

Abstract
Cyanine fluorophores are commonly used in single-molecule FRET experiments with nucleic acids. We have previously shown that indocarbocyanine fluorophores attached to the 5′-termini of DNA and RNA via three-carbon atom linkers stack on the ends of the helix, orienting their transition moments. We now investigate the orientation of sulfoindocarbocyanine fluorophores tethered to the 5′-termini of DNA via 13-atom linkers. Fluorescence lifetime measurements of sulfoindocarbocyanine 3 attached to double-stranded DNA indicate that the fluorophore is extensively stacked onto the terminal basepair at 15°C, with properties that depend on the terminal sequence. In single molecules of duplex DNA, FRET efficiency between sulfoindocarbocyanine 3 and 5 attached in this manner is modulated with helix length, indicative of fluorophore orientation and consistent with stacked fluorophores that can undergo lateral motion. We conclude that terminal stacking is an intrinsic property of the cyanine fluorophores irrespective of the length of the tether and the presence or absence of sulfonyl groups. However, compared to short-tether indocarbocyanine, the mean rotational relationship between the two fluorophores is changed by ∼60° for the long-tether sulfoindocarbocyanine fluorophores. This is consistent with the transition moments becoming approximately aligned with the long axis of the terminal basepair for the long-linker species.
Introduction
Fluorescence resonance energy transfer (FRET) is extensively used to measure or compare distances in biological macromolecules (1), especially nucleic acids (2–5). It provides distance information in a size range suitable for biological macromolecules, and because of the sensitivity of fluorescence, it can be performed on single molecules (6). However, the interpretation of FRET data is potentially complicated by effects arising from the orientation of the fluorophores used.
FRET arises from the dipolar coupling between the transition moment vectors of fluorophores, a function of distance and orientation. The efficiency of FRET (EFRET) as a function of fluorophore separation (R) is given by Förster (7) as
| (1) |
where R0 is the distance at which energy transfer is 50% efficient. R0 depends upon the spectroscopic properties of the fluorophores and the medium, given by
| (2) |
where the units of R0 and the wavelength λ are cm. The value ϕD is the quantum yield of the donor, N is the Avogadro number, n is the index of refraction of the medium, and J(λ) is the spectral overlap between donor emission and acceptor absorption, given by
| (3) |
where ϕD(λ) is the spectral shape of donor emission and εA(λ) the extinction coefficient for acceptor absorption (M−1 cm−1). The value κ2 describes the relative orientation of the fluorophores. It is given by
| (4) |
where the angles ΘT, ΘD, and ΘA describe the relative orientation of the donor and acceptor transition dipole moments (Fig. 1 A). Uncertainty in the value of κ2 (8–10) provides the largest potential difficulty in the extraction of distance information from EFRET. The value κ2 can take values between 0 and 4, or between 0 and 1 if the transition moments are coaxial and constrained to parallel planes. If the fluorophores undergo isotropic reorientation within the excited state lifetime of the donor, then κ2 = 2/3 (9), thus simplifying the extraction of distance information. In many studies this is assumed to apply, explicitly or otherwise, but this may not necessarily be warranted. There has been renewed interest in this problem recently (11–13).
Figure 1.

Orientation of the transition moments of cyanine fluorophores attached to DNA. (A) The orientation parameter κ2. The transition dipole vectors for the coupled donor and acceptor fluorophore are indicated (arrows), labeled D and A. Vector A′ is generated by the in-plane translation of vector A to share its origin with vector D. The definition of κ2, given in Eq. 4, is based upon the angles shown. (B) Parallel-eye stereoscopic view of a model of a duplex with Cy3 and Cy5 fluorophores attached to the 5′-termini via C3 linkers. This was generated using our NMR structures of Cy3 and Cy5 attached to duplex DNA. The fluorophores lie in approximately parallel planes, and thus the angular relationship between them (and thus their transition moments) will depend on the length of the DNA helix, and its helical periodicity. (C) Schematic to illustrate the principle of the experiment in which a series of DNA duplexes terminally labeled with Cy3 and Cy5 is used. The cyanine fluorophores are represented as flat disks, with the direction of the transition moments indicated (arrows). As the helix length is increased there is a rotation of the relative angle between the two transition dipoles. The term κ2 takes the value 1 when they are parallel, and 0 when perpendicular.
The cyanine fluorophores Cy3 and Cy5 are extensively used as a FRET donor acceptor pair attached to nucleic acids, particularly in single-molecule studies (14). We have shown that the fluorophores Cy3 and Cy5 stack on the ends of DNA double helices when attached to the 5′ termini via three-methylene carbon (C3) tethers generated by coupling as phosphoramidites (15,16) (Fig. 1 B). This generates a significant orientation of the fluorophores, so that an assumption of κ2 = 2/3 is not justified. This was demonstrated by making a series of DNA and DNA-RNA duplexes labeled at their 5′-termini with Cy3 and Cy5 (12). We observed that the efficiency of energy transfer from Cy3 donor to Cy5 acceptor as a function of the length of the helix was significantly modulated in a periodic manner, with maxima and minima occurring twice per helix rotation.
