

Abstract
Eukaryotic DNA ligases are ATP-dependent DNA strand-joining enzymes that participate in DNA replication, repair, and recombination. Whereas mammalian cells contain several different DNA ligases, encoded by at least three distinct genes, only one DNA ligase has been detected previously in either budding yeast or fission yeast. Here, we describe a newly identified nonessential Saccharomyces cerevisiae gene that encodes a DNA ligase distinct from the CDC9 gene product. This DNA ligase shares significant amino acid sequence homology with human DNA ligase IV; accordingly, we designate the yeast gene LIG4. Recombinant LIG4 protein forms a covalent enzyme-AMP complex and can join a DNA single-strand break in a DNA/RNA hybrid duplex, the preferred substrate in vitro. Disruption of the LIG4 gene causes only marginally increased cellular sensitivity to several DNA damaging agents, and does not further sensitize cdc9 or rad52 mutant cells. In contrast, lig4 mutant cells have a 1000-fold reduced capacity for correct recircularization of linearized plasmids by illegitimate end-joining after transformation. Moreover, homozygous lig4 mutant diploids sporulate less efficiently than isogenic wild-type cells, and show retarded progression through meiotic prophase I. Spore viability is normal, but lig4 mutants appear to produce a higher proportion of tetrads with only three viable spores. The mutant phenotypes are consistent with functions of LIG4 in an illegitimate DNA end-joining pathway and ensuring efficient meiosis.
Keywords: DNA repair, illegitimate recombination, meiosis, DNA ligase IV, CDC9
Mammalian cells contain several different nuclear DNA ligases. These enzymes share related catalytic domains but in addition have unique regions that appear to be required for interactions with other proteins, therefore conferring functional specificity. DNA ligase I is an essential replication factor that accounts for joining of Okazaki fragments during lagging-strand DNA synthesis (Barnes et al. 1990; Waga et al. 1994; Petrini et al. 1995; Mackenney et al. 1997) and may also be involved in some forms of DNA repair and recombination (Barnes et al. 1992; Prigent et al. 1994). DNA ligase III (Wei et al. 1995) interacts strongly with the DNA repair protein XRCC1 and appears to take part in base excision–repair (Thompson et al. 1990; Kubota et al. 1996). An alternatively spliced, testis-specific form of DNA ligase III has also been identified that could be active in meiotic recombination (Chen et al. 1995). DNA ligase II, a smaller DNA ligase present in nonproliferating cells, may be yet another alternatively spliced form of DNA ligase III (Roberts et al. 1994). During our attempts to clone a cDNA for DNA ligase III, a separate open reading frame (ORF) was also identified that encodes a previously unrecognized enzyme, with an extended carboxy-terminal region not present in other ATP-dependent ligases, DNA ligase IV (Wei et al. 1995). Active DNA ligase IV was subsequently isolated from HeLa cell nuclei where it occurs as a heterodimer with an unidentified partner; the function of DNA ligase IV is presently unknown (Robins and Lindahl 1996).
The only mammalian DNA ligase for which a functional assignment can be made with some certainty is the replication factor DNA ligase I. For this reason, detection of yeast counterparts of DNA ligases III and/or IV, and functional investigations of mutants defective in these activities, would be of considerable interest. Unfortunately, only a single gene for DNA ligase has been discovered in either Saccharomyces cerevisiae or Schizosaccharomyces pombe, CDC9 versus cdc17+, and the sequences of their products suggest strongly that they are counterparts to mammalian DNA ligase I (Barker et al. 1985, 1987). Conditional-lethal S. cerevisiae cdc9 or S. pombe cdc17− mutants, when grown at a permissive temperature, accumulate Okazaki fragments during DNA replication, are hypersensitive to several DNA-damaging agents, exhibit enhanced mitotic intergenic recombination, and sporulate with reduced efficiency (Johnston and Nasmyth 1978; Sipiczki et al. 1990). Biochemical investigations also showed that the CDC9 gene product is a functional homolog of mammalian DNA ligase I, and suggested further the presence of a second DNA ligase activity in S. cerevisiae cells (Johnston and Nasmyth 1978; Tomkinson et al. 1992; Ramos et al. 1997). Yet, even when the complete sequence of the S. cerevisiae genome initially became available in 1996, no ORF encoding a homolog of either DNA ligase III or IV could be detected. When we performed a more detailed investigation, however, sequence motifs typical of DNA ligase IV were revealed and an ORF masked by sequencing errors could be identified. Here we describe this gene, and properties of defective yeast mutants. The most striking phenotype of a S. cerevisiae LIG4 disruption is a greatly impaired ability to recircularize a transformed linearized plasmid by DNA double-strand break repair.
Results
The LIG4 gene and its product
The DNA sequence initially deposited in the yeast genome database described two adjacent ORFs (Sterky et al. 1996) close to the centromere on the left arm of chromosome XV, one of them (UND407) showing homology to the unique carboxy-terminal region of human DNA ligase IV (Wei et al. 1995), and the other (UNE452) to part of the catalytic domain of ATP-dependent DNA ligases. This information prompted us to re-examine the DNA sequence of the entire region covering the two putative ORFs. Sequence analysis of multiple, independently subcloned DNA fragments amplified from genomic S. cerevisiae DNA revealed one continuous ORF, which is present in the current S. cerevisiae genome database as ORF YOR005C. On chromosome XV, this ORF extends from an ATG translation start at nucleotide position 337343 (Crick strand) to an in-frame TGA translation stop 2833 bp downstream (position 334511) and has coding potential for a 109-kD protein of 944 amino acids. The deduced amino acid sequence of the S. cerevisiae LIG4 protein (scLIG4) displays significant homology to human DNA ligase IV (hsLIG4, 25% identity score) and shares less, but still significant, sequence homology with CDC9 of S. cerevisiae (scCDC9, 21% identity), human DNA ligase I (hsLIG1, 21% identity) and human DNA ligase III (hsLIG3, 19% identity). Recently, a Candida albicans gene encoding a DNA ligase was identified and called CDC9 (Andaluz et al. 1996). However, the protein encoded by this gene is only distantly related to S. cerevisiae CDC9 (20% identity) and shows greater homology to scLIG4 (33% identity). The presence of a characteristic extended carboxy-terminal region also strongly indicates that this caLIG is a homolog of scLIG4 and hsLIG4. These relationships are displayed in Figure 1A.
Figure 1.


Relationship between the S. cerevisiae LIG4 gene product and those of other eukaryotic ATP-dependent DNA ligases. Sources of proteins compared are caLIG (C. albicans CDC9 EMBL accession no. X95001); scLIG4 (S. cerevisiae LIG4, SGD, YOR005C); hsLIG4 (Homo sapiens DNA ligase IV, EMBL accession no. X83441); hsLIG1 (Homo sapiens DNA ligase I, GenBank accession no. M36067); scCDC9 (S. cerevisiae CDC9, EMBL accession no. X03246); hsLIG3 (Homo sapiens DNA ligase III, EMBL accession no. X84740). The amino acid sequences were aligned and the phylogenetic relationship calculated with the PILEUP and BESTFIT programs of the Genetic Computer Group (Devereux et al. 1984); gap weight, 5; gap length weight, 0.1. (A) Phylogenetic relationship of the scLIG4 protein to eukaryotic DNA ligase I, III, and IV type enzymes. (B) Amino acid sequence alignment of the yeast and human DNA ligase I and IV type enzymes. Identical residues are boxed. Shaded boxes denote positions of sequence motifs that are highly conserved between all ATP-dependent DNA ligases. The position of the adenylated lysine residue is residue 282 in scLIG4. Underscored are the carboxy-terminal regions that define the BRCT domains as identified by hydrophobic cluster analysis.
