a combined data set.
In addition, we showed (17) that rate constants obtained as described here from lipid mixing, content mixing, and content leakage data also accounted for the time dependence of two additional observables related to membrane structural changes, light scattering, and pyrene cholesterol fluorescence (25). All this demonstrates that our kinetic model provides a robust description of our data that yields well-defined rate constants that are capable of accounting for additional observations.
The process of fitting three data sets simultaneously while adjusting nine model parameters is a daunting task. To facilitate this, we broke the task down into a series of steps in which a limited number of parameters were determined at each step using SigmaPlot (Systat Software, San Jose, CA):
-
Step 1.
The lipid-mixing time course (LM(t)) was fit to obtain the rates k1 and k2, the lipid mixing probabilities β1 and β2 = (1−β1), and the total fraction of lipid mixing at long time, fLM.
-
Step 2.
Using k1, k2, and β1 from Step 1, the raw fluorescence data for leakage and content mixing time courses were fitted globally to obtain k3, λ0, λ1, λ2, α1, and α2. The long-time total fraction of content mixing, fCM, is obtained from the parameters derived in this fit.
-
Step 3.
The fluorescence data were then simulated using these parameters to obtain plots of observed and predicted fluorescence time courses. Residual plots obtained from these time courses were examined to test for systematic fitting errors.
Our kinetic model is based on a structural model of fusion involving a series of semistable intermediates (modified-stalk model (22) for which there is considerable evidence (26,27)). This structural model posits that mixing of trapped contents will occur only in the final FP state, but we must account for the experimental reality that every system we have examined showed evidence of contents mixing before final pore formation. We argue that the structural model is consistent with this observation only if we allow (17,24) that content mixing occurs with finite probabilities (α-values) in semistable structural intermediates via fluctuations in their already stressed structures (17). The existence of transitory or flickering small pores is widely described for electrophysiological measurements of fusion events in patch-clamped cells (e.g., see Alvarez de Toledo et al. (28)) and has been noted even in model membranes (29). In addition, we clearly observed, using extremely nonleaky small unilamellar vesicles (SUVs) composed of an 85:15 mixture of dioleoyl-phosphatidylcholine and dilinoleoyl-phosphatidylcholine two classes of pores,
-
1.
Small and reversible pores that passed protons in the hemifused state (here I1 and I2) and
-
2.
Larger and irreversible pores that passed much larger solutes (DPX and ANTS) and appear only late in the fusion process (24).
Thus, it is quite clear that contents can mix between compartments both early and late in the fusion process, although the natures of the structures that allow these two processes are likely quite different.
Results
Binding of HA wild-type and mutant peptides to PC/PE/SM/CH (35/30/15/20) SUV
We have already reported binding of HA wild-type peptide to DOPC and phosphatidylcholine (PC)/ phosphatidylethanolamine (PE)/Sphingomyelin (SM)/Cholesterol (CH) SUV as observed by
-
1.
Monitoring tryptophan fluorescence intensity changes after addition of vesicles, and
-
2.
Monitoring fluorescence intensity of membrane-bound DPH as a result of energy transfer from bound peptide (5,8).
Binding constants by both methods were consistent. We used the former method here to determine the binding constants of wild-type HA, G1E, and G13L peptides. We performed the experiments at two different concentrations of the peptide, and data were fitted globally to a standard surface-binding model (30). The binding parameters (Kd in μM and N (lipids per bound peptide)) for the three peptides are: 0.51 ± 0.06, 28 ± 5; 0.67 ± 0.05, 21 ± 5; and 0.89 ± 0.07, 12 ± 4 for the wild-type, G1E, and G13L peptides, respectively, where the error in Kd is the standard deviation derived from global fitting of binding data obtained at two different peptide concentrations. We conclude that the wild-type peptide bound more tightly and occupied more membrane surface than either the G13L or G1E mutant, with the latter occupying remarkably little membrane surface.
Effect of HA wild-type and mutant peptides on vesicles in the absence of PEG
We reported previously single-time-point observations of wild-type (X-31 strain) fusion peptide-induced content leakage and lipid mixing but not content mixing between DOPC SUVs in a peptide-concentration-dependent fashion (20). Here we report in Fig. 1 the initial rates of lipid mixing and leakage of DOPC/DOPE/SM/CH (35:30:15:20) SUVs plotted for different lipid/peptide ratios (L/Ps) for wild-type and mutant peptides in the absence of PEG at 37°C. While all three fusion peptides significantly increased the rates of content leakage at high peptide/lipid ratios (P/Ls), none of them induced any mixing of contents between SUVs in the absence of PEG. Wild-type peptide triggered some lipid mixing between SUVs at all P/Ls, as shown in Fig. 1. Data obtained at a P/L of 1:200 were also fit to a double exponential to obtain an exponential constant comparable to values obtained in the presence of PEG.
Figure 1.