The profiles of EFRET as a function of helix length could be well simulated assuming standard B- and A-form helical geometries and terminally stacked fluorophores that can undergo a significant degree of lateral mobility about a mean position deduced from NMR studies (12). Because the calculated transition moments for the π to π∗ transitions lie in the plane of the indole rings close to the poly-methine linker (16), the fluorophore positions are close to satisfying the conditions for a variation of 0 ≤ κ2 ≤ 1, and the dipoles should be perpendicular twice per helical period (i.e., κ2 = 0) (Fig. 1 C). In principle, the orientational effects could be greater if the fluorophores planes are not parallel, for example when terminally attached to a helix that is bent by some discontinuity such as a junction or bulge. In that case, κ2 could vary over its full range from 0 to 4.
In our previous studies, the cyanine fluorophores were linked to 5′-termini via a C3 linker. This is the same number of carbon atoms that exist when linked via a ribose ring, and it is possible that this constrains the fluorophores to stack on the end of the helix in the manner of an additional nucleotide base. A commonly-used alternative approach is to conjugate the fluorophores as N-hydroxysuccinimide esters to a primary amine that is linked to the 5′-terminus by a C6 tether, generating a total tether length of 13 atoms. The structures created by use of the C3-linked phosphoramidite and conjugation using the C6 linker are compared in Fig. 2.
Figure 2.
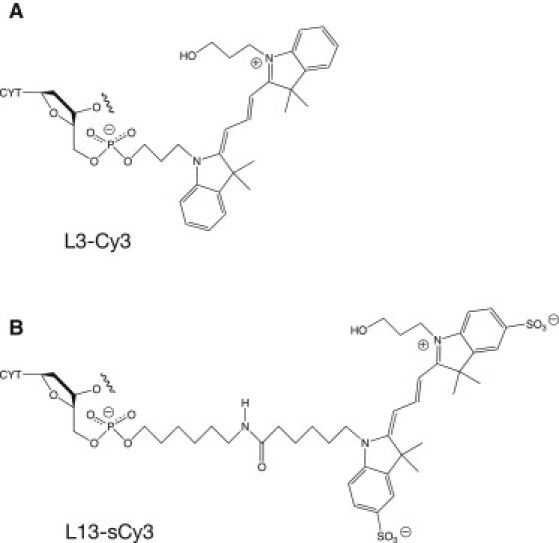
Comparison of cyanine fluorophores and their linkages to DNA. (A) L3-Cy3. Indocarbocyanine 3 attached to DNA as a phosphoramidite during synthesis is tethered via a three-methylene carbon linker. (B) L13-sCy3. Sulfoindocarbocyanine 3 contains a sulfonyl substitution in the six-membered rings of the indole systems to increase solubility. In our experiments they are attached to six-carbon 5′-amino-linker groups on the DNA by coupling as N-hydroxysuccinimide esters postsynthetically to generate the linkage shown, containing a total of 13 atoms.
In principle, the longer linker could provide much greater flexibility than the C3 tether. The structure of the fluorophores used in the two methods also differs. The Cy3 and Cy5 N-hydroxysuccinimide esters carry sulfonyl groups at the 5 and 5′ positions of the indole rings. The presence of these negative charges could affect the stacking of the fluorophores (17). The longer linker and altered structure of the fluorophore might conceivably remove a constraint that requires it to stack on the terminal basepair. On the other hand, perhaps the tendency to stack is intrinsic to the cyanine fluorophores in general, and will occur irrespective of the nature of the linkage and substitution in the indole ring. We have therefore set out to answer this question, which is of great practical significance for the interpretation of FRET experiments using these fluorophores.
We have used a 10–24 bp duplex series with 5′ sulfoindocarbocyanine 3 and 5 attached via long tethers to examine whether the orientation observed with the shorter tether is maintained. We find significant modulation of EFRET with helix length measured in single-molecule experiments, leading to the conclusion that the orientation of the cyanine fluorophores on the ends of the helix is not a consequence of a particular linker, but is intrinsic to the nature of these fluorophores. We also find that the sequence of the terminal basepair has significant yet different effects on the fluorescent lifetimes and extent of stacking of the two Cy3 fluorophores.