The predicted amino acid sequence of scLIG4, aligned with human DNA ligase I and IV and the S. cerevisiae CDC9 gene product, is shown in Fig. 1B. The ATG337343 is likely to be the translation start: (1) presence of multiple translation stops in all frames between this and the next in-frame upstream ATG; (2) translation starting at the next downstream ATG (residue 60) would remove conserved amino-terminal peptide sequence (23% identity to the amino terminus of the C. albicans LIG protein). The scLIG4 protein shares a common core domain with both human DNA ligases and scCDC9 that includes a number of highly conserved motifs characteristic of nucleotidyl transferases such as DNA ligases and mRNA capping enzymes (Shuman and Schwer 1995). Among them is the minimal active site consensus for all ATP-dependent ligases, K-DG-R (residues 282–287 of scLIG4) (Tomkinson et al. 1991), which together with the conserved regions around Glu-340, Phe-382, and Lys-464 of scLIG4 forms the basis of the nucleotide binding pocket (Subramanya et al. 1996). scLIG4 also shows six of 16 amino acid residues that define a highly conserved consensus peptide; three of them form the motif R-DK (residues 624–627 of scLIG4), which is found in all ATP-dependent DNA ligases. Outside the core domain, DNA ligases I and IV are different (Fig. 1B) and unlike DNA ligase I and scCDC9, human DNA ligase IV and the scLIG4 have long carboxy-terminal extensions. Although their similarity is not striking at the primary structure level, hydrophobic cluster analyses have revealed that of both these carboxy-terminal regions contain a tandem array of two “BRCT” sequence motifs, a feature identified in several DNA repair factors including S. cerevisiae RAD9 and REV1 and S. pombe Rad4 (Cut5), as well as in human BRCA1 (Callebaut and Mornon 1997). Such motifs have been directly implicated in specific protein–protein interactions (Nash et al. 1997).
S. cerevisiae LIG4 encodes a protein with catalytic properties of a DNA ligase
The reaction mechanism of DNA ligases is well characterized. In the first step of the reaction, eukaryotic enzymes use ATP to form a covalent ligase–adenylate complex through the active site lysine residue (Lindahl and Barnes 1992). The activated AMP moiety of the DNA ligase–adenylate intermediate is then transferred to the 5′ phosphoryl end of a strand break in double-stranded DNA to form a DNA–AMP complex. Finally, the generation of a phosphodiester bond is catalyzed by unadenylated DNA ligase with concomitant release of the AMP residue. To establish such catalytic properties for the scLIG4 gene product, it was overexpressed in Escherichia coli and purified as an amino-terminal histidine-tagged version of the protein (His–LIG4). Bacterial overexpression of His–LIG4 in different systems produced mainly insoluble matter (>98%), but small amounts of soluble, active protein could be recovered after growth at low temperatures. The soluble lysate fractions of E. coli cells expressing His–LIG4 contained a number of polypeptides which bound tightly to a nickel–agarose affinity matrix, and were recovered specifically by elution with buffer containing 500 mm imidazole (Fig. 2A, lanes 3–5) after extensive washing (Fig. 2A, lanes 1,2). In parallel control fractionations of lysates from E. coli grown with the expression vector only, no detectable protein was recovered (Fig. 2A, lane 6). Two major His–LIG4 polypeptides of apparent masses of 108 and 75 kD were present in the imidazole eluate, the larger being close to the calculated molecular mass of the full-length His–LIG4 protein (111 kD), the lower most likely being a carboxy-terminal truncation of ∼300 amino acids. On incubation with [α-32P]ATP, both polypeptides formed a covalent enzyme–AMP complex as revealed by SDS-PAGE (Fig. 2A, lanes 9–11). No such activity was observed in corresponding vector control fractions (Fig. 2A, lane 12). Moreover, E. coli DNA ligase uses NAD instead of ATP as a cofactor and would not be detected in these experiments, and assays with [32P]NAD confirmed the absence of E. coli DNA ligase in the His–LIG4 imidazole eluate.
Figure 2.
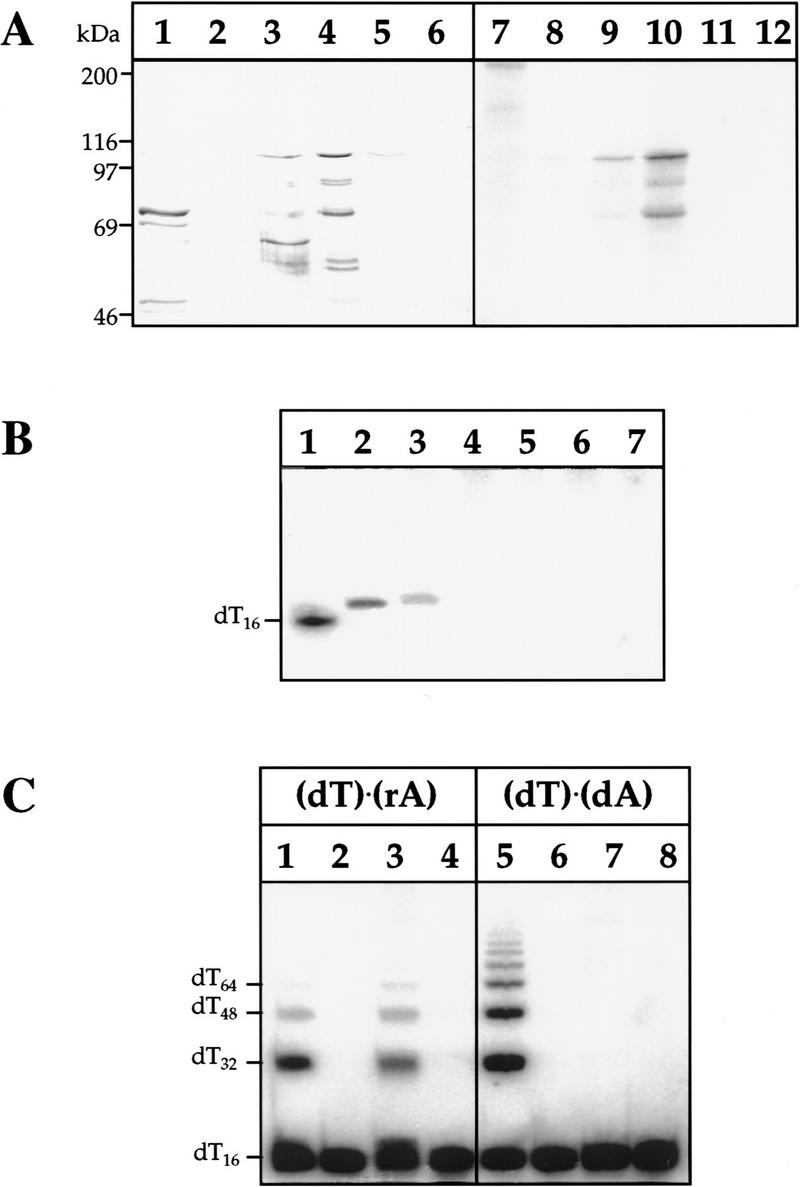
Recombinant scLIG4 protein shows the catalytic properties of an ATP-dependent DNA ligase. (A) Co-purification of histidine-tagged scLIG4 protein with enzyme-adenylate forming activity. The left panel (lanes 1–6) shows the protein content in relevant nickel column fractions (17 μl aliquots), by silver-staining after SDS–polyacrylamide gel electrophoresis (7.5%). The right panel (lanes 7–12) shows the same fractions (4 μl aliquots) assayed for enzyme–AMP complex formation as revealed after autoradiography. (Lanes 1,7) First 80 mm imidazole wash; (lanes 2,8) second 80 mm imidazole wash; (lanes 3,9) first 500 mm imidazole elution; (lanes 4,10) second 500 mm imidazole elution; (lanes 5,11) third 500 mm imidazole elution; (lanes 6,12) second 500 mm imidazole elution (corresponding to lanes 4 and 10) of a parallel vector control purification from E. coli BL21(DE3) cells with the pET16b vector alone. (B) Formation of DNA–adenylate intermediate. Reactions were carried out as described in Materials and Methods, and products were analyzed by denaturing polyacrylamide gel electrophoresis. (Lane 1) 32P-labeled molecular mass standard (dT)16; (lane 2), 1 mU of T4 DNA ligase incubated with oligo (dT)16 ⋅ poly(rA); (lanes 3–7) 6 μl of scLIG4 incubated with oligo (dT)16 ⋅ poly(rA); [5′-OH] oligo (dT)16 ⋅ poly(rA); oligo (dT)16 ⋅ poly(dA); [5′-OH] oligo (dT)16 ⋅ poly(dA); and oligo (dT)16, respectively. (C) Ligation of polynucleotide substrates. Reactions were carried out as described in Materials and Methods. Lanes 1–4 show ligation assayed with [5-32P] oligo (dT)16 ⋅ poly(rA), lanes 5–8 with [5-32P] oligo (dT)16 ⋅ poly(dA). Enzymes were added as follows; (lanes 1,5) T4 DNA ligase controls (1 mU and 0.1 mU, respectively); (lanes 2,6) no enzyme; (lanes 3,7) 6 μl scLIG4; (lanes 4,8) 6 μl of corresponding vector control fractions.