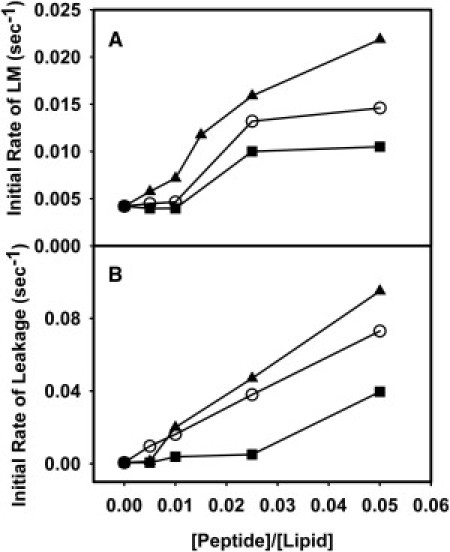
Effect of HA wild-type and mutant peptides on (A) lipid mixing and (B) leakage in PC/PE/SM/CH (35/30/15/20) SUVs in the presence of wild-type fusion peptide (solid triangle), G13L (open circle) peptide, and G1E (solid squares) peptide, in the absence of PEG. Measurements were carried out in 10 mM 2-(n-morpholino)ethanesulfonic acid (MES), 100 mM NaCl, 1 mM CaCl2, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 5.0 at 37°C. Total lipid concentration was 0.2 mM. Values presented here are the average from three experiments such as shown in Fig. 2.
Because rapid lipid exchange requires close approach of membranes (31), it is not surprising that the k1 values obtained in the absence of PEG (10, 4, and 2 × 10−4 s−1 for wild-type, G13L, and G1E peptides, respectively) were an order-of-magnitude slower than seen in the presence of PEG (Table 1). However, the order of these rates was approximately the same in the presence and absence of PEG (WT > G13L ∼ G1E). Both G13L and G1E triggered almost imperceptible lipid exchange between vesicles at low P/Ls (indeed, G1E seems totally ineffective), but produced some lipid mixing above P/L = 1:100. Wild-type and G13L peptides produced significant leakage at all P/Ls, but, consistent with a previous report (18), wild-type was more effective. Again, the G1E peptide was less effective than wild-type or G13L in triggering leakage.
Table 1.
Rate constants and probabilities of intermediate states of PC/PE/SM/CH SUVs (control) fusion and leakage in the presence of HA wild-type and G1E and G13L mutants at 26°C
| Content mixing |
Lipid mixing |
Leakage |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| System | k1 × 103 (s−1) | k2 × 103 (s−1) | k3 × 103 (s−1) | α1 (I1) | α2 (I2) | α3 (FP) | fCM | β1 (I1) | β2 (I2) | fLM | λ0 × 104 (s−1) (I0) | λ1 × 104 (s−1) (I1) | λ2 × 104 (s−1) (I2) |
| Control (PEG 5 wt%) | 10.3 ± 0.8 | 2.5 ± 0.2 | 1.06 ± 0.02 | 0.29 ± 0.04 | 0.40 ± 0.02 | 0.31 ± 0.02 | 0.15 ± 0.01 | 0.50 ± 0.06 | 0.50 ± 0.06 | 0. 54 ± 0.02 | 3.07 ± 0.02 | 1.78 ± 0.02 | 0.39 ± 0.02 |
| Wild-type HA | 20.1 ± 1.25 | 3.67 ± 0.16 | 1.40 ± 0.04 | 0.45 ± 0.05 | 0.23 ± 0.01 | 0.32 ± 0.05 | 0.21 ± 0.02 | 0.63 ± 0.02 | 0. 37 ± 0.04 | 0.46 ± 0.03 | 7.18 ± 0.11 | 3.64 ± 0.05 | 1.07 ± 0.03 |
| PEG (8 wt %) | 18.0 ± 0.63 | 3.03± 0.07 | 1.20 ± 0.01 | 0.50 ± 0.01 | 0.23 ± 0.01 | 0.27 ± 0.02 | 0.22 ± 0.02 | 0.56 ± 0.03 | 0.40 ± 0.03 | 0.52 ± 0.04 | 4.92 ± 0.16 | 2.06 ± 0.07 | 0.30 ± 0.05 |
| G1E mutant | 11.8 ± 0.81 | 2.41 ± 0.12 | 1.08 ± 0.08 | 0.34 ± 0.06 | 0.36 ± 0.02 | 0.30 ± 0.02 | 0.12 ± 0.01 | 0.57 ± 0.01 | 0.43 ± 0.01 | 0. 49 ± 0.02 | 2.68 ± 0.05 | 1.59 ± 0.03 | 0.48 ± 0.03 |
| G13L mutant | 12.4 ± 0.83 | 2.44 ± 0.13 | 0.85 ± 0.03 | 0.20 ± 0.06 | 0.34 ± 0.02 | 0.46 ± 0.06 | 0.13 ± 0.02 | 0.57 ± 0.01 | 0.43 ± 0.01 | 0.51 ± 0.02 | 2.06 ± 0.05 | 1.37 ± 0.03 | 0.44 ± 0.02 |
Parameters were obtained by fitting lipid mixing, content mixing, and content leakage time courses to a sequential two-intermediate model as described in the text. All the experiments were performed at least three times, with analysis carried out on the combined time courses. The errors presented in the Table are the parameter uncertainties from this combined analysis, although parameters from analysis of all experiments normally fell within this range of uncertainty.
Effect of HA wild-type and mutant peptides on PEG-mediated SUV fusion
We showed in an earlier publication that PEG had no influence on wild-type gp41 peptide Trp fluorescence intensity, membrane binding, and depth of penetration into membranes (16). As controls, we showed insensitivity of Trp fluorescence and binding of wild-type HA fusion peptide and its G1E and G13L mutants to membranes was independent of the presence of PEG (data not shown), confirming that the low concentration of PEG used in our experiments is unlikely to affect either formation of the peptide-membrane complex or the physical state of the peptide in solution.