Materials and Methods
Synthesis of oligonucleotides and attachment of fluorophores
DNA oligonucleotides were made by chemical synthesis using phosphoramidite chemistry. Oligonucleotides were made with C6 5′ amino-linkers, and Cy3 and Cy5 fluorophores were conjugated as N-hydroxysuccinimide esters (Amersham Biosciences, Piscataway, NJ) to make the L13-sCy3 and L13-sCy5 species, respectively. L3-Cy3 and L3-Cy5 oligonucleotides were made by coupling Cy3 and Cy5 as phosphoramidites as the final step of synthesis. All oligonucleotides were purified by gel electrophoresis in polyacrylamide, and recovered from gel fragments by electroelution or diffusion in buffer followed by ethanol precipitation. Fluorescently-labeled DNA was subjected to further purification by reversed-phase HPLC on a C18 column using an acetonitrile gradient with an aqueous phase of 100 mM triethylammonium acetate (pH 7.0). Equimolar quantities of complementary strands were mixed in 90 mM Tris·borate (pH 7.0), 25 mM NaCl, and annealed by heating at 85°C and cooled slowly to 4°C. The hybridized duplexes were purified by electrophoresis in 20% polyacrylamide in 90 mM Tris·borate (pH 8.3), 25 mM NaCl (see Fig. S1 in the Supporting Material) and recovered by electroelution and ethanol precipitation.
Phospholipid encapsulation
A 100:1 molar ratio of unmodified/biotinylated phospholipids was prepared by evaporating a number of aliquots of a mixture of 2.5 mg L-α-phosphatidylcholine and 35 μg 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (Avanti Polar Lipids, Alabaster, AL) from chloroform in a stream of argon. The aliquots were hydrated in 250 μL of 50 mM NaCl, 10 mM Tris·HCl (pH 8.1) (TN50 buffer) without (one aliquot, used for surface coating of slides) or with an addition of 200 nM of a given fluorescent DNA duplex and 10 mM MgCl2.
The DNA was encapsulated in phospholipid vesicles by repeated extrusion through a polycarbonate membrane containing 200 nm pores using a mini-extruder (both Avanti Polar Lipids), creating 200-nm diameter unilamellar vesicles (18,19). The ratio of DNA/phospholipid resulted in most vesicles being empty, and thus most encapsulations involved a single DNA molecule. Slides were prepared by injection of DNA-free phospholipid vesicles into a narrow channel made between a quartz microscope slide and a No. 1.5 coverslip (VWR International, Radnor, PA) using double-sided adhesive tape, and left at 4°C for 1 h to allow supported bilayer formation. The sample chamber was then washed with TN50 buffer, treated with 0.2 mg/ml neutravidin (Pierce, Rockford, IL) for 10 min, and washed. A 20th dilution of a chosen encapsulated DNA was injected and allowed to bind to the neutravidin for 15 min. Imaging was performed under the same buffer conditions used for vesicle preparation with an oxygen-scavenging system consisting of 1.6 mg/ml of glucose oxidase, 0.2 mg/ml of catalase, 6% (w/v) glucose, and 1 mM trolox.
Total internal reflection single-molecule microscopy
Fluorescence intensities were acquired from single DNA duplexes encapsulated in phospholipid vesicles using prism-based total internal reflection fluorescence microscopy. A microscope quartz slide containing the sample was mounted on the stage of an inverted microscope (IX70; Olympus America, Center Valley, PA). The sample chamber was chilled to 10 ± 2°C and an argon flow directed at the prism to prevent condensation.
Fluorescent emission was collected by a 1.2 NA 60× water immersion objective (Olympus America) and the incident excitation light removed by a 550-nm long-pass filter. The donor and acceptor fluorescence emissions were separated by a 645-nm dichroic mirror (Chroma Technology, Bellows Falls, VT). These two channels were focused side by side into a −60°C-cooled, back-illuminated, electron-multiplying charge-coupled device camera (iXON; Andor Technology, Belfast, Northern Ireland) (20). A 645-nm long-pass filter was present in the acceptor channel. Up to several hundred molecules could be recorded simultaneously using an image area of 8.2 × 8.2 mm (512 × 512 active pixels). Data were acquired at 10 frames s−1 using software written in Visual C++ (Microsoft, Redmond, WA). Molecules were identified and the uncorrected intensity data for donor and acceptor channels were extracted with an IDL (ITT Visual Information Solutions, Boulder, CO) routine. The data for single molecules were then analyzed in MATLAB (The MathWorks, Natick, MA), where appropriate correction factors were applied (12) as detailed in the Supporting Material.