For further analysis of the second and third steps of the ligation reaction, we used the dialyzed peak fraction of the His–LIG4 imidazole eluate and the vector control material (Fig. 2A, lanes 4,10 and 6,12). The His–LIG4 produced a DNA–AMP intermediate with the hybrid substrate oligo (dT)16 ⋅ poly(rA) (Fig. 2B, lane 3). No intermediates could be trapped with the same substrate lacking the 5′ phosphate in the (dT)16 component (Fig. 2B, lane 4), using an oligo (dT)16 ⋅ poly(dA) substrate (Fig. 2B, lanes 5,6), or single-strand (dT)16 (Fig. 2B, lane 7). Similarly, scLIG4 ligated (dT)16 to multimers when annealed to a poly(rA) complementary strand (Fig. 2C, lane 3), but not detectably so with a poly(dA) strand (Fig. 2C, lane 7). Phage T4 DNA ligase, used as a control, could ligate both substrates (Fig. 2C, lanes 1 and 5). The overexpressed scLIG4 enzyme did not join detectably EcoRI-cut plasmid DNA (data not shown); these data differ from in vivo results on efficient rejoining of linearized plasmid DNA by scLIG4 (see below, Fig. 4). Possibly, like human DNA ligase IV, scLIG4 occurs as a heterodimer in vivo (Robins and Lindahl 1996) so that only partial biochemical activity is observed with the monomer component.
Figure 4.

S. cerevisiae lig4 mutant cells are deficient in recircularizing linearized plasmid DNA. Plasmid transformation assays were carried out as detailed in Materials and Methods using either supercoiled, EcoRI-digested, or SmaI-digested pBTM116 plasmid DNA for transformation. (A) Schematic map of the yeast replicative plasmid pBTM116, carrying Amp for selection in E. coli and the yeast TRP1 gene as a selectable yeast marker. A multiple cloning sequence including the relevant EcoRI and SmaI sites is located within a stretch of DNA sequence which shares no homology with S. cerevisiae genomic sequences. This DNA is flanked by transcription control elements of yeast ADH1 (pADH1, tADH1). Numbers below the map indicate the distance in base pairs of relevant plasmid elements from the EcoRI cut site, and the arrows mark the annealing sites of the PCR primers used for analysis of plasmid repair events. (B) Graphic illustration of relative transformation efficiencies (ratios cut/uncut plasmids) obtained with EcoRI- (left) and SmaI-cut plasmid. Data are averages from at least three independent transformation experiments in which matching test and control strains were treated in parallel. Strains used LIG4, FF18734; lig4, PRSY003,1; rad52, FF18743; lig4 rad52; PRSY005; lig4/pLIG4, PRSY003,1 carrying a LIG4-expressing plasmid (pPRS156); lig4/pYES2, PRSY003,1 carrying the expression vector only.
LIG4 is not essential and mutants are only marginally hypersensitive to DNA damage
A heterozygous LIG4 gene disruption was generated in a diploid strain (PRSY001) by replacing the LIG4 ORF (amino acid residues 10–931) with a kanamycin resistance module employing the short homology PCR strategy described by Wach et al. (1994). Geneticin resistant transformants were examined for correct heterozygous gene replacement by PCR and Southern blot analysis (data not shown) and then sporulated to produce haploid meiotic progeny. Thirty of 42 dissected tetrads from two independent isolates (PRSY002,1-2) produced four viable spores, with the Geneticin resistance segregating 2:2 in all cases, suggesting a nonessential function for the LIG4 gene. The mutant phenotypes of haploid lig4 segregants (PRSY003,1, PSRY004,2) were investigated further and the same strains were used for construction of lig4 rad52 and cdc9 lig4 double mutants by crossing (see Materials and Methods, Table 2, below).
Table 2.
S. cerevisiae strains used
| Strain
|
Genotype
|
Source
|
|---|---|---|
| FF18734 | α leu2-3 trp1-289 ura3-52 his7-2 lys1-1 | F. Fabre (Institut Curie, Paris, France) |
| FF18743 | α lue2-3 trp1-289 ura3-52 his7-2 lys1-1 rad52::URA3 | F. Fabre |
| FF18984 | a leu2-3 ura3-52 his7-1 lys2-1 | F. Fabre |
| PRSY001 | diploid, FF18743/FF18984 | FF18743 × FF18984 |
| PRSY002,n | PRSY001 but LIG4/lig4::kanMX4 | PRSY001 transformants |
| PRSY003,1 | α leu2-3 trp1-289 ura3-52 his7 lys1-1 lig4::kanMX4 | PRSY002,1 spore clone |
| PRSY004,2 | a leu2-3 ura3-52 his7 lys2-1 lig4::kanMX4 | PRSY002,2 spore clone |
| PRSY005 | α leu2-3 trp1-289 ura3-52 his7 lys1-1 lig4::kanMX4 rad52::URA3 | FF18743 × PRSY004,2 |
| PRSY006 | a leu2-3 ura3-52 his7 lys2-1 lig4::kanMX4 rad52::URA3 | FF18743 × PRSY004,2 |
| L22-12C | α ade1/2 cdc9-1 his1 lys2 | L. Johnston (National Institute for Medical Research, London, UK) |
| PRSY009 | α leu2 his7-2 lys1-1 cdc9-1 | PRSY004,2 × L22-12C (× FF18734 > 87% FF18734) |
| PRSY011 | α leu2 his7-2 lys1-1 cdc9-1 lig4::kanMX4 | PRSY004,2 × L22-12C (× FF18734 > 87% FF18734) |
lig4 single mutants did not show any effect on mitotic growth or on cell cycle progression as established by parallel examination of growth rates and distribution patterns of unbudded, small budded, premitotic, and postmitotic large budded cells in vegetative cultures (data not shown). They showed no temperature-sensitive phenotype up to 37°C nor any cold sensitivity. We also examined the effect of the LIG4 disruption in exponentially growing haploid cells on resistance to DNA damage induced by exposure to ultraviolet light (UV), γ-ray irradiation or by incubation in the presence of methyl methanesulfonate (MMS). lig4 mutant cells were slightly, but insignificantly more sensitive to DNA damage induced by high doses of UV light (>120 J/m2), ionizing radiation, or MMS than isogenic wild-type cells (Fig. 3). Also, cdc9 lig4 double mutants were not significantly more sensitive to the DNA-damaging agents than cdc9 or lig4 single mutants, indicating an absence of synergism between the two DNA ligases (Fig. 3). Because MMS- and ionizing radiation-induced DNA damage may be repaired predominantly by homologous recombination in yeast, we investigated the possibility that repair defects associated with the lack of the LIG4 function might be manifest only in genetic backgrounds where homologous recombination is nonfunctional. As expected, rad52 single mutant cells were highly sensitive to MMS and ionizing radiation. Additional inactivation of lig4 in rad52 mutants did not result in further sensitization of the cells, however (Fig. 3B,C); in fact, rad52 appears to be epistatic to lig4.
Figure 3.

Sensitivity of lig4 mutant cells to DNA damaging agents. Cells were exposed to UV, MMS, or ionizing radiation as indicated. Survival data are shown from at least three independent experiments. Standard deviations are shown where relevant for judgment of differences between strains. Matching test and control strains were always assayed in parallel as detailed in Materials and Methods. The strains used are listed with genotypes in Table 2 and are referred to in the figure as (wt) FF18734 and FF18984 (▪); (lig4) PRSY003,1 and PRSY004,2 (□); (rad52) FF18743 (♦); (lig4 rad52) PRSY005, PRSY006 (⋄); (cdc9) PRSY009 (•); and (cdc9 lig4) PRSY011 (○).