Fig. 2 shows time courses of PEG-mediated lipid mixing (LM) (Fig. 2 A), contents mixing (CM) (Fig. 2 B), and content leakage in DOPC/DOPE/SM/CH SUVs (Fig. 2 C) in the presence of wild-type and mutant peptides (L/P = 200:1) at 5% PEG at 26°C. All three data sets (LM, CM, and leakage) were fitted globally to the model in Scheme 1 to obtain the rate constants of each step and probabilities of lipid mixing and content mixing in each intermediate state (17). Wild-type peptide increased by 40% the total extent of contents mixing, doubled the rate of initial intermediate formation, increased by somewhat less the k2 and k3 rate constants (∼30 and 50%, respectively), and increased the probability that content mixing and lipid mixing occur in state I1 at the expense of content mixing in I2 (Table 1). The two mutant peptides produced none of these effects and may have slightly decreased the extent of content mixing. Finally, of the three peptides, only the wild-type significantly increased the rate of content leakage.
Figure 2.
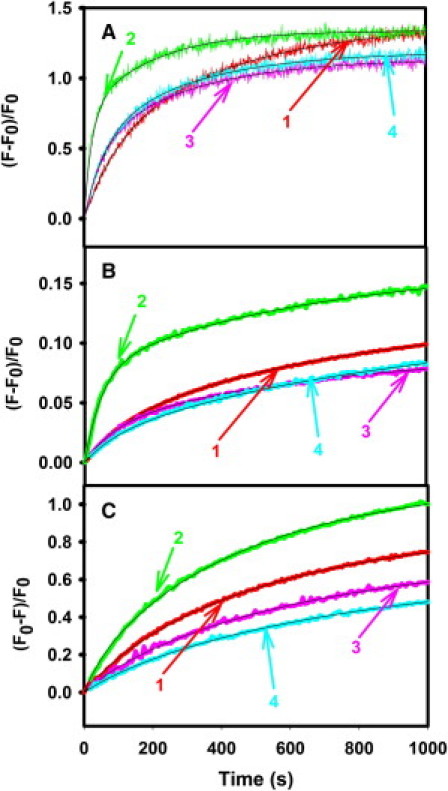
Effect of HA wild-type (2), G1E (3), and G13L (4) peptides, compared to the absence of peptide, i.e., control (1), on kinetics of (A) lipid mixing, (B) contents mixing, and (C) leakage in PC/PE/SM/CH (35:30:15:20 mol %) SUVs in the presence of 5% PEG at 26°C. Results (observed fluorescence intensity changes) are shown for an L/P of 200:1. Measurements were carried out in 10 mM MES, 100 mM NaCl, 1 mM CaCl2, 1 mM EDTA, pH 5.0 at a total lipid concentration was 0.2 mM. Smooth curves drawn through the data show a two-state sequential model global fit. The fitting parameters shown in Table 1 were obtained by combining data taken on three different samples on different days.
Scheme 1.
Effect of HA wild-type and mutant peptides on bilayer properties
We examined the bilayer properties of membranes using three different fluorescent probes, C6NBDPC, DPH, and TMA-DPH, to characterize membrane structural properties. DPH reports order in the interior of the lipid bilayer. TMA-DPH locates to the interfacial region (32) and probes order and/or thermal motions in this region (33). C6NBDPC partitions between the upper region of the bilayer (34) and micelles (34) with its average lifetime reflecting the partition coefficient between these two phases (33), making C6NBDPC a sensitive reporter of free volume and lipid packing within membrane outer leaflets (33,35). We measured the phase shifts and modulation ratios of frequency-modulated C6NBDPC fluorescence at different P/Ls in DOPC/DOPE/SM/CH SUVs and analyzed these data in terms of three lifetime components to obtain the mole fractions of probe in both environments (33). The changes in the average lifetime of the membrane-located C6NBDPC probe and its partitioning into the membrane are shown in Fig. S1, A and B (see the Supporting Material), respectively, as a function of P/L. The average lifetime of C6NBDPC located in PC/PE/SM/CH SUVs increased with increasing concentration of peptides, with G13L having by far the biggest effect and G1E almost no effect. The increase of C6NBDPC lifetime in the membrane upon addition of peptide suggests either that
-
1.
The peptide inhibited motions and increased packing order in the neighborhood of the probe (the carboxyl backbone region (34)),
-
2.
The peptide caused C6NBDPC to partition deeper into the membrane outer leaflet, or
-
3.
The peptide prevented water from penetrating to the neighborhood of the probe in the membrane outer leaflet.
Wild-type HA or gp41 fusion peptides do not result in a change in the ratio of TMA-DPH lifetimes in water and D2O buffers (8), nor do the HA mutants investigated here (results not shown). This could mean that these peptides do not alter water penetration into the bilayer interfacial region or that they all induce deeper penetration of the probe into the bilayer (33). Because the wild-type and mutant peptides had very different effects on both C6NBDPC partitioning (see Fig. S1 B) and TMA-DPH anisotropy, it indicates that these have very different effects on free volume and order within the upper regions of the bilayer. Thus, we could not expect all three peptides to have the same effect on positioning of TMA-DPH in the bilayer.
We conclude that none of the peptides at the P/Ls we examined had a significant effect on water penetration into the bilayer. Thus, decreased water penetration is not likely the cause of the increased lifetime of C6NBDPC seen in Fig. S1 A. Increased interfacial order and reduced thermal motions likely plays a role in the increased C6NBDPC lifetime for G13L in Fig. S1 A, because it produced a dramatic increase in TMA-DPH anisotropy (Fig. S2 A). The G1E mutant has little perturbing effect on any region of the bilayer (see Fig. S1 and Fig. S2). We conclude that only the wild-type peptide significantly fills bilayer free volume (see Fig. S1 B) and increases order in the bilayer interior (see Fig. S2 B). The increased C6NBDPC lifetime that it causes thus likely reflects this ability to fill space and decrease thermal motion in the deeper regions of the bilayer. Based on these observations, we conclude that
-
1.
