

Abstract
The 18.5-kDa myelin basic protein (MBP), the most abundant isoform in human adult myelin, is a multifunctional, intrinsically disordered protein that maintains compact assembly of the sheath. Solution NMR spectroscopy and a hydrophobic moment analysis of MBP's amino-acid sequence have previously revealed three regions with high propensity to form strongly amphipathic α-helices. These regions, located in the central, N- and C-terminal parts of the protein, have been shown to play a role in the interactions of MBP with cytoskeletal proteins, Src homology 3-domain-containing proteins, Ca2+-activated calmodulin (Ca2+-CaM), and myelin-mimetic membrane bilayers. Here, we have further characterized the structure-function relationship of these three domains. We constructed three recombinant peptides derived from the 18.5-kDa murine MBP: (A22–K56), (S72–S107), and (S133–S159) (which are denoted α1, α2, and α3, respectively). We used a variety of biophysical methods (circular dichroism spectroscopy, isothermal titration calorimetry, transmission electron microscopy, fluorimetry, and solution NMR spectroscopy and chemical shift index analysis) to characterize the interactions of these peptides with actin and Ca2+-CaM. Our results show that all three peptides can adopt α-helical structure inherently even in aqueous solution. Both α1- and α3-peptides showed strong binding with Ca2+-CaM, and both adopted an α-helical conformation upon interaction, but the binding of the α3-peptide appeared to be more dynamic. Only the α1-peptide exhibited actin polymerization and bundling activity, and the addition of Ca2+-CaM resulted in depolymerization of actin that had been polymerized by α1. The results of this study proved that there is an N-terminal binding domain in MBP for Ca2+-CaM (in addition to the primary site located in the C-terminus), and that it is sufficient for CaM-induced actin depolymerization. These three domains of MBP represent molecular recognition fragments with multiple roles in both membrane- and protein-association.
Introduction
Multiple sclerosis is the major demyelinating disease of humans characterized by degradation of the myelin sheath due to autoimmune attack (1). The most studied candidate autoantigen is the classic 18.5-kDa myelin basic protein (MBP) isoform (the most abundant in adult myelin) (2–6). This isoform exists as a number of charge components or isomers (denoted C1–C8) due to a wide variety of posttranslational modifications, such as methylation, phosphorylation, and deimination (7). The least posttranslationally modified isomer with a net positive charge of +19 at neutral pH is the focus of this study, which we will henceforth refer to as “MBP” or “rmC1” (the unmodified recombinant murine C1 charge component of MBP) (8). Due to its high net positive charge and conformational adaptability to its environment, MBP is an intrinsically disordered protein (2,5,6,9). MBP is an essential structural protein in the central nervous system, where it maintains the tight multilamellar assembly of the myelin sheath by adhesion of the apposing cytoplasmic leaflets of the oligodendrocyte membrane (10,11), and is multifunctional (3,4,6), like other intrinsically disordered proteins.
It has been shown in different in vitro studies that MBP interacts directly with cytoskeletal proteins (actin and tubulin) (12–17), Ca2+-activated calmodulin (Ca2+-CaM) (18–22), Src homology 3 (SH3)-domain containing proteins (23,24), and divalent metal cations (25), and either directly or indirectly with voltage-operated calcium channels (26). Moreover, MBP has been shown to interact simultaneously with actin and lipid vesicles, thus supporting its suggested role in the organization of the actin cytoskeleton, and in tethering it to the inner leaflet of the oligodendrocyte membrane (12,13,27). In oligodendrocytes cultured from the shiverer mouse (which lacks MBP), actin filaments appeared to be disorganized, and the cell processes were smaller than normal with a larger cell body (28–30). Our recent studies with cultured oligodendrocytes have shown that the 18.5-kDa MBP isoform associates with the cytoskeleton during phorbol ester-induced membrane ruffling (31), and that SH3-ligand mutations of MBP result in attenuated affinity for Fyn kinase and alter process differentiation and protein localization (32). Altogether, these diverse observations suggest that the interaction of MBP with cytoskeletal and signaling proteins is crucial for oligodendrocyte function, and myelin formation and maintenance.
In aqueous solution, MBP adopts an extended conformation and circular dichroism (CD) spectroscopy indicates a mainly random coil conformation. However, different segments of MBP gain ordered secondary structure upon interaction with its biological partners (reviewed in (2,5,6)), such as lipids (9,33–36), actin (16), and Ca2+-activated CaM (19,22). These intermolecular associations may involve preformed structural elements such as α-helices, and/or an induced fit, i.e., coupled folding and binding. This idea has been encapsulated in the Molecular Recognition Fragments (MoRF, specifically α-MoRF) hypothesis for intrinsically disordered proteins (37–39). In this conceptual framework, a hydrophobic moment analysis of MBP's amino-acid sequence reveals three regions in the MBP sequence with high propensity to form strongly amphipathic α-helices located in the N- and C-termini, and in the central part of the protein (see Fig. S1 in the Supporting Material) (5,6). This analysis is strongly supported by experimental studies using electron paramagnetic resonance (EPR) and solution and solid-state NMR spectroscopy, which showed that these regions interact with myelin-mimetic lipid membranes and adopt an α-helical structure (19,33,34,36,40,41). Additionally, these regions were shown to play a role in the interactions of MBP with other partners. The central region of MBP represents a primary immunodominant epitope (Pro82−Pro93) and the proline-rich region immediately following it forms a transient poly-proline type II conformation that represents a ligand for SH3-domain binding proteins (5,24,32,42–44). Recently, the N- and C-terminal regions of MBP were shown to play key roles in actin polymerization and binding (16,17), and the C-terminus in binding to Ca2+-activated CaM (19).
Here, we have focused on the characterization of the three described amphipathic α-helical MBP domains in terms of the relationship between their potential structure and function. For the purpose of this study, we constructed three recombinant peptides derived from the 18.5-kDa murine MBP sequence: (A22–K56), (S72–S107), and (S133–S159), which are denoted α1, α2, and α3, respectively. In our numbering convention, the N-terminal methionine is not counted because it is cleaved (45). Our results here show that all three peptides can adopt α-helical structure inherently, even in aqueous solution. Although both the N-terminal α1- and C-terminal α3-peptides showed strong binding with Ca2+-CaM, only α1 exhibited actin polymerization and bundling activity. Actin that was polymerized by α1 could subsequently be depolymerized by Ca2+-CaM. Solution NMR spectroscopy showed major structural reorganizations of both the α1- and α3-peptides upon binding to Ca2+-CaM, but with different degrees of interaction.