LIG4 is involved in nonhomologous double-strand break joining
To investigate whether LIG4 might be involved in nonhomologous joining of double-strand breaks in vivo, we performed plasmid rescue assays (Boulton and Jackson 1996b). Briefly, the yeast plasmid pBTM116 was digested with restriction enzymes in a region without sequence homology to chromosomal DNA (Fig. 4A), and completion of digestion was verified by Southern blotting (data not shown). Competent yeast cells were then transformed in parallel with limiting amounts of cut or uncut plasmid DNA, and the number of transformants was determined after selection for a plasmid-expressed genetic marker (TRP1). As plasmid replication and establishment of a TRP+ phenotype depends on successful religation of the cut plasmids, the transformation efficiency obtained with linear versus circular plasmid is a relative measure of the double-strand break repair capacity of a yeast strain.
Transformation data with EcoRI-digested plasmid DNA are illustrated in Figure 4B. LIG4 wild-type cells were highly efficient in recircularizing linearized plasmids with 5′ overlapping cohesive ends. In contrast, lig4 mutant cells showed a dramatic 300-fold reduced relative transformation efficiency with EcoRI-digested plasmid, whereas transformation with supercoiled plasmid was as efficient as in the LIG4 wild-type cells. The same result was obtained in several different experiments and was independent of the overall transformation efficiency when limiting amounts of DNA were used. The deficiency in recircularizing EcoRI-cut plasmid was fully complemented in lig4 mutant cells by expression of the His–LIG4 fusion ORF carried on a yeast episomal vector (pPRS156) under the control of a GAL1 promoter. No complementation was observed in parallel experiments with cells carrying the expression vector only (pYES2, Fig. 4B). Homologous recombination does not engage in joining of EcoRI ends in vivo, as rad52 mutants did not show significantly reduced transformation efficiencies with linear plasmid. rad52 lig4 double mutants, however, behaved as lig4 single mutants and joined EcoRI ends with a similarly reduced efficiency (Fig. 4B).
Transformation experiments with SmaI-digested blunt-ended plasmid DNA produced essentially similar results, but the effects were much less pronounced (Fig. 4B); this was mainly because LIG4 wild-type cells were reduced 42-fold in relative transformation efficiency with blunt-ended plasmids as compared with cohesive-ended plasmids. Interestingly, this difference depended on the LIG4 function as lig4 mutants joined cohesive- and blunt-ended plasmid substrates with equal efficiency, but were still significantly (fivefold) less efficient in blunt-end joining than LIG4 wild-type cells (Fig. 4B). RAD52-dependent recombination events may contribute more to blunt-end than to cohesive-end ligation, as the difference between RAD52 wild-type and mutant cells increased slightly when rescue of SmaI-cut plasmid was assayed. Again, rad52 lig4 double mutant cells produced relative transformation efficiencies with blunt-ended plasmids indistinguishable from those of the lig4 single mutants.
To characterize the molecular nature of events that led to the establishment of tryptophan prototrophy after transformation of cells with EcoRI digested pBTM116, we analyzed 35 LIG4 wild-type and 48 lig4 mutant transformants by PCR amplification of a plasmid segment spanning the putative EcoRI junction, EcoRI digestion, and DNA sequencing of the fragments, and by plasmid stability assays and Southern blotting (data not shown). All LIG4 wild-type transformants analyzed had acquired an unstable TRP+ phenotype by correct religation of the plasmid. The situation in the lig4 mutants was strikingly different and only 23% of the rare lig4 mutant transformants analyzed showed a correct religation of the EcoRI-cut plasmid. In 4%, plasmid rejoining was associated with exonucleolytic degradation of DNA ends resulting in the loss of the EcoRI site and flanking nucleotides. In 61% of the mutant transformants, the entire ORF of the ADH1 gene had been converted into the plasmid by homologous recombination through ADH1 promoter and terminator sequences (Fig. 4A). Associated with this process was the loss of the heterologous sequences immediately flanking the EcoRI site on either side. Stability assays and Southern analysis of these transformants showed further that half of them (28% of all) contained a plasmid that circularized by homologous double-strand gap repair, whereas the other half (33% of all) carried an integrated copy of the gap-repaired plasmid at the ADH1 locus (data not shown). Interestingly, these events appeared to be RAD52 dependent and were not detected in transformants of the lig4 rad52 double mutant. The remaining 12% of the lig4 mutant transformants analyzed represented more complex events. Some of them were stable but had no integrated plasmid sequence suggesting that they were TRP1 convertants.
LIG4-deficient diploids are delayed in meiosis I
DNA ligase functions are required not only in the vegetative state but also during meiotic differentiation of S. cerevisiae a/α diploids. To assess meiotic effects of a lig4 disruption, we examined the sporulation capacity of homozygous and heterozygous LIG4 wild-type and mutant diploids in meiotic time-course experiments, and viability of meiotic progeny in spore tetrads. Progression through meiosis was monitored by withdrawing aliquots of cells from sporulating populations at different times post-induction, and establishing distribution patterns of meiocytes containing 1, 2, 3, or 4 DAPI-staining bodies in at least 200 randomly counted cells per time point. There were significant differences between wild-type and mutant diploids before meiosis I (one DAPI body) and past meiosis II (four DAPI bodies), whereas the distribution of cells with two and three DAPI bodies was the same. Comparison of the cell class distribution showed that lig4 mutant diploids are specifically delayed in processes before meiosis I, but once they have passed this stage they appear to be able to complete meiosis without difficulty. The meiosis I delay is best illustrated by comparing the accumulated fractions of cells past meiosis I (two, three, and four DAPI bodies) (Fig. 5). Moreover, lig4/lig4 homozygotes sporulated less efficiently (53% completed meiosis after 72 hr) than isogenic homozygous LIG4/LIG4 (87% after 72 hr), or heterozygous LIG4/lig4 (81% after 72 hr) diploids (Fig. 5). Spore viability in the sporulating fraction of homozygous lig4 mutant cells was normal (Table 1) and heterozygous marker alleles segregated 2:2. However, examination of the distribution of tetrads with 4, 3, 2, 1, and 0 viable spores in homozygous LIG4 wild-type and mutant crosses revealed that LIG4-deficient diploids produced an increased proportion of tetrads with only three viable spores correlated with a decreased fraction of tetrads with four viable products (Table 1).
Figure 5.

LIG4-deficient diploids are delayed in meiosis I. Fractions of cells in sporulating populations that have passed meiosis I, as evidenced by DAPI staining, are shown as a function of time postinduction. Analyses were performed as described in Materials and Methods and averages from two independent experiments are given. The values represent accumulated fractions of cells with two or more DAPI staining bodies in the sporulating populations. Strains are referred to as follows: (LIG4/LIG4) diploid FF18734/FF18984 (▪); (LIG4/lig4) diploids FF18734/PRSY004,2 and PRSY003,1/FF18984 (□); (lig4/lig4) diploid PRSY003,1/PRSY004,2 (♦).
Table 1.
Spore viability in tetrad analysis
| Cross
|
Relevant genotype
|
Viable : dead spores in tetrads
|
Spore viability (%)
|
|||||
|---|---|---|---|---|---|---|---|---|
|
n
|
4 : 0
|
3 : 1
|
2 : 2
|
1 : 3
|
0 : 4
|
|||
| FF18734 × FF18984 | LIG4/LIG4 | 98 | 87* | 6* | 4 | 1 | 0 | 96 |
| 89% | 6.1% | 4.1% | 1.0% | |||||
| PRSY003 × PRSY004 | lig4/lig4 | 106 | 84* | 16* | 5 | 1 | 0 | 93 |
| 79% | 15% | 4.7% | 0.9% | |||||
Spore tetrads from two independent diploids were analyzed. Full genotypes of strains are listed in Table 2. An asterisk (*) indicates tetrad classes with a significantly different distribution in wild-type and mutant diploids (χ2 = 5.122, 2 × 2 tables, χ2P0.05 = 3.84).
Discussion
A previously unrecognized S. cerevisiae gene encoding a protein whose predicted amino acid sequence shows a high degree of homology to ATP-dependent DNA ligases has been identified. We designate the gene LIG4 because it forms a distinct group of DNA ligases together with human DNA ligase IV and a C. albicans DNA ligase, whereas it is related more distantly to CDC9, the yeast homolog of human DNA ligase I. LIG4 encodes an enzyme catalyzing reactions characteristic of ATP-dependent DNA ligases, and disruption of the gene causes a phenotype that is consistent with a function of the gene product in nonhomologous joining of DNA double-strand breaks.