Wild-type HA peptide occupies available free volume, increases order, and somewhat reduces thermal motion in the membrane interior, while the mutant peptides do not;
-
2.
G13L occupies interfacial space more than wild-type or G1E; and
-
3.
G1E mutant had almost no effect on bilayer structure, and certainly much less than either the wild-type or G13L mutant peptides.
Possible structures of the peptide-membrane complexes
Wild-type peptide
The secondary structure of wild-type HA fusion peptide and mutants were examined by both circular dichroism (CD) and polarized attenuated total internal reflection Fourier-transform infrared (PATIR-FTIR) spectroscopy. Fig. 3 shows the CD spectra of all three peptides in PC/PE/CH (2:1:1) vesicles. Analysis using CD Pro (http://lamar.colostate.edu/∼sreeram/CDPro/main.html) showed the wild-type fusion peptide to be highly helical (∼42%), with some turn (∼25%) and unordered structure (∼33%) but no β-structure. Fig. 4 shows PATIR-FTIR amide I′ spectra of wild-type and mutant peptides in PC/PE/SM/CH monolayers supported on a silanized germanium internal reflection crystal exposed to bulk aqueous buffer at pH 5.0. Spectra were collected with a polarizer oriented parallel (Fig. 4) and perpendicular (not shown) to the plane of incidence, and these were analyzed to obtain the dichroic ratios in Table S1 (see the Supporting Material).
Figure 3.
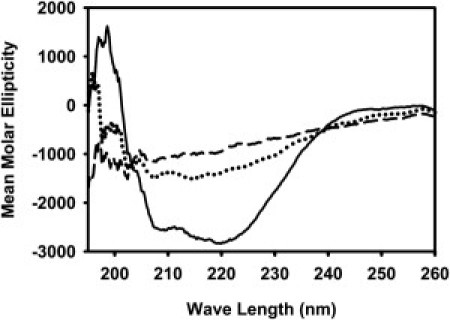
CD spectra of HA wild-type (solid line), G1E (dotted line), and G13L (dashed line) in the presence of PC/PE/CH (2:1:1) SUVs. The peptide concentration was 80 μM, and the lipid concentration was 2 mM. Small amounts of peptide solution in dimethylsulfoxide were dried in thin films on the surface of 3.5 mL brown vials. The films were frozen using dry ice, and maintained overnight under high vacuum. Preformed SUVs (in 10 mM phosphate buffer at pH 5.0) were added to the vials and vortexed for 30 min to transfer to the membranes.
Figure 4.
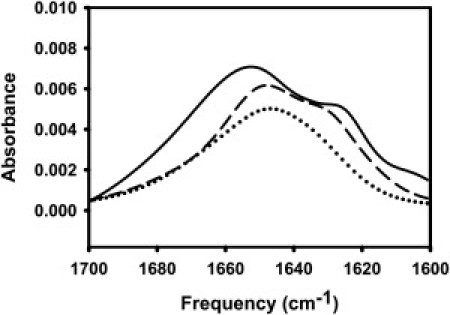
PATIR-FTIR amide I′ spectra of HA wild-type (solid line), G1E (dotted line), and G13L (dashed line) on a supported monolayer of PC/PE/SM/CH. Parallel polarized (shown) and perpendicular polarized spectra were collected for secondary structure and peptide orientation in membrane at pH 5.0.
As previously reported (8), wild-type HA peptide exhibited a split amide I′ band, with the bulk of the absorption occurring in a broad band centered at 1649 cm−1. This band has a dichroic ratio of ∼1.7, which is significantly less than the isotropic ratio for this system (Riso = 2.34), indicating a slight tendency for backbone peptide groups to be oriented parallel to the membrane surface. The wild-type peptide also has a prominent shoulder centered at 1625 cm−1, a position commonly associated with extended β secondary structure but possibly reflecting a more complex structure. It is significant that this component has a dichroic ratio of 1.2, close to the minimum possible ratio (Rmin = 0.9), indicating that a preponderance of the backbone peptide groups associated with this secondary structure are very nearly parallel to the membrane surface. Because this component is absent in spectra taken with peptide associated with DOPC monolayers (8), the nature of this structural element depends on the membrane with which it associates. There is no evidence of β-structure in our CD spectrum of wild-type peptide, so it seems unlikely that this secondary structure element is an extended sheet. The NMR-based structure of wild-type peptide reports a considerably disordered 3(10) helical C-terminal structure, but also no β-structure (7).
Analysis of a 3(10) helical peptide shows a single weak CD trough at ∼208 nm (36), which we would have difficulty resolving because significant α-helix is present. In addition, the NMR structure was determined with a wild-type peptide covalently linked to a solubilizing peptide and bound to DPC micelles and thus may not contain the structural elements present in the wild-type peptide bound to the DOPC/DOPE/SM/CH vesicles used in this study. Taken together, these results suggest that the C-terminus of HA fusion peptide used in this study is a weakly ordered H-bonded structure whose peptide units lie roughly parallel to the bilayer. The fact that the wild-type peptide reduced free volume and decreased thermal motions in the hydrophobic region of the bilayer, taken with the CD and FTIR data, suggests that the largely hydrophobic N-terminal helix of the wild-type peptide must protrude significantly into the bilayer. This is consistent with our previously published model (8) and with the model based on solution NMR of the wild-type peptide joined to a soluble peptide carrier (7).