Time-resolved fluorescence measurements
Time-resolved fluorescence intensity measurements were performed by time-correlated single-photon counting using a LifeSpec-II spectrometer (Edinburgh Instruments, Edinburgh, UK). The excitation source was a SC400-PP supercontinuum white light laser (Fianium, Hamble, Southampton, Hampshire, UK) operating at 2 MHz. Fluorescence emission was detected using a monochromator and a cooled microchannel plate photomultiplier tube. All data were collected at 15°C. The excitation wavelength was 530 nm and the emission wavelength was 565 nm with a 20-nm bandwidth. The excitation polarizer was set at 0° and the emission polarizer at 54.7°. A 550-nm long-pass filter was used to exclude scattered incident light.
An instrument response function was recorded before data collection. Fluorescence decay curves of 1 μM solutions of each duplex were recorded on a timescale of 20 ns, resolved into 4096 channels, to a total of 100,000 counts in the peak channel. Decay curves were analyzed by using a standard iterative reconvolution method in the F900 (Edinburgh Instruments) software package, using a multiexponential decay function. Fluorescence decays of the free fluorophores were fitted to two exponential functions. Fluorescence decays of the labeled DNA duplex species were globally fitted to three exponential functions, where the short lifetime (corresponding to the unstacked fluorophore) was linked globally for the duplexes labeled with the same fluorophore type (i.e., L3-Cy3 or L13-sCy3).
Results
Conjugation of cyanine fluorophores to duplex DNA
In this study we have covalently tethered cyanine fluorophores to six-carbon amino-linkers attached to the 5′ phosphate groups of duplex DNA postsynthetically using N-hydroxysuccinimide esters (Fig. 2). This differs from our previous study in two ways. The tethers comprise a total of 13 atoms, compared to just three atoms for the fluorophores coupled as phosphoramidites. The longer tether is a chain of methylene carbon atoms with an amide linkage close to the center, and would be expected to be flexible. Another difference lies in the structure of the fluorophores themselves, that have sulfonyl group substitution in the phenyl rings. Although the terms “Cy3” and “Cy5” are frequently applied to fluorophores both with and without the sulfonyl groups, here we shall refer to the nonsulfonated fluorophores (also known as “DiIC2(3)” and “DiIC2(5)”) attached as phosphoramidites as “L3-Cy3” and “L3-Cy5”, and the sulfoindocarbocyanine fluorophores attached by the 13-atom tethers as “L13-sCy3” and “L13-sCy5”. We have synthesized a series of duplex species between 10- and 24-bp in length with 5′-terminally attached L13-sCy3 and L13-sCy5 fluorophores. The terminal sequences are kept constant for each duplex, preserving the environment of the fluorophores. Full sequences are presented in the Supporting Material.
Fluorescent lifetimes of sulfoindocarbocyanine 3 attached by a 13-atom linker
Time-resolved fluorescence provides valuable information on the environment of cyanine fluorophores attached to dsDNA. In the free state, both sCy3 and Cy3 undergo relaxation by a cis-trans isomerization in the polymethine linker connecting the indole rings (21), giving a short fluorescent lifetime of ∼200 ps. However, this process is hindered if Cy3 is attached to the 5′-terminus of DNA and stacked onto the terminal basepair, whereupon components of longer lifetime are observed. On this basis, Sanborn et al. (22) showed that a fraction of terminally attached L3-Cy3 was hindered with a longer lifetime, whereas part had a short lifetime consistent with an unstacked environment. We have used time-resolved fluorescence to quantify the extent of stacking of L3-Cy3 onto the 5′-terminus of dsDNA (12). We have now compared the time-resolved fluorescence properties of L13-sCy3 and L3-Cy3 attached to the 5′-terminus of dsDNA. For this, we chose a 16-bp duplex with a 5′-cytosine. The duplexes were analyzed using time-correlated single photon counting, and the decay curves obtained were each fitted to a multiexponential decay function. The lifetimes and amplitudes are tabulated in Table 1.
Table 1.