The LIG4 protein is an ATP-dependent DNA ligase
The LIG4 protein overexpressed in E. coli forms a covalent enzyme–AMP complex when incubated with ATP, a characteristic first step of the DNA ligation reaction of eukaryotic DNA ligases. Size determination of the radioactively labeled enzyme–AMP complex by SDS-PAGE showed that LIG4 is a larger protein than the 87-kD CDC9 DNA ligase, and that LIG4 migrated at the rate expected for the 109-kD protein predicted from the ORF shown in Figure 1B (111 kD for the His-tagged protein). The enzyme could also transfer the AMP residue to a DNA strand break and ligate the nick, as expected for a DNA ligase. However, LIG4 only showed efficient activity in vitro when the complementary strand in the double helix was a polyribonucleotide rather than a polydeoxyribonucleotide; this most likely explains the failure of several research groups to detect a separate DNA ligase activity in extracts of S. cerevisiae cdc9 mutant cells, when a standard substrate of nicked DNA was employed for enzyme assays. The unusual substrate specificity in vitro of purified monomeric LIG4 may not be fully representative of the enzyme activity in vivo but does not appear to be a consequence of the amino-terminal His–tag, as the tagged enzyme expressed in lig4 mutant yeast cells was able to complement their plasmid repair deficiency (Fig. 4B).
Role for LIG4 in RAD52-independent double strand break repair and in progression through meiotic prophase I
The LIG4 gene is not essential; lig4 mutant haploid and diploid cells grow at normal rates, are respiratory-competent, and show normal cell cycle progression. Therefore, it may be concluded that this DNA ligase is not involved in DNA replication, nor in maintenance of mitochondria in S. cerevisiae. Surprisingly, lig4 single mutants display only marginally increased sensitivity to UV, the alkylating agent MMS, or ionizing radiation. S. cerevisiae predominantly repairs UV damage by the nucleotide excision repair pathway represented by the RAD3 epistasis group of genes, whereas MMS- and ionizing radiation-induced lesions are predominantly repaired by the homologous recombination pathway controlled by the RAD52 epistasis group (for review, see Petes et al. 1991; Haber 1992a; Friedberg et al. 1995). The absence of significantly increased sensitivities in lig4 mutant cells suggests that LIG4 does not have an important role in either of these pathways, and the fact that no synergistic effect on cellular sensitivity was observed in a cdc9 lig4 double mutant indicates that back-up compensation of a LIG4 defect by the CDC9 DNA ligase in these pathways does not occur. As rad52 single and lig4 rad52 double mutants are equally sensitive to ionizing radiation, there is no redundancy with the recombinational double-strand break repair pathway. In conclusion, LIG4 does not appear to be active in excision-repair and mitotic homologous recombination processes in S. cerevisiae.
A strong phenotype was revealed when lig4 mutant cells were challenged with a specific DNA substrate for ligation; cells lacking the LIG4 function were dramatically impaired in their capacity to rejoin nonhomologous DNA ends of linearized replicative plasmids on transformation, and this repair pathway is RAD52 independent. Nonhomologous, RAD52-independent double-strand break repair modes in S. cerevisiae have been observed previously, and it was shown that they require the functions of at least three genes, RAD50, XRS2, and MRE11 (Schiestl and Petes 1991; Kramer et al. 1994; Mezard and Nicolas 1994; Schiestl et al. 1994; Moore and Haber 1996). At least two pathways appear to exist for nonhomologous recircularization of linearized plasmids on transformation; one of them accurately joins ends without the loss of nucleotide sequence and engages the yeast Ku70 and Ku80 homologs as accessory factors (illegitimate end-joining), whereas the other one results in the loss of variable amounts of terminal DNA sequence (illegitimate recombination) (Mezard and Nicolas 1994; Boulton and Jackson 1996a,b). Our analysis of the double-strand break repair events in the LIG4 wild-type background showed that all transformants contained precisely religated plasmids independent of whether the linearized plasmids had cohesive- or blunt-ended termini or whether the host cells were RAD52 wild-type or mutant. In contrast, only 23% of the scarce lig4 mutant transformants contained accurately religated plasmids, which might be accounted for by inefficient background ligation provided by the CDC9 DNA ligase, and most (77%) were apparently channeled into alternative double-strand break repair pathways. In conclusion, our data are consistent with a critical role for the LIG4 DNA ligase in a RAD52-independent illegitimate DNA end-joining pathway, which shows a strong preference for DNA ends with short homologies such as the single-stranded protruding sequences generated by some restriction enzymes.
Homozygous lig4 mutants sporulate less efficiently than isogenic wild-type diploids. Reduced sporulation efficiency is a common phenotype of many genes encoding components of meiotic structures, or factors involved in cell division cycle control or DNA replication, repair, and homologous recombination; among them CDC9, the yeast DNA ligase I homolog and its fission yeast counterpart cdc17+ (Simchen 1974; Sipiczki et al. 1990). The fact that lig4 mutants are specifically impaired before meiosis I could suggest a function of LIG4 in premeiotic DNA replication or meiotic prophase I. However, premeiotic DNA replication must have been completed accurately in the 42% of lig4 diploids that produce asci with four viable haploid spores. Therefore, it seems more likely that LIG4 functions in the subsequent meiotic prophase I, where homologous pairing of chromosomes is accompanied by initiation of high levels of homologous recombination by introduction of specific transient double-strand breaks and synaptonemal complex formation (for review, see Petes et al. 1991; Nicolas and Petes 1994; Roeder 1995). The formation and rejoining of specific double-strand breaks is essential for correct alignment of homologous chromosomes, without which proper segregation cannot occur, and it might engage more than one double-strand break repair pathway. It appears that LIG4 is not essential for meiosis but beneficial to its efficiency and accuracy. A minor deficiency in chromosome segregation would be consistent with the lig4 mutant phenotype; more detailed investigations of meiosis in lig4 mutants will clarify this point.
Comparison with Ku of S. cerevisiae and mechanistic implications
There are interesting parallels and differences between the mutant phenotypes of the LIG4 gene and those of the recently identified S. cerevisiae homologs of the mammalian Ku70 and Ku80 proteins (Feldmann and Winnacker 1993; Boulton and Jackson 1996a,b; Mages et al. 1996; Milne et al. 1996; Siede et al. 1996; Tsukamoto et al. 1996; Barnes and Rio 1997). Ku mutants display marked hypersensitivity to DNA damage in the absence of functional homologous recombination. In a plasmid repair assay, Ku mutants are only partly deficient in recircularizing a plasmid with cohesive ends and rejoin a blunt-ended plasmid more efficiently. If both LIG4 and Ku are involved in illegitimate DNA end-joining, one possible explanation of the apparent discrepancies in their mutant phenotypes could be that Ku proteins have a nonspecific DNA end protection role, whereas LIG4 might recognize a much more limited range of DNA substrates; if such specific DNA end arrangements are only rarely generated in consequence to DNA-damaging treatment with UV, MMS, or ionizing radiation, it would be expected that the contribution of LIG4 to repair of such damage is unimportant. Recently, Teo and Jackson (1997) showed that yku70 lig4 double mutant strains are not further impaired in rejoining of DNA double-strand breaks compared with lig4 or yku70 single mutants, suggesting that both proteins may function together in some forms of nonhomologous recombination.
Several DNA metabolic processes are known to generate DNA structural intermediates that could be substrates for LIG4. One is mating type switching, a mitotic gene conversion process initiated by introduction of a specific transient double-strand break in the chromosome, which is then repaired by means of homologous recombination (for review, see Haber 1992b). Another possibility is meiotic recombination, which is associated with introduction of specific double-strand breaks at recombination hotspots (for review, see Nicolas and Petes 1994); the meiotic phenotypes described here could be consistent with an involvement of LIG4 in repair of such double-strand breaks. Furthermore, a cdc17 DNA ligase-defective mutant of S. pombe with impaired DNA replication, excision–repair, and mitotic recombination exhibits both unaffected meiotic recombination and mating type switching (Sipiczki et al. 1990), suggesting that a separate DNA ligase is involved. A third possibility for a role of LIG4 is in site-specific recombination, in particular RNA-mediated transposition of Ty elements. The preferential joining of DNA strands in a DNA/RNA hybrid double helix by the purified enzyme would be compatible with such a role. Retrotransposition is known to involve both the formation of DNA/RNA hybrid duplexes during the process of reverse transcription, and integration of cDNA into chromosomes mediated by precise cutting and rejoining of DNA double strands (Derr et al. 1991).