Materials and Methods
Fully detailed Materials and Methods are provided in the Supporting Material available online. Briefly, three different regions of murine 18.5-kDa MBP coding for amino acids (A22–K56), (S72–S107), and (S133–S159) were cloned into the Champion pET Small Ubiquitin MOdifier (SUMO) Expression System (Invitrogen Life Technologies, Burlington, ON) and were named α1, α2, and α3, respectively. The plasmids coding for these SUMO-MBP-α-peptide fusion proteins were transformed into Escherichia coli BL21-CodonPlus (DE3)-RP cells (Stratagene, La Jolla, CA) and were expressed and purified as detailed in the Supporting Material. CD spectroscopy was used to evaluate the secondary structure of each peptide in aqueous solution, with trifluoroethanol to stabilize α-helices, and in dodecyl phosphocholine (DPC) micelles as a membrane-mimetic condition. Isothermal titration calorimetry was used to probe the interaction of Ca2+-activated CaM (Ca2+-CaM) with each peptide.
Actin polymerization induced by each peptide was followed by measuring an increase in fluorescence intensity of pyrene-labeled actin via an automated microplate fluorescence reader, and overall morphology evaluated by transmission electron microscopy. Actin bundling was assessed by a centrifugation and gel electrophoretic assay, and the structural changes of two MBP-derived peptides (α1 and α3) interacting with Ca2+-CaM were assessed by CD and solution NMR spectroscopy.
Results and Discussion
Expression and purification of α1, α2, and α3
Three peptides α1 (A22-K56), α2 (S72–S107), and α3 (S133–S159) from the 18.5-kDa murine MBP amino-acid sequence were designed to include three regions (T33-D46, V83-T92, and V142-L154) that had been previously suggested to adopt a strongly amphipathic α-helical structure (see Fig. S1). One of the methods to prevent in vivo degradation of recombinant short polypeptides during expression in prokaryotic cells (E. coli) is to express them as fusion proteins from which target peptides can be released by enzymatic cleavage. This strategy also enables stable isotope-labeling for NMR spectroscopy (46). Here, we used the small ubiquitin-related modifier (SUMO) linked with a hexahistidine tag to overexpress MBP-α-peptides in E. coli BL21-CodonPlus (DE3)-RP cells.
The advantages of this expression system lie in its simplicity and ability to produce isotopically labeled peptides at relatively high yield, and without any additions related to the purification protocol (47,48). Using this system, we were able to express up to 130 mg of SUMO-peptide-fused products per liter of culture and, using a native lysis procedure followed by immobilized metal affinity chromatography (using Ni-NTA resin), purify them from the whole bacterial lysates (see Fig. S2, A, C, and E, left-hand side). In the subsequent step, SUMO was enzymatically removed in a reaction with specific SUMO protease I (Ulp1, kindly provided by Dr. C. D. Lima, Sloan-Kettering Institute, NY).
To achieve complete cleavage, we applied 1 μg of protease to 1 mg of fused proteins, and the reaction mixtures were incubated with protease at 30°C for 3 h. The efficiency of proteolytic cleavage was assessed by Tricine-polyacrylamide gel electrophoresis (see Fig. S2, A, C, and E, right-hand side), and the peptides were further purified by subtractive chromatography using the same Ni-NTA resin once again. During this chromatography step, the cleaved SUMO-tag along with the SUMO protease bound to the column, because they both contained hexahistidine affinity tags, whereas the untagged peptides of interest were recovered in the flowthrough and wash fractions. Peptides were further purified by high-performance liquid chromatography (HPLC) and the final purity was assessed by rechromatography using HPLC (see Fig. S2, B, D, and F). The retention times from the HPLC column for the α1-, α2-, and α3-peptides appeared to be 19.6 min (35.6% acetonitrile), 18.7 min (34.7% acetonitrile), and 16.1 min (32.1% acetonitrile), respectively, and the purity of the peptides was 99.5%, 98.9%, and 98.7%, respectively. This protocol yielded ∼13–18 mg of α2- and α3-peptides, and a slightly lower amount (∼6–12 mg) of α1 per liter of culture. To confirm the molecular mass of the purified peptides, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry was used, yielding 3822.99 Da (calculated 3823.3 Da), 3969.98 Da (calculated 3970.4 Da), and 2841.38 Da (calculated 2840.1 Da) for α1, α2, and α3, respectively.
Secondary structure analysis of MBP-α-peptides in different environments
CD spectroscopy of MBP in aqueous solution has shown that its conformation is predominantly a random coil, but the protein undergoes some conformational changes and adopts a certain degree of ordered secondary structure in the presence of detergents or lipids (2,40,49). As stated above, the primary role of MBP is the maintenance of the compact myelin assembly surrounding axons by adhesion of the apposing cytoplasmic faces of the oligodendrocyte membranes. Therefore, a physiologically more relevant environment of 18.5-kDa MBP is membranous; however, most solution spectroscopic methods used to analyze protein structure are not applicable to MBP in its native-mimetic environment because protein-lipid aggregation produces semisolid assemblies (see (2,9,40)). Two widely accepted ways to overcome this problem are the use of 2,2,2-trifluoroethanol (TFE), a membrane-mimetic organic solvent (50–52), or dodecyl phosphocholine (DPC), a micelle-forming lysolipid. One of the goals of this study was to confirm that the MBP segments that have been proposed to adopt an α-helical conformation in the full-length protein, based on diverse experimental evidence, indeed do so in aqueous and/or membrane-mimetic environments.