Fluorescent lifetime data for indocarbocyanine 3- and sulfoindocarbocyanine 3-labeled DNA duplexes
| Sulfoindocarbocyanine |
Indocarbocyanine |
|||||||
|---|---|---|---|---|---|---|---|---|
| τ | α | fi | χ2 | τ | α | fi | χ2 | |
| Free | 0.24 | 92 | 84.7 | 1.15 | 0.16 | 98 | 95.8 | 1.78 |
| 0.48 | 8 | 15.3 | 0.43 | 2 | 4.2 | |||
| L13-sCy3 | L3-Cy3 | |||||||
| 5′ base | τ | α | fi | χ2 | τ | α | fi | χ2 |
| 0.31 | 32 | 12.1 | 0.33 | 45 | 21.3 | |||
| C | 0.97 | 63 | 76.9 | 1.25 | 0.90 | 50 | 64.8 | 1.14 |
| 1.80 | 5 | 10.9 | 1.81 | 5 | 14.0 | |||
| 0.31 | 40 | 18.6 | 0.33 | 41 | 24.5 | |||
| G | 0.85 | 57 | 73.0 | 1.27 | 0.83 | 55 | 66.5 | 1.21 |
| 2.01 | 3 | 8.4 | 2.02 | 4 | 9.0 | |||
| 0.31 | 37 | 11.3 | 0.33 | 47 | 20.9 | |||
| T | 0.97 | 36 | 34.2 | 1.27 | 0.78 | 50 | 65.4 | 1.24 |
| 2.03 | 27 | 54.5 | 2.00 | 3 | 13.7 | |||
| 0.31 | 45 | 16.6 | 0.33 | 28 | 11.1 | |||
| A | 0.90 | 33 | 35.6 | 1.08 | 0.90 | 59 | 63.2 | 1.21 |
| 1.81 | 22 | 47.9 | 1.65 | 13 | 25.7 | |||
Fluorescence lifetimes of indocarbocyanine 3-phosphate and sulfoindocarbocyanine 3-NHS ester as free dyes compared to their lifetimes attached to DNA duplexes with different terminal sequences. The labels τ, α, and fi are the fluorescent lifetime (ns), lifetime amplitude (%), and fractional intensity (%) for each component, respectively, and χ2 is the chi-squared statistic.
As expected, the free fluorophores exhibited only short lifetimes (Fig. 3). However, for both fluorophores we observed longer lifetimes when tethered to dsDNA and the decays were more complex, with a significant component having a fluorescence lifetime ∼900 ps. A small fraction of each fluorophore has a lifetime close to the radiative lifetime of Cy3 of 2 ns (22). The longer lifetimes indicate that, for a significant fraction of the fluorophore relaxation by cis-trans, isomerization is hindered; this is true for both forms of the fluorophore and attachment, suggesting that L13-sCy3 makes an intimate interaction with the end of the DNA helix. In fact, the fraction of fluorophore with a short lifetime (which we assume to be the unstacked form) is lower for L13-sCy3 than for L3-Cy3, suggesting that the interaction may be stronger despite the long tether.
Figure 3.

Time-resolved fluorescence decay of cyanine fluorophores free and attached to DNA. Decay curves are presented for the free fluorophores indocarbocyanine 3 phosphate (Cy3) and the sulfoindocarbocyanine 3-NHS ester (sCy3) and the same species attached to DNA, L3-Cy3 (shaded) and L13-Cy3 (solid), respectively. The instrument response function is shown (light shaded).
To examine the influence of the terminal sequence, we made three further 16-bp duplexes in which the terminal C⋅G basepair was replaced by G⋅C, T⋅A, and A⋅T. To each was attached L3-Cy3 or L13-sCy3 (Fig. S8). We observed a fraction of longer lifetimes for all combinations of sequence and fluorophore (Table 1), suggesting that each fluorophore was significantly stacked onto the end of the DNA. In each case, the short lifetime was found to be very similar for the same form of fluorophore, consistent with its assignment as the unstacked species (data not shown). Thus, the data were fitted assuming a common short lifetime. The possibility that other lifetimes could be common for different terminal sequences was considered but rejected due to significantly poorer fits to the data.
FRET efficiency between the ends of duplex DNA as a function of length exhibits modulation
We have measured FRET efficiency for each duplex 5′-labeled with L13-sCy3 and L13-sCy5 as single molecules encapsulated within phospholipid vesicles at 10 ± 2°C, as previously employed for the combination of L3-Cy3 and L3-Cy5 (12). This approach ensures potentially mishybridized molecules do not contribute to the data used to calculate EFRET, and the encapsulation obviates the need for tethering the molecules directly to the surface. There is no free 5′-terminus available for this purpose, and any tether might interact with the fluorophores, potentially complicating the results.
The measured values of EFRET as a function of duplex length are plotted in Fig. 4. Inspection immediately reveals that the data do not fit a simple distance dependence with a constant value of κ2 = 2/3. Instead, there is a clear modulation, with maxima at 16 and 21 bp, and minima at 14 and 19 bp. The modulation has twice the periodicity of a B-form helix, as would be expected if the fluorophores are partly stacked onto the ends of the helix such that the angle between their transition moments changes with helix length. From this, we conclude that despite the longer linkers, both cyanine fluorophores are at least partially stacked onto the ends of the helices, consistent with the fluorescent lifetime data.
Figure 4.