The present data on yeast lig4 mutants suggest that mammalian DNA ligase IV may also be a nonessential DNA ligase involved in specific recombination processess. Construction of knockout mice deficient in this enzyme should elucidate further the physiological roles of the type IV group of DNA ligases, now identified both in yeast and in mammalian cells.
Materials and methods
Genetic methods
Yeast complete medium (YPD), pre-sporulation medium, and synthetic drop-out media were prepared as described (Sherman et al. 1982). Media for selection of respiration-proficient cells and sporulation medium have been described by Bähler et al. (1994). For selection of Geneticin resistance, 0.02% Geneticin G-418 (Gibco BRL) was employed in YPD. Strains were propagated and sporulated at 30°C except for cdc9 mutants, which were grown at 25°C. Transformations were performed by a slight modification of the high-efficiency lithium acetate method (Gietz and Schiestl 1991).
Yeast strains
The S. cerevisiae strains used in this study are listed in Table 2. With the exception of L22–12C, they are all isogenic derivatives of two closely related, congenic series represented by FF18734 and FF18984 in an A364A background (F. Fabre, pers. comm.) and were obtained by transformation and crossing within the set as indicated in Table 2. cdc9 mutants (PRSY009, PRSY011) were obtained by crossing L22–12C with PRSY004,2 and made congenic by backcrossing three times with FF18734. Appropriate genotypes were isolated from dissected spore tetrads using standard replica-plating techniques. The LIG4 gene disruption was generated by standard gene replacement techniques using a PCR-derived disruption construct consisting of the kanMX4 module (Wach et al. 1994) flanked by 60 bp of homologous DNA sequences from the 5′ and 3′ ends of the LIG4 ORF. This was gel-purified and 1 μg was used for transformation of PRSY001. Geneticin-resistant transformants were genotyped by replica-plating onto diagnostic media, by standard genomic PCR, and by Southern blot analysis. Verified transformants carrying the expected heterozygous replacement of the LIG4 ORF with the kanMX4 cassette (PRSY002,n) were then sporulated and haploid progeny with appropriate genotypes identified in dissected spore tetrads using standard genetic and PCR genotyping techniques.
Plasmids
pPRS150 is pGEM–5Zf(+) (Promega) carrying the original PCR-amplified LIG4 ORF (as described below). pPRS154 was used for expression of recombinant scLIG4 protein in E. coli with an amino-terminal histidine tag and was constructed by subcloning the entire LIG4 ORF (the 2.8-kb NdeI fragment of pPRS150) into the NdeI site of pET16b (Novagen). pPRS156 was used for complementation of the plasmid repair phenotype in lig4 mutants and was constructed by subcloning the NcoI–BamHI fragment of pPRS154, containing the entire His–LIG4 fusion ORF, into the HindIII–BamHI sites of the yeast expression vector pYES2 (Invitrogen), using an appropriate synthetic HindIII–NcoI adapter. Plasmid pFA6a–kanMX4 (Wach et al. 1994) was provided by Dr. W.D. Heyer (University of Bern, Switzerland) and was used as template for PCR-amplification of the LIG4 disruption construct. pBTM116 was provided by Dr. S. Jackson (Wellcome/Cancer Research Campaign Institute, Cambridge, UK) and was used for plasmid rescue assays; it is an E. coli–yeast shuttle vector carrying 2μ sequences for stable maintenance, the TRP1 gene as a selectable marker, and the promoter and terminator sequences of the ADH1 gene flanking a multiple cloning sequence (P. Bartel and S. Fields, unpubl.).
PCR cloning and sequence analyses
The entire LIG4 ORF was amplified by PCR of genomic DNA isolated from FF18743. The primers were designed (sense primer 5′-GCATGCATCATATGATATCAGCACTAGATTCTATACC-3′; antisense primer, 5′-GCATGCATCATATGTCAGTAGTTGACTACGGGG-3′) to anneal across the LIG4 translation start and stop (underscored), respectively, and NdeI restriction sites were added (italic) to facilitate subcloning of the PCR products. PCR products were then purified (QIAquick PCR Purification Kit, Qiagen), NdeI-digested, and subcloned into pGEM–5Zf(+) (Promega) after agarose gel purification. The resultant construct (pPRS150) was verified by automated DNA sequencing using an ABI 377 DNA Sequencer. DNA and protein sequence analyses were performed with the Genetics Computer Group program package, Version 8, 1994 (Devereux et al. 1994). For general database searches and comparisons, we used the BLAST, FASTA, and ENTREZ services provided at NCBI’s world wide web page; for yeast genome database searches we accessed MIPS and SGD through their web pages.
Bacterial expression and purification of scLIG4
Recombinant, histidine-tagged scLIG4 protein was expressed in E. coli BL21(DE3) from pPRS154 under control of the T7 RNA polymerase promoter. Precultures of 120-ml Luria broth containing 100 μg/ml ampicillin were inoculated with six freshly transformed BL21(DE3)/pPRS154 colonies, incubated overnight at 30°C with vigorous shaking, and used for inoculation of 6 liters of culture. Cultures were grown at 30°C to an OD600 of 0.7–0.8, then shifted to 16°C and incubated further for 24 hr. Cells were harvested by centrifugation and stored as pellets at −80°C. Frozen cell pellets were thawed on ice, resuspended in three volumes of lysis buffer (50 mm Tris-HCl, 300 mm NaCl, 10% glycerol, 0.25% Tween 20, 5 mm β-mercaptoethanol, 1 mm imidazole, 1 mm phenylmethanesulfonyl fluoride, at pH 8.0) and sonicated on ice for five cycles of 30 sec with intermittent chilling. After centrifugation of the cell lysate (20 min, 4°C, 15,000g), soluble proteins in the supernatant were batch adsorbed to 1 ml of pre-equilibrated (lysis buffer) Ni-NTA agarose (Qiagen) at 4°C for 1 hr. The Ni-NTA resin was packed into a disposable column (BioRad) and washed twice with 15 volumes of lysis buffer supplemented with 80 mm imidazole. Finally, bound proteins were eluted with lysis buffer containing 500 mm imidazole. LIG4 peak fractions were identified by SDS–polyacrylamide gel electrophoresis and dialyzed against 50 mm Tris-HCl, 60 mm NaCl, 1 mm EDTA, 5 mm dithiothreitol, 10% glycerol (pH 7.5).
Enzyme assays
Ligase–AMP formation
scLIG4 protein fractions (4 μl) or T4 DNA ligase were adenylated in reaction mixtures (20 μl) containing 60 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 50 μg/ml of bovine serum albumin, 5 mm dithiothreitol, and 1 μCi of [α-32P]ATP (3000 Ci/mmole; Amersham) at 20°C for 15 min, and then analyzed by electrophoresis in SDS-7.5% polyacrylamide gels followed by autoradiography or phosphorimaging.
DNA–AMP formation Reaction mixtures (15 μl) as above were supplemented with 0.5 μg nonradio-labeled substrate DNA and scLIG4 protein fractions (6 μl) or T4 DNA ligase (Fig. 2B). Reactions were stopped by adding three volumes of 90% formamide-dyes, and heating to 95°C for 5 min. The reaction products were separated in 20% denaturing polyacrylamide gels and identified by autoradiography or phosphorimaging.
Ligation
DNA ligation was assayed with the double-stranded polymer substrates [5′-32P]oligo(dT)16⋅poly(dA) and [5′-32P]oligo(dT)16⋅poly(rA). The substrates were made as described (Prigent et al. 1994). Samples (6 μl) of scLIG4 fractions were incubated in reaction mixtures (15 μl) containing 60 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 50 μg/ml bovine serum albumin, 5 mm dithiothreitol, 1 mm ATP, and 0.1 μg of polynucleotide substrate (20,000–50,000 cpm) at 25°C. After 3 hr incubation, the reactions were stopped by adding 90% formamide-dyes, heating at 95°C for 5 min and immediately chilling on ice. 32P-labeled oligo(dT)16 multimers were resolved by electrophoresis through denaturing 15% polyacrylamide gels and detected by autoradiography or phosphorimaging.