G13L peptide
The CD spectrum of the G13L peptide was less clearly interpretable, although CD Pro (http://lamar.colostate.edu/∼sreeram/CDPro/main.html) analysis suggested that this mutation increased unordered contributions at the expense of helical content (∼32%), leaving turn structure unchanged. The G13L mutant also exhibited a split amide I′ band. While the main peak and the shoulder were more narrowly split than for wild-type HA, and thus more difficult to resolve, the 1649 cm−1 band was reduced relative to other bands, consistent with the decrease in helical content suggested by CD. The low frequency shoulder (1629 cm−1) also had a somewhat lower dichroic ratio (∼1.5), consistent with the behavior of the wild-type peptide—but again it is difficult to resolve bands this narrowly split, and resolution enhancement techniques are not appropriate in this setting. G13 is thought to be part of a motif that defines the N-terminal helix of the wild-type peptide (18), so replacing it with a leucine could break this helix and provide sufficient hydrophobic driving force to draw the side chain at position 13 into the membrane and thus tilt the N-terminal helix toward the bilayer interface, disrupting the inverted-V structure of the wild-type peptide. This would be consistent with the G13L mutant increasing order at the bilayer interface (see Fig. S2 A). This would also be consistent with this peptide providing a somewhat less disordered environment for C6NBDPC, explained in its increased lifetime (see Fig. S1 A). This position should not increase interior order (see Fig. S2 B) or decrease free volume (see Fig. S1 B), as does wild-type peptide. Thus, our data suggest that the G13L mutant has a shortened N-terminal helix that lies near the membrane interface.
G1E peptide
This peptide also yielded a poorly defined CD spectrum. CD Pro (http://lamar.colostate.edu/∼sreeram/CDPro/main.html) analysis suggested appearance of β-sheet structure (∼30%) at the expense of turn and α-helix (24%). The G1E mutant exhibited a broad featureless amide I′ absorption, with a dichroic ratio of 1.6:1.8, again indicating a slight tendency for backbone peptide groups to be oriented parallel to the membrane surface but with a poorly defined secondary structure or a variety of possible secondary structural elements. Replacing the N-terminal glycine with a glutamic acid should make it more difficult for the N-terminus of the fusion peptide to insert into and fill space in the bilayer, which is consistent with its lack of effect on interior membrane order and free volume (see Fig. S1 and Fig. S2). Moving the N-terminus toward the more polar interface might also be expected to destabilize the N-terminal helix, several residues of which are quite hydrophobic. If so, the β-sheet component of the CD spectrum suggests that at least some of the H-bonding potential of this helix might be satisfied through formation of some intermolecular β-sheet. A location at or even slightly above the interface would be consistent with the inability of this mutant to perturb the bilayer (see Fig. S1 and Fig. S2).
Discussion
Our results are discussed below in the context of the kinetic model summarized in Scheme 1 (17,33). In a calculation based on lipid material properties and the modified or extended stalk hypothesis (22), the initial (I1) and second (I2) intermediates in SUV fusion are defined by minima in the free energy of a hemifused vesicle pair as a function of stalk radius (14). I1 corresponds to a slightly expanded stalk and I2 to an expanded trans-membrane contact (TMC) or diaphragm. This calculation predicts the TMC structure to be a high-free-energy structure separating the stalk and expanded TMC low free energy intermediates (14). Vesicles brought into close contact by PEG (23) constitute the initial aggregated state (A) from which fusion proceeds in our experiments. The final fusion pore (FP) state is envisioned as stable pores between juxtaposed vesicles (17). Contents and lipids can mix between vesicles via transitory events (i.e., flickering pores) that occur with finite probability early in the fusion process before formation of larger stable FPs (33). Thus, fusion peptides can promote fusion by:
-
1.
Altering the probabilities of contents mixing, α, and lipid mixing, β, at each stage of the fusion process,
-
2.
By affecting the rates of intermediate or fusion pore formation, and
-
3.
By influencing the total extent of content mixing (%CM) or lipid mixing (%LM).
Each of these mechanisms can reflect different effects of the peptide on lipid structures.
In this study, we determined the effects of wild-type HA fusion peptide and its two mutants on
-
1.
The binding with the membrane,
-
2.
The different steps of PEG-induced model membrane fusion, and
-
3.
The structure of the PC/PE/SM/CH bilayers, and we also obtained data related to the structure of the peptides on the vesicles.
These studies were meant to answer three questions put forth in the Introduction:
-
1.
At what stage(s) of the fusion process does the wild-type HA fusion peptide exert its fusion-promoting influence?
-
2.
How does the wild-type peptide influence events at each stage?
-
3.
Can differences between the structures and influences on bilayer structure of HA and wild-type mutant peptides shed light on the mechanism(s) by which a wild-type peptide promotes fusion?
Finally, these peptides influence lipid phase behavior (B. G. Tenchov, D. P. Siegel, R. C. MacDonald, M. E. Haque, and B. R. Lentz, unpublished, and B. R. Lentz, University of North Carolina at Chapel Hill, personal communication, 2011), and we consider the potential importance of this effect relative to other effects considered here. In answer to the first two questions, the wild-type fusion peptide exerted three major fusion-enhancing influences: a twofold enhancement in the rate of initial intermediate formation (k1) and a 30–50% increase in k2 and k3, promotion of content mixing earlier in the fusion process (increased α1), and a 40% increase in the extent of contents mixing or fusion. In discussing these three effects, we also touch on the other two questions raised here.