For this purpose, we compared CD spectra of α1-, α2-, and α3-peptides in aqueous buffer and a membrane-mimetic (TFE and DPC) environment (see Fig. S3). The typical CD spectrum of full-length 18.5-kDa mMBP in aqueous solution is characterized by a negative peak with a minimum at 197 nm, resulting from a mostly random coil structure (8,49). However, here it was obvious that all three peptides, even in the aqueous buffer, exhibited a much higher proportion of ordered secondary structure than full-length 18.5-kDa murine MBP, because the minima for all three peptides were observed at ∼205 nm. In the experiments where we added an increased amount of TFE to the buffered peptides, a dramatic effect was observed. Even at as low as 10% TFE, one can see a shift toward increased α-helical conformation, and at higher concentrations of TFE, all three peptides exhibited well-pronounced α-helical conformations characterized by troughs at ∼207 nm and ∼222 nm.
The mechanism by which TFE stabilizes (or sometimes induces) secondary structure is based on reduction of the dielectric constant around the peptide, thus allowing formation of hydrogen bonds between residues in the polypeptide chain (50). Similarly, protein-lipid interactions stabilize and/or induce hydrogen bonds between residues by exclusion of bulk solvent from the protein surroundings (53). Thus, not surprisingly, DPC produced very similar effects on the secondary structure of all three MBP-α-peptides. However, the most pronounced effect was observed at a concentration of DPC higher than the critical micelle concentration (1 mM), and the most prominent α-helical conformations were achieved when there was at least one micelle per molecule of MBP-α-peptide (each DPC micelle contains 40–60 lipid molecules). Altogether, these considerations mean that the change in the secondary structure of the three MBP-α-peptides reflects their potential to adopt the same conformation in their native membrane environment. In general, it would be expected that the interactions of each of the α1-, α2-, and α3-peptides with lipid bilayers would be subtly different (52,54), and that posttranslational modifications in each domain would also play a role in modulating the binding (5,6,33,34,55).
Here, the α3-peptide—the C-terminal segment of MBP that was considered to contain the primary CaM-binding domain (18,19)—adopted the highest degree of α-helical conformation, particularly in the presence of TFE. This capability, in fact, can play an extremely important role in recognition of this MBP region by Ca2+-activated CaM, because in its canonical mode of interaction, Ca2+-CaM recognizes α-helical segments and closes its lobes around them (56,57). In the full-length 18.5-kDa protein, this region has been shown by EPR spectroscopy to be α-helical in association with phospholipid membranes, and to interact with Ca2+-CaM both in solution and on a membrane (33,58).
CaM binding to MBP-α-peptides
It has been previously demonstrated by numerous techniques that MBP interacts with CaM in a Ca2+-dependent manner (see (18,19,21,22), and references therein). Although the primary CaM-binding site of MBP was initially predicted to be a C-terminal segment (Lys132−Arg167), several experimental results suggested that there was more than one binding site (21,58,59). Solution NMR investigations demonstrated that the C-terminal segment (Pro120−Arg160) was the primary binding site for Ca2+-CaM in solution, as indicated by contiguous chemical shift perturbations (19). Those studies were complemented by in silico molecular modeling and EPR spectroscopy that suggested three segments of MBP (Arg41−Arg52, Pro82−Pro93, and Thr147−Asp158) modeled as α-helices could bind Ca2+-CaM with favorable energetics (58). The interaction of the central immunodominant epitope (Pro82−Pro93), however, was shown to be energetically unfavorable when modeled in the extended conformation.
The peptides that we used here were designed to include all three potential interaction sites. We used isothermal titration calorimetry to characterize the binding parameters of CaM with the MBP-α-peptides (Fig. 1, A and B). All experiments in this section were carried out in aqueous buffer, where the presence of α-helical structure was minimal. We found that only the α1- and α3-peptides bound to Ca2+-CaM, and formed 1:1 molar complexes with dissociation constants of 1.23 μM and 3.57 μM, respectively (see Table S1 in the Supporting Material). In the case of the α2-peptide (including the central membrane-anchoring motif), we did not observe any changes in heat upon its titration into the CaM solution (not shown). Moreover, as was expected, the interaction of the α1- and α3-peptides with CaM was strongly Ca2+-dependent.
Figure 1.
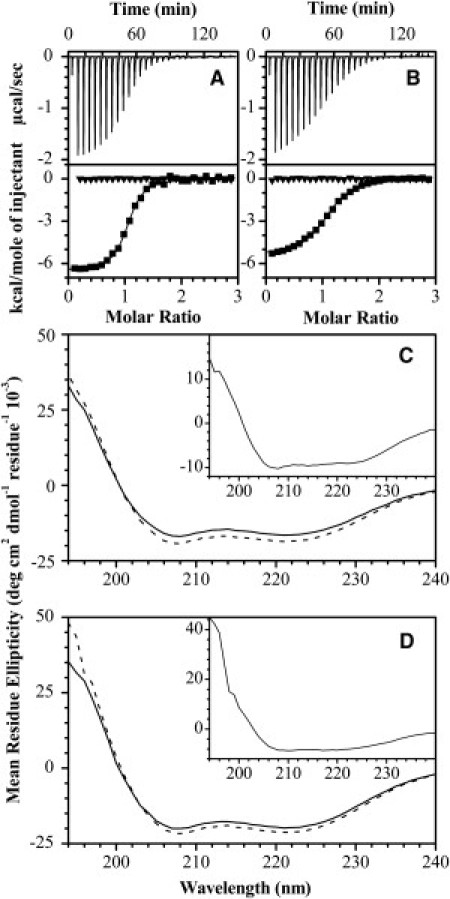
Isothermal titration calorimetry and circular dichroism spectroscopy of MBP-derived peptides interacting with Ca2+-activated CaM. A 100 μM protein solution (20 mM HEPES-KOH, pH 7.4, 100 mM KCl, 5 mM CaCl2) was titrated with 1 mM solution of α1-peptide (A) and α3-peptide (B) at 30°C (-▪-). The Ca2+-dependency of the binding was assessed in the same reaction protocol, but now in the Ca2+-depleted buffer (20 mM HEPES-KOH, pH 7.4, 100 mM KCl, 8 mM EDTA, 2 mM EGTA) (-▾-). The data were fitted to the software Origin's one-set-of-sites model (OriginLab, Northampton, MA). All parameters are summarized in Table S1. The CD spectra of 131 μM and 176 μM Ca2+-CaM in the absence (solid lines) or presence (dashed lines) of equimolar amounts of α1- or α3-peptide are shown in panels C and D, respectively. (Insets) Difference spectra of Ca2+-CaM complexes with peptides minus Ca2+-CaM.