Mitotic growth and sensitivity to DNA-damaging agents
Mitotic growth was examined in YPD cultures by inoculating 105 cells/ml from stationary phase cultures and incubating at 30°C with shaking. At indicated times, samples were removed from the cultures, and titers of viable cells were determined by plating onto YPD agar plates and incubation at 30°C for 3 days. The distribution of cells in G1, S phase, G2, and mitosis was assessed microscopically by cell morphology (budding state of cells) and by staining of nuclear DNA with DAPI (4′,6′-diamidino-2-phenylindol).
UV sensitivity
YPD cultures (20 ml) were inoculated with 3 × 106 cells/ml from stationary phase precultures and grown to mid-log phase (107 cells/ml) at 30°C with shaking. After plating appropriate dilutions onto YPD agar plates (200–400 cells/plate), cells were exposed to UV light (254 nm UV Lamp XX–1552, Ultra Violet Products Ltd., 3 Jm−2sec−1) for varying times. To avoid photoreactivation, plates were incubated in the dark immediately after irradiation. Incubations were at 30°C for 3–4 days, except for cdc9 mutants, which were either incubated at 25°C only, or for 1 hr at 37°C immediately after irradiation and then at 25°C for 3–4 days.
MMS sensitivity
Mid-log-phase cultures were grown as described above. Cells were collected by centrifugation at room temperature and washed in 70 mm sodium phosphate buffer (pH 6.5), at 30°C. They were resuspended at 107cells/ml in the same buffer containing 2% glucose. MMS was added to the desired final concentration, and the cells were incubated at 30°C, or 25°C in experiments involving cdc9 mutants. Samples were removed at intervals as indicated, diluted 104-fold in H2O and plated onto YPD agar plates 1 hr after dilution. Surviving clones were counted after 3–4 days incubation at 30°C. cdc9 mutants were either kept at 25°C throughout the procedure or incubated for 1 hr at 37°C after dilution with prewarmed H2O.
Sensitivity to γ-irradiation
Mid-log phase cells were grown and plated onto YPD agar plates as described above. Cells were exposed to different doses of γ-irradiation (137Cs γ-beam, IBL437C irradiator, CIS Bio International) at a rate of 2.82 Gy/min. Three plates were irradiated at each dose and incubated at 30°C for 3–4 days before counting surviving clones.
Plasmid rescue assays
pBTM116 DNA was digested to completion with either EcoRI or SmaI restriction enzymes (New England Biolabs) as verified by agarose gel electrophoresis and Southern blotting. Restriction enzymes were inactivated by incubation at 65°C for 20 min, and plasmid DNA was purified by extraction with phenol/chloroform/isoamyl alcohol (25:24:1) followed by ethanol precipitation. Samples of undigested plasmid were subjected to the same procedure except that restriction enzymes were omitted. Equal amounts (160 ng) of undigested and digested plasmid DNA were used for parallel transformation of competent yeast cells. Serial dilutions were plated on medium lacking tryptophan and TRP+ transformants were counted after 4 days of incubation at 30°C. Plasmids were analyzed in individual transformants by standard colony PCR amplification (Jesnowski et al. 1995) of a 1170-bp segment spanning the putative EcoRI junction, and PCR products were sequenced as described above. The same transformants were assayed for plasmid stability. Selective precultures were grown in medium lacking tryptophan. Ten milliliter (nonselective) YPD cultures were inoculated with 1% of precultures and grown for another 24 hr at 30°C. Approximately 200 cells were then plated onto YPD–agar and grown for 3 days at 30°C. After replica-plating the colonies onto selective media and incubating for another 3 days at 30°C, the stability of tryptophan prototrophy was analyzed. For Southern blot analysis, cells from the selective preculture were pelleted and total cellular DNA was prepared. DNA (5 μg), either undigested or digested with EcoRI or PstI was separated by agarose gel electrophoresis and analyzed by standard Southern blot hybridization.
Meiotic time course analysis and spore viability
Diploids were constructed freshly for each sporulation experiment by mating haploid parental strains of the lys1-1 and lys2-2 series, respectively, in 1-ml YPD liquid cultures followed by selection for LYS+ complementation on agar plates lacking lysine. Before sporulation, respiration-efficient diploids were selected in glycerol medium. Sporulation was then performed in liquid cultures at 30°C essentially as described by Padmore et al. (1991) starting from freshly isolated single colonies on YPD plates. Samples were withdrawn at different intervals, and cells fixed in 70% ethanol for DAPI staining of nuclear DNA. Examination of meiotic progression was done by bright-field and fluorescence microscopy. In parallel, spore viability was analyzed in tetrads dissected on YPD agar plates after incubation for 4–5 days at 30°C.
Acknowledgments
We thank Deborah Barnes and Christine Mezard for helpful discussions, Steve Jackson for providing plasmid pBTM116, and Francis Fabre, Wolf-Dietrich Heyer, Noel Lowndes, and Alain Nicolas for yeast strains and technical advice. P.S. was supported by an EMBO long-term fellowship and G.H. by grant He 2675/1-1 from the Deutsche Forschungsgemeinschaft.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lindahl@icrf.icnet.uk; FAX 44-0-171-269-3819.
References
- Andaluz E, Larriba G, Calderone R. A Candida albicans gene encoding a DNA ligase. Yeast. 1996;12:893–898. doi: 10.1002/(SICI)1097-0061(199607)12:9%3C893::AID-YEA973%3E3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Bähler J, Hagens G, Holzinger G, Schertan H, Heyer W-D. Saccharomyces cerevisiae cells lacking the homologous pairing protein p175SEP1 arrest at pachytene during meiotic prophase. Chromosoma. 1994;103:129–141. doi: 10.1007/BF00352322. [DOI] [PubMed] [Google Scholar]
- Barker DG, White JH, Johnston LH. The nucleotide sequence of the DNA ligase gene (CDC9) from Saccharomyces cerevisiae: A gene which is cell-cycle regulated and induced in response to DNA damage. Nucleic Acids Res. 1985;13:8323–8337. doi: 10.1093/nar/13.23.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Molecular characterisation of the DNA ligase gene, CDC17, from the fission yeast Schizosaccharomyces pombe. Eur J Biochem. 1987;162:659–667. doi: 10.1111/j.1432-1033.1987.tb10688.x. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Johnston LH, Kodama K, Tomkinson AE, Lasko DD, Lindahl T. Human DNA ligase I cDNA: Cloning and functional expression in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1990;87:6679–6683. doi: 10.1073/pnas.87.17.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Tomkinson AE, Lehmann AR, Webster ADB, Lindahl T. Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell. 1992;69:495–503. doi: 10.1016/0092-8674(92)90450-q. [DOI] [PubMed] [Google Scholar]
- Barnes G, Rio D. DNA double strand break sensitivity, DNA replication, and cell cycle arrest phenotypes of Ku-deficient Saccharomyces cerevisiae. Proc Natl Acad Sci. 1997;94:867–872. doi: 10.1073/pnas.94.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue; Roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 1996a;24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996b;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- Callebaut I, Mornon JP. From BRCA1 to RAP1: A widespread BRCT module closely associated with DNA repair. FEBS Lett. 1997;400:25–30. doi: 10.1016/s0014-5793(96)01312-9. [DOI] [PubMed] [Google Scholar]
- Chen J, Tomkinson AE, Ramos W, Mackey ZB, Danehower S, Walter CA, Schultz RA, Besterman JM, Husain I. Mammalian DNA ligase III: Molecular cloning, chromosomal localization, and expression in spermatocytes undergoing meiotic recombination. Mol Cell Biol. 1995;15:5412–5422. doi: 10.1128/mcb.15.10.5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derr LK, Strathern JN, Garfinkel DJ. RNA-mediated recombination in S. cerevisiae. Cell. 1991;67:355–364. doi: 10.1016/0092-8674(91)90187-4. [DOI] [PubMed] [Google Scholar]
- Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;16:7025–7042. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H, Winnacker EL. A putative homologue of the human autoantigen Ku from Saccharomyces cerevisiae. J Biol Chem. 1993;268:12895–12900. [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, D.C.: American Society for Microbiology; 1995. pp. 662–669. [Google Scholar]