The k1 increase brought about by wild-type fusion peptide reflects a decrease in the activation free energy for transitioning from the A to the I1 states. The transition state between A and I1 is not semistable and cannot be described in terms of material properties of lamellar or nonlamellar lipid phases, as it must involve significant rearrangements of water and lipid molecules in order to bridge the lamellar and nonlamellar geometries of the A-state and the stalk intermediate (14). Binding of wild-type HA fusion peptide must either raise the free energy of the A-state or lower the free energy of the transition state. Because the SUVs used for these studies have a positive curvature stress imposed on their exposed monolayer as well as a negative curvature stress associated with their inner monolayer, this makes them prone to fusion (37). It has been suggested that HA fusion peptide might impart a negative curvature to lipid structures (10).
Of the three peptides examined, only the wild-type peptide increased k1, filled free space in the exposed monolayer, and increased lipid order in the bilayer (Table 1 and see Fig. S2 B). It also bound more avidly and in a way that occupied more surface area. Binding of a peptide that does all this should actually stabilize state A and inhibit fusion, as has been reported for HA peptide and fusion of large unilamellar vesicles having outer monolayers with induced positive curvature stress (5). We conclude that the HA fusion peptide likely stabilizes the transition state for formation of the initial, hemifused fusion intermediate from the dehydrated lamellar phase. However, we know nothing about the structure or thermodynamics of this transition state, so it is difficult to conclude more than that the wild-type peptide stabilizes it and that the mutant peptides fail to do so.
Beyond increasing k1, wild-type fusion peptide also increased k2 and k3. In the case of k2, fusion peptide could influence either the free energy of the stalk or the free energy of the barrier between the stalk and I2. This barrier occurs just shy of the TMC structure as fusion intermediates expand from the stalk through the TMC to the extended TMC or diaphragm (see Fig. 3 of Malinin and Lentz (14)). Because all these structures have the same topology and thus Gaussian curvature, the major contributions to the relative free energies of the stalk and diaphragm intermediates arise from the increased (relative to the original state of aggregated SUVs) interstice or void free energy and the decreased bending free energy (14).
It has been suggested that HA fusion peptide might impart a negative curvature to lipid structures (10), thereby stabilizing fusion intermediates. Increasing the negative curvature of each monolayer stabilizes both the stalk and the TMC but increases the difference between their free energies (14) and thus should decrease k2. Both monolayers contribute to this effect, but increasing the negative curvature of only the contiguous monolayer of fusion intermediates should stabilize the stalk more than the TMC and thus decrease k2 even more (14). Thus, it is unlikely that fusion peptide could increase k2 by increasing the negative curvature of the contiguous monolayers. Bending free energy is the product of bending modulus times the net of intrinsic and imposed curvature stress (15), and the gp41 fusion peptide reduces the bending modulus of lipid monolayers (12).
However, SUVs are so highly curved that they have a very unfavorable bending free energy, and the structure that exists between the stalk and the TMC has lower curvature stress than either the stalk or the TMC and is disfavored primarily by its interstice free energy (14). Thus, decreasing the bending moduli of both contacting monolayers (12) should diminish the effect of the negative intrinsic curvature of our lipid mixture on both stalk and TMC by a constant factor, and therefore diminish the free energy difference between them by very little if at all. Thus, the interstice or void or mismatch free energy probably constitutes the major contribution to the free energy difference between stalk and TMC, and this difference decreases as the mismatch volume decreases (14). Neither the G13L nor G1E mutants altered either the membrane free volume (see Fig. S1 B) or k2 (Table 1), while wild-type peptide both increased k2 and decreased free volume. Thus, the wild-type HA fusion peptide appears to increase k2 according to its ability to occupy space and lower interstice free energy of the TMC structure.
Just as for stalk formation (A to I1), pore formation (I2 to FP) involves a change in topology and therefore must be accomplished through some sort of an unstable disordered transition state in which water and lipid molecules are in contact. Thus, it is reasonable to presume that the wild-type fusion peptide might stabilize such a transition state as it does for the transition state leading to I1. However, just as for I1 formation, we have too little information about the transition state for final pore formation to understand how the peptide accomplishes this. We note, however, that the wild-type peptide increased the probability of content mixing early in the fusion process (α1) and increased the extent of content mixing (%CM) while the G13L and G1E mutants had lesser, no, or opposite effects (see Table 1).
The extent of content mixing reflects all the content mixing events that occur by the time that content mixing saturates. Content mixing occurs either when a stable fusion pore forms, or when a porelike fluctuation in an intermediate allows ANTS and DPX to pass between vesicles or to escape to the external compartment (leakage). In either case, the effect of the peptide must be to stabilize pore-like structures relative to other structures comprising intermediates in the fusion process. The stalk, TMC, and porelike structures are proposed as intermediates between lamellar and either hexagonal II or cubic phases (38). We show elsewhere that HA fusion peptide promotes cubic phase over hexagonal and lamellar phase to a much greater extent than do the G13L and G1E HA mutant peptides (B. G. Tenchov, D. P. Siegel, R. C. MacDonald, M. E. Haque, and B. R. Lentz, unpublished, and B. R. Lentz, University of North Carolina at Chapel Hill, personal communication, 2011). The origin of HA fusion peptide's ability to promote cubic phase is not established. It could be due to bending modulus reduction (12), to induced negative intrinsic curvature (10), to reduced interstice volume, or to a reduction in Gaussian modulus. Despite this uncertainty, the experimental fact is that wild-type HA fusion peptide might also promote content mixing by stabilizing a final fusion pore or perhaps by stabilizing porelike fluctuations in fusion intermediates that could account for content mixing observed before formation of a final fusion pore.