Not surprisingly, both α1 and α3 exhibited an increase in proportion of secondary structure components upon binding with Ca2+-CaM (Fig. 1, C and D). Analysis of CD spectra suggests that two peptides underwent a transition to predominantly an α-helical structure representative of a standard target structure induced by interaction with Ca2+-CaM (57). We have previously modeled CaM with peptide fragments of MBP constructed to be α-helical (58), and this disorder-to-order transition is consistent with this segment of MBP being an α-helical MoRF (5,19,37). Moreover, the secondary structure propensity of full-length 18.5-kDa recombinant murine MBP complexed with Ca2+-CaM previously indicated formation of α-helical structure (19), and later with natural bovine MBP (21), but, to our knowledge, the new CD data presented here with the smaller peptides show this transition more clearly.
These results have further demonstrated that α1 contains an additional binding domain for Ca2+-CaM, which we have previously postulated (18,58,59). Although the dissociation constants for the interaction of peptides and Ca2+-CaM determined in our study are significantly higher than those reported for the full-length recombinant or bovine MBP (low nanomolar range) (18), our results are in agreement with the Kd value calculated for a different C-terminal MBP peptide designed in another study (10 μM) (20,21). Moreover, these recent studies have suggested that CaM binding to MBP is cooperative; thus, probably, the entire sequence of MBP is required to bind with higher affinity (compare (19–21)). These additional interactions explain the differences in dissociation constants observed for peptide segments and the full-length protein.
Actin polymerization induced by MBP-α-peptides and depolymerization induced by CaM
We have recently shown that there are at least two segments in the 18.5-kDa mMBP sequence that cause actin polymerization under otherwise nonpolymerizing conditions (17). We were able to conclude that one site was located in the N-terminal two-thirds of the protein, and the other in the C-terminal two-thirds of the sequence. Additionally, we have shown that the actin polymerization activity of the N-terminal part was higher than for the C-terminal one, but we could not obtain any other site-specific information about the interaction. It was previously shown by others that peptides derived by thrombin digestion of 18.5-kDa MBP purified from bovine brain, particularly the (A1–F43) and (R96–R168) segments, had strong actin-polymerizing activity (61). (These two bovine 18.5-kDa MBP segments correspond to the murine 18.5-kDa MBP segments (A1–F43) and (R94–R167), respectively.) In another recent study using Fourier transform infrared and solid-state NMR spectroscopy, we identified potential regions in MBP that might undergo changes in secondary structure upon interaction with actin (16). To gain further insight into these associations, it was important to probe the interaction with actin of all three MBP-α-peptides (α1, α2, and α3), separately.
We used an increase in the fluorescence intensity of pyrene-labeled actin as a measure of actin polymerization. We then followed the rate of actin polymerization induced by different concentrations of α1-, α2-, and α3-peptides, and compared these results to the rate of full-length 18.5-kDa MBP-induced activity (data not shown). It appeared that of these three peptides, only α1 induced actin polymerization, but at a rate much slower than that of the full-length rmMBP. Typically, for the full-length protein, the maximal fluorescence intensity was observed after ∼5 min, but it required ∼20 min in the case of α1. As shown in Fig. 2 A, the titration curve of an enhancement in fluorescence intensity upon addition of increasing concentrations of α1 had a sigmoidal shape.
Figure 2.
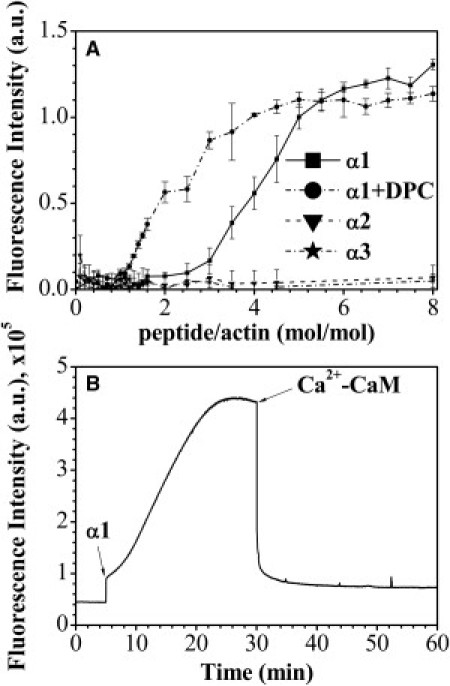
Actin polymerization and bundling induced by MBP-α-peptides and the effect of Ca2+-CaM. (A) Dependence of fluorescence enhancement caused by actin polymerization on the molar ratio of MBP-derived peptides to actin. All reactions were carried out in G-buffer at 30°C and contained 5 μM G-actin with various concentrations of α1- (solid line), α2- (dashed line), and α3- (dashed-dotted line) peptides. The α1-induced actin polymerization was probed also in the presence of DPC (dash-dot-dotted line). The mean ± SD of triplicates is shown. Data points are expressed as (Fi-F0)/F0, where Fi is the fluorescence intensity at different peptide/actin molar ratios and F0 is the fluorescence intensity of 5 μM G-actin in G-buffer. (B) Time dependence of the increase in fluorescence caused by polymerization of 1.1 nmol of G-actin induced by 13.3 nmol of α1-peptide and decrease in fluorescence due to depolymerization after the addition of 13.3 nmol of Ca2+-CaM. (Arrows) Time of α1-peptide and CaM addition. Shown is a representative experiment (out of three replicates).