- Gietz RD, Schiestl RH. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast. 1991;7:253–263. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- Haber JE. Exploring the pathways of homologous recombination. Curr Opin Cell Biol. 1992a;4:401–412. doi: 10.1016/0955-0674(92)90005-w. [DOI] [PubMed] [Google Scholar]
- ————— Mating-type gene switching in Saccharomyces cerevisiae. Trends Genet. 1992b;8:446–452. doi: 10.1016/0168-9525(92)90329-3. [DOI] [PubMed] [Google Scholar]
- Jesnowski R, Naehring J, Wolf K. A rapid and reliable method for PCR-based amplification of chromosomal and mitochodrial DNA from intact yeast cells. Curr Genet. 1995;27:318–319. doi: 10.1007/BF00352100. [DOI] [PubMed] [Google Scholar]
- Johnston LH, Nasmyth KA. Saccharomyces cerevisiae cell cycle mutant cdc9 is defective in DNA ligase. Nature. 1978;274:891–893. doi: 10.1038/274891a0. [DOI] [PubMed] [Google Scholar]
- Kramer KM, Brock JA, Bloom K, Moore K, Haber JE. Two different types of double strand breaks in Saccharomyces cerevisiae are repaired by similar RAD52 independent, nonhomologous recombination events. Mol Cell Biol. 1994;14:1293–1301. doi: 10.1128/mcb.14.2.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Nash RA, Klungland A, Schär P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase β and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Barnes DE. Mammalian DNA ligases. Annu Rev Biochem. 1992;61:251–281. doi: 10.1146/annurev.bi.61.070192.001343. [DOI] [PubMed] [Google Scholar]
- Mackenney VJ, Barnes DE, Lindahl T. Specific function of DNA ligase I in SV40 DNA replication by human cell-free extracts is mediated by the amino terminal non-catalytic domain. J Biol Chem. 1997;272:11550–11556. doi: 10.1074/jbc.272.17.11550. [DOI] [PubMed] [Google Scholar]
- Mages GJ, Feldmann HM, Winnacker EL. Involvement of Saccharomyces cerevisiae HDF1 gene in DNA double-strand break repair and recombination. J Biol Chem. 1996;271:7910–7915. doi: 10.1074/jbc.271.14.7910. [DOI] [PubMed] [Google Scholar]
- Mezard C, Nicolas A. Homologous, homeologous, and illegitimate repair of double-strand breaks during transformation of a wild-type strain and a rad52 mutant strain of Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:1278–1292. doi: 10.1128/mcb.14.2.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GT, Jin S, Shannon KB, Weaver DT. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4189–4198. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash RA, Caldecott KW, Barnes DE, Lindahl T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry. 1997;36:5207–5211. doi: 10.1021/bi962281m. [DOI] [PubMed] [Google Scholar]
- Nicolas A, Petes TD. Polarity of meiotic gene conversion in fungi: Contrasting views. Experientia. 1994;50:242–252. doi: 10.1007/BF01924007. [DOI] [PubMed] [Google Scholar]
- Padmore R, Cao L, Kleckner N. Temporal comparison of recombination and synaptonemal complex formation during meiosis in S. cerevisiae. Cell. 1991;66:1239–1256. doi: 10.1016/0092-8674(91)90046-2. [DOI] [PubMed] [Google Scholar]
- Petes TD, Malone RE, Symington LS. Recombination in yeast. In: Broach JR, Jones E, Pringle J, editors. The molecular and cellular biology of the yeast Saccharomyces: Genome dynamics, protein synthesis and energetics. Vol. 1. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1991. pp. 407–521. [Google Scholar]
- Petrini JHJ, Xiao Y, Weaver DT. DNA ligase I mediates essential functions in mammalian cells. Mol Cell Biol. 1995;15:4303–4308. doi: 10.1128/mcb.15.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent C, Satoh MS, Daly G, Barnes DE, Lindahl T. Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol Cell Biol. 1994;14:310–317. doi: 10.1128/mcb.14.1.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos W, Tappe N, Talamantez J, Friedberg EC, Tomkinson AE. Two distinct DNA ligase activities in mitotic extracts of the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 1997;25:1485–1492. doi: 10.1093/nar/25.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E, Nash RA, Robins P, Lindahl T. Different active sites of mammalian DNA ligases I and II. J Biol Chem. 1994;269:3789–3792. [PubMed] [Google Scholar]
- Robins P, Lindahl T. DNA ligase IV from HeLa cell nuclei. J Biol Chem. 1996;271:24257–24266. doi: 10.1074/jbc.271.39.24257. [DOI] [PubMed] [Google Scholar]
- Roeder GS. Sex and the single cell: Meiosis in yeast. Proc Natl Acad Sci. 1995;92:10450–10456. doi: 10.1073/pnas.92.23.10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Petes TD. Integration of DNA fragments by illegitimate recombination in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1991;88:7585–7589. doi: 10.1073/pnas.88.17.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Zhu J, Petes TD. Effect of mutations in genes affecting homologous recombination on restriction enzyme-mediated and illegitimate recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:4493–4500. doi: 10.1128/mcb.14.7.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F, Fink GK, Hicks JB. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- Shuman S, Schwer B. RNA capping enzyme and DNA ligase: A superfamily of covalent nucleotide transferases. Mol Microbiol. 1995;17:405–410. doi: 10.1111/j.1365-2958.1995.mmi_17030405.x. [DOI] [PubMed] [Google Scholar]
- Siede W, Friedl AA, Dianova I, Eckardt-Schupp F, Friedberg EC. The Saccharomyces cerevisiae Ku autoantigen homologue affects radiosensitivity only in the absence of homologous recombination. Genetics. 1996;142:91–102. doi: 10.1093/genetics/142.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchen G. Are mitotic functions required in meiosis? Genetics. 1974;76:745–753. doi: 10.1093/genetics/76.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipiczki M, Grossenbacher-Grunder AM, Bodi Z. Recombination and mating-type switching in a ligase-deficient mutant of Schizosaccharomyces pombe. Mol & Gen Genet. 1990;220:307–313. [Google Scholar]
- Sterky F, Holmberg A, Pettersson B, Uhlén M. The sequence of a 30 kb fragment on the left arm of chromosome XV from Saccharomyces cerevisiae reveals 15 open reading frames, five of which correspond to previously identified genes. Yeast. 1996;12:1091–1095. doi: 10.1002/(SICI)1097-0061(199609)12:10B%3C1091::AID-YEA22%3E3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Subramanya HS, Doherty AJ, Ashford SR, Wigley DB. Crystal structure of an ATP-dependent DNA ligase from bacteriophage T7. Cell. 1996;85:607–615. doi: 10.1016/s0092-8674(00)81260-x. [DOI] [PubMed] [Google Scholar]
- Teo, S.-H. and S.P. Jackson. 1997. Identification of Saccharomyces cerevisiae DNA ligase IV: Involvement in DNA double-strand-break repair. EMBO J. (in press). [DOI] [PMC free article] [PubMed]
- Thompson LH, Brookman KW, Jones NJ, Allen SA, Carrano AV. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol Cell Biol. 1990;10:6160–6171. doi: 10.1128/mcb.10.12.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkinson AE, Totty NF, Ginsburg M, Lindahl T. Location of the active site for enzyme-adenylate formation in DNA ligases. Proc Natl Acad Sci. 1991;88:400–404. doi: 10.1073/pnas.88.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkinson AE, Tappe NJ, Friedberg EC. DNA ligase I from Saccharomyces cerevisiae: Physical and biochemical characterization of the CDC9 gene product. Biochemistry. 1992;31:11762–11771. doi: 10.1021/bi00162a013. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Kato J-i, Ikeda H. Hdf1, a yeast Ku-protein homologue, is involved in illegitimate recombination, but not in homologous recombination. Nucleic Acids Res. 1996;24:2067–2072. doi: 10.1093/nar/24.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Waga S, Bauer G, Stillman B. Reconstitution of complete SV40 DNA replication with purified replication factors. J Biol Chem. 1994;269:10923–10934. [PubMed] [Google Scholar]
- Wei Y-F, Robins P, Carter K, Caldecott K, Pappin DJC, Yu G-L, Wang R-P, Shell BK, Nash RA, Schär P, Barnes DE, Haseltine WA, Lindahl T. Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination. Mol Cell Biol. 1995;15:3206–3216. doi: 10.1128/mcb.15.6.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]