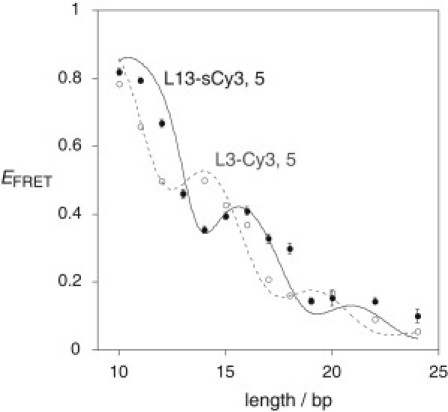
Variation of FRET efficiency between L13-sCy3 and L13-sCy5 terminally attached to DNA as a function of helix length. EFRET was measured for each duplex species as phospholipid vesicle-encapsulated single molecules. The EFRET values are plotted (solid circles) as a function of helix length, with estimated errors. The data have been simulated (solid line) using a model in which 50% of the molecules had one or both fluorophores unstacked (κ2 = 2/3) whereas the remaining 50% were stacked. The best fit was obtained where there was no mean rotation of the fluorophores relative to the terminal basepair. However, lateral rotation of the fluorophores was permitted about the mean, with a half-width of 20°. Standard B-form geometry of the DNA helix was used, with 10.5 bp/turn and a helical rise of 3.6 Å/bp step. These calculations were based upon a measured value of the spectral overlap integral J(λ) = 5.4 × 10−13 M−1 cm3, refractive index n = 1.33, and a quantum yield for L13-sCy3 of ϕD = 0.35. The data (open circles) and simulation (broken line) for a duplex series with L3-Cy3 and L3-Cy5 are plotted, taken from our earlier study (12). In this simulation the mean position of each fluorophore is rotated by 31° relative to the terminal basepair, accounting for the phase difference between the two curves.
As observed previously for the combination of L3-Cy3 + L3-Cy5, the minima do not reach EFRET = 0, suggesting that the interaction with the ends of the helix may exhibit some lateral flexibility that locally averages the data. The extent of modulation observed here is similar to that obtained using the short tethers. However, comparison with the previously-observed L3-Cy3 + L3-Cy5 series (plotted as open circles fitted with a broken line in Fig. 4) reveals a striking difference. Although the periodicity remains unchanged (consistent with the geometry of a B-form helix—see below), there is a significant phase shift between the two data sets. For the L3-Cy3 + L3-Cy5 duplex species, we observed maxima at 14 and 19 bp (12). This indicates that although the fluorophores interact with the helical termini for both linkers, the manner of this interaction differs such that the angle between the transition moments and the long axis of the terminal basepairs is significantly altered for the L13-sCy3 + L13-sCy5 species, with a change in the mean angle between the two fluorophores of ∼60° compared to the L3-Cy3 + L3-Cy5 duplex series.
Simulation of FRET efficiency data based upon the interaction of the cyanine fluorophores with the helical termini
As in our study of the L3-Cy3 + L3-Cy5-labeled species (12), we have simulated the data for the L13-sCy3 + L13-sCy5-labeled species using models that involve lateral angular motion of the fluorophores while remaining essentially stacked onto the terminal basepairs, with a Gaussian distribution consistent with Boltzmann population of rotational species based on energies determined by a torsional dependence according to Hooke's law. We have used two varieties of the model. In one, the fluorophores are wholly constrained to remain stacked on the helical termini. In the other model, the fraction of fluorophore determined to be unstacked according to the fluorescent lifetime measurements is given a constant value of κ2 = 2/3, and the remainder is treated as in the first model. The latter model is clearly more consistent with the fluorescent lifetime data.
The simulation based on full stacking is shown in Fig. S7. The simulation according to the second model is plotted with the experimental data in Fig. 4, where the fraction of molecules with one or both fluorophores unstacked is 0.5, based on the lifetime data (extrapolating from the measurements of Cy3 to assume that the fraction of unstacked species is 0.3 for both fluorophores). We have observed that the quantum yield for L13-sCy3 is higher than that for L3-Cy3, and we have used a value of 0.35 in the simulation (consistent with the longer lifetime). The value of the overlap integral J(λ) used is presented in the Supporting Material. The simulation is based on standard B-form DNA geometry (with no adjustable parameters), which gives an excellent fit to the periodicity of the data. However, to fit the phase of these data we found that we had to rotate the mean angular position of the fluorophores in the plane, so that the transition moments are aligned parallel to the long axis of the terminal basepairs.
This is a significant difference from the L3-Cy3 + L3-Cy5-labeled DNA, when both fluorophores were rotated by ∼31° relative to the terminal basepairs (virtually in the manner of an additional basepair), consistent with the NMR data of the corresponding species (15,16). The half-width of the distribution of lateral mobility required to fit the data was 20° about the mean. The model based on full stacking gave a significantly worse fit to the amplitude of the data, although the modulation is well reproduced (see Fig. S7). This required a distribution of lateral mobility with a half-width of 45°.