At a molar ratio of ∼6.5 (α1 to actin), saturation in the level of actin polymerization was reached. We also examined the morphology of MBP-α-peptide/F-actin assemblies by transmission electron microscopy (see Fig. S4). We collected electron micrographs from samples where the α-peptide/actin molar ratio was 4.5. Only with α1 did we observe bundling of actin filaments, which appeared to be morphologically indistinguishable from those assembled by full-length protein (17). Yet, one can see that there were many unbundled actin filaments in the background, which typically were not present with the full-length protein. Additionally, because we observed that under membrane-mimetic conditions (presence of DPC at concentrations higher than its critical micelle concentration), the α1-peptide adopted a higher degree of secondary structure (see Fig. S3 D), we decided to test the ability of this peptide to polymerize actin and bundle filaments in the presence of DPC micelles (Fig. 2 A and see Fig. S5). Clearly, the presence of micelles enhanced the actin polymerization activity of the peptide. The saturation in actin polymerization level was achieved at an α1-peptide/actin molar ratio of ∼4.5. However, the presence of DPC did not appear to affect the bundling ability of the peptide. Even at a molar ratio of peptide/actin as high as eight, actin partially existed in its unbundled filamentous form as derived from analysis of the sedimentation assay (see Fig. S5 B).
Another important question for investigation was to detect whether the α1-peptide tethered DPC micelles to actin upon interaction. Because the only source of phosphorus in the actin bundles could be DPC, we used a phosphorus assay to quantify it in the actin-peptide complex after the sedimentation assay, as we have done previously (17). We found that even though the α1-peptide represents only a small part of MBP, it tethered DPC to the actin bundles at a ratio of one micelle per one peptide molecule, as ascertained by the phosphorus assay (data not shown). Furthermore, using the same sedimentation assay, we quantified the amount of bound and free peptide, after sedimentation of bundled actin from the reaction mixtures with different initial actin/peptide molar ratios, in the presence or absence of DPC. By plotting results of fractional saturation [α1bound]/[actintotal] versus [α1free] (see Fig. S5 C), we estimated that the affinity of peptide binding to actin, in the presence and absence of DPC, was in a low micromolar range. The dissociation constants appeared to be 103-fold higher than those reported for the full-length 18.5-kDa MBP (17), thus indicating weaker binding. The shape of the binding curves suggested that there was binding at a ratio of four peptide molecules per actin molecule in the presence of DPC. However, in the absence of DPC, the shape of the binding isotherm appeared to be sigmoidal, thus signifying again that concentrations of peptide higher than critical (to allow actin polymerization) were required for binding.
Interestingly, the other two MBP-α-peptides (α2 and α3)—which included the segments of 18.5-kDa mMBP that, based on a previous solid-state NMR study (16), were expected to interact with actin, such as Pro82−Phe86 and Ile90−Pro96 (included in the α2-peptide); and Ser133−His135 and Ala141−Leu148 (included in the α3-peptide)—did not induce any polymerization of actin. This result suggests that the interaction between MBP and actin is not only charge-based, but involves primarily the N-terminal domain. It also could be that the α2- and α3-peptides were not long enough, or did not include the other residues responsible for actin polymerization caused by other segments of 18.5-kDa MBP. It is very likely that, in vivo, the synergistic action of charge-based and region-specific interactions between MBP and actin is required to polymerize actin efficiently, especially in the environment of the oligodendrocyte membrane, when parts of the protein are embedded in the lipid bilayer, but not the charged residues that are available for the interaction.
It has been shown in several previous studies, that Ca2+-activated CaM could reverse polymerization of actin by MBP (12,13,15,62,63). Although we have previously shown that the primary CaM-binding domain is located at the C-terminus of MBP (18,19), in this study we have shown that both α1- and α3-peptides bound Ca2+-CaM (Fig. 1 and see Table S1), and thus we examined here the ability of Ca2+-CaM to reverse α1-polymerized actin assembly (Fig. 2 B). We found that, indeed, Ca2+-CaM instantly caused actin depolymerization. The rate appeared to be much faster than in the case of full-length 18.5-kDa rmMBP (27). These results suggest that Ca2+-CaM caused dissociation of α1 from actin, because it alone is not directly involved in either actin polymerization or depolymerization.
The rate of this dissociation was noticeably faster than for the full-length MBP, probably because there is more than one binding site in MBP to actin. In contrast to the binding of full-length MBP to actin with the dissociation constant in the low nanomolar range, peptide binding was characterized by Kd in the micromolar range. Thus, Ca2+-CaM can effectively compete with actin for peptide binding. Altogether, these results proved not only that the α1-peptide contains an additional binding domain for Ca2+-CaM, but also that this binding is important for CaM-induced actin depolymerization. Similar control mechanisms have been observed for other proteins like myristoylated alanine-rich C-kinase substrate (64,65). Interestingly, another group has recently shown that the N-terminal region of MBP inhibits the formation of amyloid-β fibrils (66).
Solution NMR spectroscopy
As of the time of this writing, we are using solution NMR spectroscopy to investigate further the interactions of the three peptides with other partners, namely Ca2+-CaM (9,19) and SH3-domains (32,44), and solid-state NMR spectroscopy to study the α1-actin interaction (16). We will only present here and in the Supporting Material initial results on the interaction of the α3-peptide with Ca2+-CaM. Fig. 3 is an overlay of the 1H-15N-HSQC spectra of α3 alone and in association with Ca2+-CaM at a 1:1 molar ratio, showing chemical shift perturbations and peak broadening upon binding. These data indicate that almost the entire α3-peptide is subject to dramatic changes in its immediate chemical environment and/or structural state as a result of the interaction, and that the complexes are in fast conformational or chemical exchange with their components. Sequence-specific assignments of backbone resonances of the α3-peptide alone (e.g., see Fig. S7, Table S3, and Table S4) were used to identify the residues subject to these changes, and the data in Fig. 3 indicate that the interaction takes place along the whole peptide.
Figure 3.
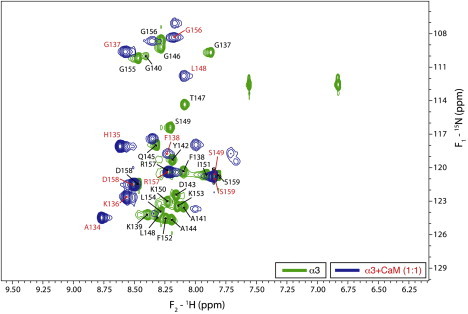
Overlays of the 1H-15N-HSQC spectra for the α3-peptide alone (green contours), and the α3-peptide-Ca2+-CaM complex (blue contours) at a 1:1 molar ratio. Peak labels denote assignments for the free peptide (black) and peptide in complex with Ca2+-CaM (red).