In summary, the effects of HA fusion peptide on the kinetics of content mixing (thus fusion) are likely attributed to:
-
1.
Stabilization of transition states between the A and I1 or the I2 and FP states;
-
2.
Filling hydrophobic volume (thus lowering interstice free energy) in the high free energy TMC structure and increasing k2; and
-
3.
Stabilizing porelike fluctuations in fusion intermediates or the final FP state, as suggested by its ability to promote cubic phase formation (B. G. Tenchov, D. P. Siegel, R. C. MacDonald, M. E. Haque, and B. R. Lentz, unpublished, and B. R. Lentz, University of North Carolina at Chapel Hill, personal communication, 2011).
In support of this proposal, the mutant peptides G1E and G13L do not exhibit effects 1 or 2, and promote cubic phase to a much smaller extent than does the wild-type peptide (B. G. Tenchov, D. P. Siegel, R. C. MacDonald, M. E. Haque, and B. R. Lentz, unpublished, and B. R. Lentz, University of North Carolina at Chapel Hill, personal communication, 2011). Confirmation of the first effect will require further experiments to establish the effects of fusion peptide on thermodynamics of transition states between fusion intermediates.
Acknowledgments
We thank Drs. Howard Fried and Michael Bruno for critical reading of the article. Thanks also to Dr. David Siegel for useful and lively discussions on the topic of lipid phase behavior and pore formation.
This research was supported by the National Institute of General Medical Sciences (grant No. 32707 to B.R.L.).
Footnotes
Md. Emdadul Haque's present address is Department of Biology, Duke University, Durham, NC.
Tilen Koklic's present address is Laboratory of Biophysics (EPR Center), Jozef Stefan Institute, Ljubljana, Slovenia.
Supporting Material
References
- 1.White J.M. Viral and cellular membrane fusion proteins. Annu. Rev. Physiol. 1990;52:675–697. doi: 10.1146/annurev.ph.52.030190.003331. [DOI] [PubMed] [Google Scholar]
- 2.Leikina E., Markovic I., Kozlov M.M. Delay of influenza hemagglutinin refolding into a fusion-competent conformation by receptor binding: a hypothesis. Biophys. J. 2000;79:1415–1427. doi: 10.1016/S0006-3495(00)76393-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiley D.C., Skehel J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 4.Gething M.J., Doms R.W., White J. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J. Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haque M.E., McCoy A.J., Lentz B.R. Effects of hemagglutinin fusion peptide on poly(ethylene glycol)-mediated fusion of phosphatidylcholine vesicles. Biochemistry. 2001;40:14243–14251. doi: 10.1021/bi011308l. [DOI] [PubMed] [Google Scholar]
- 6.Epand R.M., Epand R.F., Ruysschaert J.M. Membrane interactions of mutated forms of the influenza fusion peptide. Biochemistry. 2001;40:8800–8807. doi: 10.1021/bi0107187. [DOI] [PubMed] [Google Scholar]
- 7.Han X., Bushweller J.H., Tamm L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 8.Haque M.E., Koppaka V., Lentz B.R. Properties and structures of the influenza and HIV fusion peptides on lipid membranes: implications for a role in fusion. Biophys. J. 2005;89:3183–3194. doi: 10.1529/biophysj.105.063032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorieau J.L., Louis J.M., Bax A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc. Natl. Acad. Sci. USA. 2010;107:11341–11346. doi: 10.1073/pnas.1006142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epand R.M., Epand R.F. Relationship between the infectivity of influenza virus and the ability of its fusion peptide to perturb bilayers. Biochem. Biophys. Res. Commun. 1994;202:1420–1425. doi: 10.1006/bbrc.1994.2089. [DOI] [PubMed] [Google Scholar]
- 11.Zemel A., Ben-Shaul A., May S. Modulation of the spontaneous curvature and bending rigidity of lipid membranes by interfacially adsorbed amphipathic peptides. J. Phys. Chem. B. 2008;112:6988–6996. doi: 10.1021/jp711107y. [DOI] [PubMed] [Google Scholar]
- 12.Tristram-Nagle S., Nagle J.F. HIV-1 fusion peptide decreases bending energy and promotes curved fusion intermediates. Biophys. J. 2007;93:2048–2055. doi: 10.1529/biophysj.107.109181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozlovsky Y., Kozlov M.M. Stalk model of membrane fusion: solution of energy crisis. Biophys. J. 2002;82:882–895. doi: 10.1016/S0006-3495(02)75450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malinin V.S., Lentz B.R. Energetics of vesicle fusion intermediates: comparison of calculations with observed effects of osmotic and curvature stresses. Biophys. J. 2004;86:2951–2964. doi: 10.1016/S0006-3495(04)74346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel D.P. The Gaussian curvature elastic energy of intermediates in membrane fusion. Biophys. J. 2008;95:5200–5215. doi: 10.1529/biophysj.108.140152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haque M.E., Lentz B.R. Influence of gp41 fusion peptide on the kinetics of poly(ethylene glycol)-mediated model membrane fusion. Biochemistry. 2002;41:10866–10876. doi: 10.1021/bi020269q. [DOI] [PubMed] [Google Scholar]
- 17.Weinreb G., Lentz B.R. Analysis of membrane fusion as a two-state sequential process: evaluation of the stalk model. Biophys. J. 2007;92:4012–4029. doi: 10.1529/biophysj.106.090043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumoto T. Membrane destabilizing activity of influenza virus hemagglutinin-based synthetic peptide: implications of critical glycine residue in fusion peptide. Biophys. Chem. 1999;79:153–162. doi: 10.1016/s0301-4622(99)00051-4. [DOI] [PubMed] [Google Scholar]
- 19.Reference deleted in proof.