Discussion
The pronounced modulation observed over the duplex series using the 13-atom linker clearly shows that the cyanine fluorophores are oriented on the ends of the helix despite the long, flexible tethers. We therefore conclude that terminal stacking is an intrinsic propensity of these fluorophores, not a result of constraint imposed by the linker. This is consistent with observations of interactions between free cyanine fluorophores and nucleobases (17).
However, the phase shift between the sets of data with different linker lengths reveals a marked reorientation of the fluorophores in this data. The periodicity remains close to 10.5-bp/turn, i.e., consistent with B-form DNA structure, but the fluorophores are repositioned with respect to the terminal basepairs. This could result from the change in the chemical nature of the fluorophores, i.e., the addition of the sulfonyl groups (which should interact electrostatically with the phosphate groups), or a change in flexibility of the linker, or a combination of both. However, despite this there is no indication that the fluorophores themselves are any less constrained. We have initiated NMR studies of a DNA duplex with sulfoindocarbocyanine 3 tethered by the 13-atom linker to provide a structural description of the relationship between the fluorophore and the DNA.
To guide the modeling of the data, we determined the extent of unstacking of the fluorophores using time-resolved fluorescence measurements. Our data were fitted to three lifetimes of ∼0.3, 0.9, and 2.0 ns. The shortest lifetime is close to that observed for the free fluorophore, and we assume this is the lifetime of the unstacked fluorophore, where rotation about the polymethine chain is expected to be least hindered. The longest lifetime is close to the radiative lifetime of Cy3 (22), and thus is likely to reflect a fully stacked conformation where no rotation is possible. The intermediate lifetime may arise due to partially stacked conformations, in which rotation about the polymethine chain is possible but significantly hindered. Intermediate states amenable to isomerization were observed during a recent computational study by Spiriti et al. (23). They observed a variety of conformers where the proximal indole ring of L3-Cy3 was stacked over the 5′ nucleobase and the distal ring projected over the sugar backbone or minor groove. Our observation of a fraction of L13-sCy3 of short lifetime is fully consistent with the good fit obtained with the FRET efficiencies in the duplex series based on the model in which a fraction of one or other fluorophore is unstacked.
The extent of stacking of both L3-Cy3 and L13-sCy3 fluorophores was influenced by the identity of the terminal basepair. Our data for L3-Cy3 are in good agreement with those of Spiriti et al. (23), with the A⋅T basepair showing the greatest extent of stacking and a significantly longer average lifetime than the other sequences. However, we observe a significantly different sequence dependence for L13-sCy3. In this case, the T⋅A basepair is the best stacked, followed by the A⋅T. Spiriti et al. suggested that the relatively weak interaction between L3-Cy3 and T⋅A may be due to the out-of-plane C-H bonds of the methyl group of thymidine. If so, our data suggest that the longer linker permits the fluorophore to adopt an orientation that avoids this steric hindrance, perhaps by a 180° rotation of the fluorophore about its long axis to stack on its opposite face so that the dimethyl groups of L13-sCy3 lie in the minor rather than the major groove.
The intrinsic orientation of the cyanine fluorophores means that an experimentalist cannot simply use a longer linker and be assured that a simple κ2 = 2/3 is then valid. Moreover, the potential error resulting from this assumption would be even greater if the transition moments are not rotating within coaxial, parallel planes, because the orientation could then cover its full range of 0 ≤ κ2 ≤ 4. For example, using constrained internal cyanine fluorophores in DNA duplexes, Ranjit et al. (24) observed a value of κ2 = 3.2. However, if the orientation effects can be fully understood, they could provide a valuable source of angular data in nucleic acids.
Acknowledgments
We thank Dr. Dirk Nather (Edinburgh Instruments) for access to time-resolved fluorimetric equipment and software, Xinsheng Zhao, Sinan Arslan, and Taekjip Ha for discussion, and Scott McPhee for chemical synthesis of DNA oligonucleotides.
We thank Cancer Research UK for financial support.