Titration with the chelating agents EDTA and EGTA proved the interaction of the basic peptide with acidic CaM to be Ca2+-dependent and thus specific (and not simply electrostatic), even if relatively weak (see Fig. S7). Preliminary data on the interaction of the α1-peptide with Ca2+-CaM also show chemical shift perturbations indicative of a structural reorganization of this Ca2+-CaM-target (see Fig. S8). In comparison with the α3-peptide, there is somewhat less peak broadening for the α1-peptide in association with Ca2+-CaM, and the peak shifts appear more linear as opposed to curved (data not shown). Taken together, these preliminary data suggest a more complex and dynamic association of the α3-peptide with Ca2+-CaM than for the α1-peptide, which warrants further investigation. Studies are continuing in our group to delineate further the nature of these disorder-to-order transitions for both the α1- and α3-peptides, as well as the synergistic association of the whole protein, with Ca2+-CaM, actin, and SH3-domains.
Conclusions
The inherent structural flexibility of MBP is directly associated with its myriad membrane and protein associations, and thus its multifunctionality (2,3,5,6,9). The interactions of other intrinsically disordered proteins with various binding partners have revealed themes of preformed structural elements, “moonlighting” of specific binding segments, and “fuzziness” (or structural plasticity) of the complexes (67–71). The “fuzziness” has been previously demonstrated in that much of full-length 18.5-kDa MBP is mobile even in association with phospholipid membranes (34,35) or with actin bundles (16,17).
Here, we have focused on three segments of MBP, which we show to have an inherent α-helical structure even in aqueous solution. Two of these regions (α2 and α3) have previously been demonstrated experimentally to form amphipathic α-helices in association with membranes (33,34,36). Because the C-terminal α3-peptide has also been demonstrated to be a Ca2+-CaM-binding target (19,20), it represents an example of “moonlighting” wherein it has different associations and presumably different conformations in each environment (68). Although the N-terminal segment of 18.5-kDa MBP encompassing the α1-peptide associates with lipid bilayers (72), to our knowledge it has yet to be characterized structurally in this state. Here, we have shown that α1 is the minimal MBP segment required to assemble actin, and that it also binds Ca2+-CaM—another example of “moonlighting”.
Acknowledgments
We are grateful to Mrs. Janine Voyer-Grant and Ms. Stina Nilsson for excellent technical assistance, to Mr. Robert Harris (Manager, Guelph Regional Microscopy Facility) for his help with sample preparation and transmission electron microscopy, and to Ms. Valerie Robertson and Mr. Peter Scheffer of the University of Guelph NMR Centre for instrumental support. We thank Dr. Joan Boggs (Department of Molecular Structure and Function, Hospital for Sick Children, Toronto), Dr. Vladimir Ladizhansky (Physics, Guelph), and Dr. David Libich (Laboratory of Chemical Physics, National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health, Bethesda, MD) for helpful discussions and comments on the article.
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (No. RG121541 to G.H.). The NMR studies were initially and partially supported by an Operating Grant from the Canadian Institutes of Health Research (MOP No. 74468 to G.H. and Vladimir Ladizhansky). V.V.B. was the recipient of a Postdoctoral Fellowship, and G.S.T.S. and M.D.A. were the recipients of Doctoral Studentships from the Multiple Sclerosis Society of Canada.
Footnotes
Mumdooh A. M. Ahmed's present address is Department of Physics, Faculty of Science at Suez, Suez Canal University, Suez, Egypt.
Supporting Material
References
- 1.Martin R., McFarland H.F., McFarlin D.E. Immunological aspects of demyelinating diseases. Annu. Rev. Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 2.Harauz G., Ishiyama N., Farès C. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503–542. doi: 10.1016/j.micron.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Boggs J.M. Myelin basic protein: a multifunctional protein. Cell. Mol. Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boggs J.M. Nova Science Publishers; Hauppauge, NY: 2008. Myelin Basic Protein. [Google Scholar]
- 5.Harauz G., Libich D.S. The classic basic protein of myelin—conserved structural motifs and the dynamic molecular barcode involved in membrane adhesion and protein-protein interactions. Curr. Protein Pept. Sci. 2009;10:196–215. doi: 10.2174/138920309788452218. [DOI] [PubMed] [Google Scholar]
- 6.Harauz G., Ladizhansky V., Boggs J.M. Structural polymorphism and multifunctionality of myelin basic protein. Biochemistry. 2009;48:8094–8104. doi: 10.1021/bi901005f. [DOI] [PubMed] [Google Scholar]
- 7.Kim J.K., Mastronardi F.G., Moscarello M.A. Multiple sclerosis: an important role for post-translational modifications of myelin basic protein in pathogenesis. Mol. Cell. Proteomics. 2003;2:453–462. doi: 10.1074/mcp.M200050-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Bates I.R., Matharu P., Harauz G. Characterization of a recombinant murine 18.5-kDa myelin basic protein. Protein Expr. Purif. 2000;20:285–299. doi: 10.1006/prep.2000.1307. [DOI] [PubMed] [Google Scholar]
- 9.Libich D.S., Ahmed M.A.M., Harauz G. Fuzzy complexes of myelin basic protein—NMR spectroscopic investigations of a polymorphic organizational linker of the central nervous system. Biochem. Cell Biol. 2010;88:143–155. doi: 10.1139/o09-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Omlin F.X., Webster H.D., Cohen S.R. Immunocytochemical localization of basic protein in major dense line regions of central and peripheral myelin. J. Cell Biol. 1982;95:242–248. doi: 10.1083/jcb.95.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Readhead C., Takasashi N., Hood L. Role of myelin basic protein in the formation of central nervous system myelin. Ann. N. Y. Acad. Sci. 1990;605:280–285. doi: 10.1111/j.1749-6632.1990.tb42401.x. [DOI] [PubMed] [Google Scholar]