- 20.Haque M.E., McIntosh T.J., Lentz B.R. Influence of lipid composition on physical properties and PEG-mediated fusion of curved and uncurved model membrane vesicles: “nature's own” fusogenic lipid bilayer. Biochemistry. 2001;40:4340–4348. doi: 10.1021/bi002030k. [DOI] [PubMed] [Google Scholar]
- 21.Lentz B.R., McIntyre G.F., Massenburg D. Bilayer curvature and certain amphipaths promote poly(ethylene glycol)-induced fusion of dipalmitoylphosphatidylcholine unilamellar vesicles. Biochemistry. 1992;31:2643–2653. doi: 10.1021/bi00125a003. [DOI] [PubMed] [Google Scholar]
- 22.Siegel D.P. The modified stalk mechanism of lamellar/inverted phase transitions and its implications for membrane fusion. Biophys. J. 1999;76:291–313. doi: 10.1016/S0006-3495(99)77197-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgess S.W., McIntosh T.J., Lentz B.R. Modulation of poly(ethylene glycol)-induced fusion by membrane hydration: importance of interbilayer separation. Biochemistry. 1992;31:2653–2661. doi: 10.1021/bi00125a004. [DOI] [PubMed] [Google Scholar]
- 24.Lee J., Lentz B.R. Evolution of lipidic structures during model membrane fusion and the relation of this process to cell membrane fusion. Biochemistry. 1997;36:6251–6259. doi: 10.1021/bi970404c. [DOI] [PubMed] [Google Scholar]
- 25.Malinin V.S., Lentz B.R. Pyrene cholesterol reports the transient appearance of nonlamellar intermediate structures during fusion of model membranes. Biochemistry. 2002;41:5913–5919. doi: 10.1021/bi011924h. [DOI] [PubMed] [Google Scholar]
- 26.Chernomordik L.V., Kozlov M.M. Membrane hemifusion: crossing a chasm in two leaps. Cell. 2005;123:375–382. doi: 10.1016/j.cell.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 27.Yang L., Huang H.W. Observation of a membrane fusion intermediate structure. Science. 2002;297:1877–1879. doi: 10.1126/science.1074354. [DOI] [PubMed] [Google Scholar]
- 28.Alvarez de Toledo G., Fernandez-Chacon R., Fernandez J.M. Release of secretory products during transient vesicle fusion. Nature. 1993;363:554–558. doi: 10.1038/363554a0. [DOI] [PubMed] [Google Scholar]
- 29.Chanturiya A., Chernomordik L.V., Zimmerberg J. Flickering fusion pores comparable with initial exocytotic pores occur in protein-free phospholipid bilayers. Proc. Natl. Acad. Sci. USA. 1997;94:14423–14428. doi: 10.1073/pnas.94.26.14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koppaka V., Lentz B.R. Binding of bovine factor Va to phosphatidylcholine membranes. Biophys. J. 1996;70:2930–2937. doi: 10.1016/S0006-3495(96)79863-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J.R., Lentz B.R. Mechanism of poly(ethylene glycol)-induced lipid transfer between phosphatidylcholine large unilamellar vesicles: a fluorescent probe study. Biochemistry. 1991;30:6780–6787. doi: 10.1021/bi00241a022. [DOI] [PubMed] [Google Scholar]
- 32.Stubbs C.D., Ho C., Slater S.J. Fluorescence techniques for probing water penetration into lipid bilayers. J. Fluor. 1995;5:19–28. doi: 10.1007/BF00718779. [DOI] [PubMed] [Google Scholar]
- 33.Lee J., Lentz B.R. Outer leaflet-packing defects promote poly(ethylene glycol)-mediated fusion of large unilamellar vesicles. Biochemistry. 1997;36:421–431. doi: 10.1021/bi9622332. [DOI] [PubMed] [Google Scholar]
- 34.Chattopadhyay A., London E. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 1987;26:39–45. doi: 10.1021/bi00375a006. [DOI] [PubMed] [Google Scholar]
- 35.Slater S.J., Kelly M.B., Stubbs C.D. The modulation of protein kinase C activity by membrane lipid bilayer structure. J. Biol. Chem. 1994;269:4866–4871. [PubMed] [Google Scholar]
- 36.Biron Z., Khare S., Anglister J. A monomeric 3(10)-helix is formed in water by a 13-residue peptide representing the neutralizing determinant of HIV-1 on gp41. Biochemistry. 2002;41:12687–12696. doi: 10.1021/bi026261y. [DOI] [PubMed] [Google Scholar]
- 37.Suurkuusk J., Lentz B.R., Thompson T.E. A calorimetric and fluorescent probe study of the gel-liquid crystalline phase transition in small, single-lamellar dipalmitoylphosphatidylcholine vesicles. Biochemistry. 1976;15:1393–1401. doi: 10.1021/bi00652a007. [DOI] [PubMed] [Google Scholar]
- 38.Siegel D.P., Green W.J., Talmon Y. The mechanism of lamellar-to-inverted hexagonal phase transitions: a study using temperature-jump cryo-electron microscopy. Biophys. J. 1994;66:402–414. doi: 10.1016/s0006-3495(94)80790-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.