Supporting Material
References
- 1.Stryer L., Haugland R.P. Energy transfer: a spectroscopic ruler. Proc. Natl. Acad. Sci. USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clegg R.M. In: Fluorescence Imaging Spectroscopy and Microscopy. Wang X.F., Herman B., editors. John Wiley & Sons; New York: 1996. pp. 179–252. [Google Scholar]
- 3.Lilley D.M.J., Wilson T.J. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr. Opin. Chem. Biol. 2000;4:507–517. doi: 10.1016/s1367-5931(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 4.Clegg R.M. FRET tells us about proximities, distances, orientations and dynamic properties. J. Biotechnol. 2002;82:177–179. [PubMed] [Google Scholar]
- 5.Lilley D.M.J. The structure and folding of branched RNA analyzed by fluorescence resonance energy transfer. Methods Enzymol. 2009;469:159–187. doi: 10.1016/S0076-6879(09)69008-X. [DOI] [PubMed] [Google Scholar]
- 6.Joo C., Balci H., Ha T. Advances in single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- 7.Förster T. Intermolecular energy transfer and fluorescence [Zwischenmolekulare energiewanderung und fluoreszenz] Ann. Phys. 1948;2:55–75. [Google Scholar]
- 8.Haas E., Katchalski-Katzir E., Steinberg I.Z. Effect of the orientation of donor and acceptor on the probability of energy transfer involving electronic transitions of mixed polarization. Biochemistry. 1978;17:5064–5070. doi: 10.1021/bi00616a032. [DOI] [PubMed] [Google Scholar]
- 9.Dale R.E., Eisinger J., Blumberg W.E. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 1979;26:161–193. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu P., Brand L. Orientation factor in steady-state and time-resolved resonance energy transfer measurements. Biochemistry. 1992;31:7939–7947. doi: 10.1021/bi00149a027. [DOI] [PubMed] [Google Scholar]
- 11.Lewis F.D., Zhang L., Zuo X. Orientation control of fluorescence resonance energy transfer using DNA as a helical scaffold. J. Am. Chem. Soc. 2005;127:10002–10003. doi: 10.1021/ja0524402. [DOI] [PubMed] [Google Scholar]
- 12.Iqbal A., Arslan S., Lilley D.M. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nucleic acids. Proc. Natl. Acad. Sci. USA. 2008;105:11176–11181. doi: 10.1073/pnas.0801707105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Börjesson K., Preus S., Wilhelmsson L.M. Nucleic acid base analog FRET-pair facilitating detailed structural measurements in nucleic acid containing systems. J. Am. Chem. Soc. 2009;131:4288–4293. doi: 10.1021/ja806944w. [DOI] [PubMed] [Google Scholar]
- 14.Levitus M., Ranjit S. Cyanine dyes in biophysical research: the photophysics of polymethine fluorescent dyes in biomolecular environments. Q. Rev. Biophys. 2011;44:123–151. doi: 10.1017/S0033583510000247. [DOI] [PubMed] [Google Scholar]
- 15.Norman D.G., Grainger R.J., Lilley D.M. Location of cyanine-3 on double-stranded DNA: importance for fluorescence resonance energy transfer studies. Biochemistry. 2000;39:6317–6324. doi: 10.1021/bi992944a. [DOI] [PubMed] [Google Scholar]
- 16.Iqbal A., Wang L., Norman D.G. The structure of cyanine 5 terminally attached to double-stranded DNA: implications for FRET studies. Biochemistry. 2008;47:7857–7862. doi: 10.1021/bi800773f. [DOI] [PubMed] [Google Scholar]
- 17.Harvey B.J., Levitus M. Nucleobase-specific enhancement of Cy3 fluorescence. J. Fluoresc. 2009;19:443–448. doi: 10.1007/s10895-008-0431-1. [DOI] [PubMed] [Google Scholar]
- 18.Boukobza E., Sonnenfeld A., Haran G. Immobilization in surface-tethered lipid vesicles as a new tool for single biomolecule spectroscopy. J. Phys. Chem. B. 2001;105:12165–12170. [Google Scholar]
- 19.Okumus B., Wilson T.J., Ha T. Vesicle encapsulation studies reveal that single molecule ribozyme heterogeneities are intrinsic. Biophys. J. 2004;87:2798–2806. doi: 10.1529/biophysj.104.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ha T. Single-molecule fluorescence resonance energy transfer. Methods. 2001;25:78–86. doi: 10.1006/meth.2001.1217. [DOI] [PubMed] [Google Scholar]
- 21.Chibisov A.K., Zakharova G.V., Tolmachev A.I. Photorelaxation processes in covalently linked indocarbocyanine and thiacarbocyanine dyes. J. Phys. Chem. 1995;99:886–893. [Google Scholar]
- 22.Sanborn M.E., Connolly B.K., Levitus M. Fluorescence properties and photophysics of the sulfoindocyanine Cy3 linked covalently to DNA. J. Phys. Chem. B. 2007;111:11064–11074. doi: 10.1021/jp072912u. [DOI] [PubMed] [Google Scholar]
- 23.Spiriti J., Binder J.K., van der Vaart A. Cy3-DNA stacking interactions strongly depend on the identity of the terminal basepair. Biophys. J. 2011;100:1049–1057. doi: 10.1016/j.bpj.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ranjit S., Gurunathan K., Levitus M. Photophysics of backbone fluorescent DNA modifications: reducing uncertainties in FRET. J. Phys. Chem. B. 2009;113:7861–7866. doi: 10.1021/jp810842u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.