- 12.Boggs J.M., Rangaraj G. Interaction of lipid-bound myelin basic protein with actin filaments and calmodulin. Biochemistry. 2000;39:7799–7806. doi: 10.1021/bi0002129. [DOI] [PubMed] [Google Scholar]
- 13.Boggs J.M., Rangaraj G., Harauz G. Effect of arginine loss in myelin basic protein, as occurs in its deiminated charge isoform, on mediation of actin polymerization and actin binding to a lipid membrane in vitro. Biochemistry. 2005;44:3524–3534. doi: 10.1021/bi0473760. [DOI] [PubMed] [Google Scholar]
- 14.Hill C.M.D., Libich D.S., Harauz G. Assembly of tubulin by classic myelin basic protein isoforms and regulation by post-translational modification. Biochemistry. 2005;44:16672–16683. doi: 10.1021/bi050646+. [DOI] [PubMed] [Google Scholar]
- 15.Hill C.M.D., Harauz G. Charge effects modulate actin assembly by classic myelin basic protein isoforms. Biochem. Biophys. Res. Commun. 2005;329:362–369. doi: 10.1016/j.bbrc.2005.01.151. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed M.A.M., Bamm V.V., Ladizhansky V. Induced secondary structure and polymorphism in an intrinsically disordered structural linker of the CNS: solid-state NMR and FTIR spectroscopy of myelin basic protein bound to actin. Biophys. J. 2009;96:180–191. doi: 10.1016/j.bpj.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bamm V.V., Ahmed M.A., Harauz G. Interaction of myelin basic protein with actin in the presence of dodecyl phosphocholine micelles. Biochemistry. 2010;49:6903–6915. doi: 10.1021/bi100308d. [DOI] [PubMed] [Google Scholar]
- 18.Libich D.S., Hill C.M.D., Harauz G. Interaction of the 18.5-kD isoform of myelin basic protein with Ca2+-calmodulin: effects of deimination assessed by intrinsic Trp fluorescence spectroscopy, dynamic light scattering, and circular dichroism. Protein Sci. 2003;12:1507–1521. doi: 10.1110/ps.0303603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Libich D.S., Harauz G. Backbone dynamics of the 18.5 kDa isoform of myelin basic protein reveals transient α-helices and a calmodulin-binding site. Biophys. J. 2008;94:4847–4866. doi: 10.1529/biophysj.107.125823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majava V., Petoukhov M.V., Kursula P. Interaction between the C-terminal region of human myelin basic protein and calmodulin: analysis of complex formation and solution structure. BMC Struct. Biol. 2008;8:10. doi: 10.1186/1472-6807-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majava V., Wang C., Kursula P. Structural analysis of the complex between calmodulin and full-length myelin basic protein, an intrinsically disordered molecule. Amino Acids. 2010;39:59–71. doi: 10.1007/s00726-009-0364-2. [DOI] [PubMed] [Google Scholar]
- 22.Wang C., Neugebauer U., Kursula P. Charge isomers of myelin basic protein: structure and interactions with membranes, nucleotide analogues, and calmodulin. PLoS ONE. 2011;6:e19915. doi: 10.1371/journal.pone.0019915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homchaudhuri L., Polverini E., Boggs J.M. Influence of membrane surface charge and post-translational modifications to myelin basic protein on its ability to tether the Fyn-SH3 domain to a membrane in vitro. Biochemistry. 2009;48:2385–2393. doi: 10.1021/bi8022587. [DOI] [PubMed] [Google Scholar]
- 24.Polverini E., Rangaraj G., Harauz G. Binding of the proline-rich segment of myelin basic protein to SH3 domains: spectroscopic, microarray, and modeling studies of ligand conformation and effects of posttranslational modifications. Biochemistry. 2008;47:267–282. doi: 10.1021/bi701336n. [DOI] [PubMed] [Google Scholar]
- 25.Smith G.S.T., Chen L., Harauz G. The interaction of zinc with membrane-associated 18.5 kDa myelin basic protein: an attenuated total reflectance-Fourier transform infrared spectroscopic study. Amino Acids. 2010;39:739–750. doi: 10.1007/s00726-010-0513-7. [DOI] [PubMed] [Google Scholar]
- 26.Smith G.S.T., Paez P.M., Harauz G. Classical 18.5-and 21.5-kDa isoforms of myelin basic protein inhibit calcium influx into oligodendroglial cells, in contrast to Golli isoforms. J. Neurosci. Res. 2011;89:467–480. doi: 10.1002/jnr.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boggs J.M., Rangaraj G., Heng Y.M. Effect of phosphorylation of myelin basic protein by MAPK on its interactions with actin and actin binding to a lipid membrane in vitro. Biochemistry. 2006;45:391–401. doi: 10.1021/bi0519194. [DOI] [PubMed] [Google Scholar]
- 28.Dyer C.A., Philibotte T.M., Billings-Gagliardi S. Myelin basic protein mediates extracellular signals that regulate microtubule stability in oligodendrocyte membrane sheets. J. Neurosci. Res. 1994;39:97–107. doi: 10.1002/jnr.490390112. [DOI] [PubMed] [Google Scholar]
- 29.Dyer C.A., Philibotte T.M., Wolf M.K. Cytoskeleton in myelin-basic-protein-deficient shiverer oligodendrocytes. Dev. Neurosci. 1995;17:53–62. doi: 10.1159/000111273. [DOI] [PubMed] [Google Scholar]
- 30.Dyer C.A., Phillbotte T., Billings-Gagliardi S. Regulation of cytoskeleton by myelin components: studies on shiverer oligodendrocytes carrying an Mbp transgene. Dev. Neurosci. 1997;19:395–409. doi: 10.1159/000111237. [DOI] [PubMed] [Google Scholar]
- 31.Smith G.S.T., Petley-Ragan L.M., Harauz G. Classic isoforms of myelin basic protein associate with the cytoskeleton in oligodendroglial cells during ruffling. Trans. Am. Soc. Neurochem. 2010 41st Annual Meeting, OP05-01. [Google Scholar]
- 32.Smith G.S.T., De Avila M., Harauz G. Proline substitutions and threonine pseudo-phosphorylation of the SH3-ligand of 18.5 kDa myelin basic protein decrease affinity for the Fyn-SH3-domain and alter process development and protein localization in oligodendrocytes. J. Neurosci. Res. 2011 doi: 10.1002/jnr.22733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Homchaudhuri L., De Avila M., Boggs J.M. Secondary structure and solvent accessibility of a calmodulin-binding C-terminal segment of membrane-associated myelin basic protein. Biochemistry. 2010;49:8955–8966. doi: 10.1021/bi100988p. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed M.A.M., Bamm V.V., Ladizhansky V. Solid-state NMR spectroscopy of membrane-associated myelin basic protein—conformation and dynamics of an immunodominant epitope. Biophys. J. 2010;99:1247–1255. doi: 10.1016/j.bpj.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong L., Bamm V.V., Ladizhansky V. Solid-state NMR spectroscopy of 18.5 kDa myelin basic protein reconstituted with lipid vesicles: spectroscopic characterization and spectral assignments of solvent-exposed protein fragments. Biochim. Biophys. Acta. 2007;1768:3193–3205. doi: 10.1016/j.bbamem.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bates I.R., Feix J.B., Harauz G. An immunodominant epitope of myelin basic protein is an amphipathic α-helix. J. Biol. Chem. 2004;279:5757–5764. doi: 10.1074/jbc.M311504200. [DOI] [PubMed] [Google Scholar]
- 37.Oldfield C.J., Cheng Y., Dunker A.K. Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 38.Mohan A., Oldfield C.J., Uversky V.N. Analysis of molecular recognition features (MoRFs) J. Mol. Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 39.Vacic V., Oldfield C.J., Dunker A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Libich D.S., Harauz G. Solution NMR and CD spectroscopy of an intrinsically disordered, peripheral membrane protein: evaluation of aqueous and membrane-mimetic solvent conditions for studying the conformational adaptability of the 18.5 kDa isoform of myelin basic protein (MBP) Eur. Biophys. J. 2008;37:1015–1029. doi: 10.1007/s00249-008-0334-8. [DOI] [PubMed] [Google Scholar]
- 41.Farès C., Libich D.S., Harauz G. Solution NMR structure of an immunodominant epitope of myelin basic protein. Conformational dependence on environment of an intrinsically unstructured protein. FEBS J. 2006;273:601–614. doi: 10.1111/j.1742-4658.2005.05093.x. [DOI] [PubMed] [Google Scholar]
- 42.Bessonov K., Bamm V.V., Harauz G. Misincorporation of the proline homologue Aze (azetidine-2-carboxylic acid) into recombinant myelin basic protein. Phytochemistry. 2010;71:502–507. doi: 10.1016/j.phytochem.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 43.Polverini E., Coll E.P., Harauz G. Conformational choreography of a molecular switch region in myelin basic protein—molecular dynamics shows induced folding and secondary structure type conversion upon threonyl phosphorylation in both aqueous and membrane-associated environments. Biochim. Biophys. Acta. 2011;1808:674–683. doi: 10.1016/j.bbamem.2010.11.030. [DOI] [PubMed] [Google Scholar]
- 44.De Avila M., Ahmed M.A.M., Harauz G. Modes of SH3-domain interactions of 18.5 kDa myelin basic protein in vitro and in oligodendrocytes. 55th Annual Meeting of the Biophysical Society, Baltimore, MD, 5–9 March. Biophys. J. 2011 Abstract B164. [Google Scholar]
- 45.Hill C.M.D., Haines J.D., Harauz G. Terminal deletion mutants of myelin basic protein: new insights into self-association and phospholipid interactions. Micron. 2003;34:25–37. doi: 10.1016/s0968-4328(02)00058-6. [DOI] [PubMed] [Google Scholar]
- 46.Rodríguez-Castañeda F., Coudevylle N., Griesinger C. 1H, 13C and 15N resonance assignments of the Calmodulin-Munc13-1 peptide complex. Biomol. NMR Assign. 2010;4:45–48. doi: 10.1007/s12104-009-9204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malakhov M.P., Mattern M.R., Butt T.R. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J. Struct. Funct. Genomics. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 48.Satakarni M., Curtis R. Production of recombinant peptides as fusions with SUMO. Protein Expr. Purif. 2011;78:113–119. doi: 10.1016/j.pep.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 49.Polverini E., Fasano A., Cavatorta P. Conformation of bovine myelin basic protein purified with bound lipids. Eur. Biophys. J. 1999;28:351–355. doi: 10.1007/s002490050218. [DOI] [PubMed] [Google Scholar]
- 50.Roccatano D., Colombo G., Mark A.E. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: a molecular dynamics study. Proc. Natl. Acad. Sci. USA. 2002;99:12179–12184. doi: 10.1073/pnas.182199699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Povey J.F., Smales C.M., Howard M.J. Comparison of the effects of 2,2,2-trifluoroethanol on peptide and protein structure and function. J. Struct. Biol. 2007;157:329–338. doi: 10.1016/j.jsb.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 52.Doig A.J. Stability and design of α-helical peptides. Prog. Mol. Biol. Transl. Sci. 2008;83:1–52. doi: 10.1016/S0079-6603(08)00601-6. [DOI] [PubMed] [Google Scholar]
- 53.Otzen D.E., Sehgal P., Nesgaard L.W. Alternative membrane protein conformations in alcohols. Biochemistry. 2007;46:4348–4359. doi: 10.1021/bi700091r. [DOI] [PubMed] [Google Scholar]
- 54.Drin G., Antonny B. Amphipathic helices and membrane curvature. FEBS Lett. 2010;584:1840–1847. doi: 10.1016/j.febslet.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 55.Musse A.A., Boggs J.M., Harauz G. Deimination of membrane-bound myelin basic protein in multiple sclerosis exposes an immunodominant epitope. Proc. Natl. Acad. Sci. USA. 2006;103:4422–4427. doi: 10.1073/pnas.0509158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoeflich K.P., Ikura M. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 2002;108:739–742. doi: 10.1016/s0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 57.Lukas T.J., Burgess W.H., Watterson D.M. Calmodulin binding domains: characterization of a phosphorylation and calmodulin binding site from myosin light chain kinase. Biochemistry. 1986;25:1458–1464. doi: 10.1021/bi00354a041. [DOI] [PubMed] [Google Scholar]
- 58.Polverini E., Boggs J.M., Cavatorta P. Electron paramagnetic resonance spectroscopy and molecular modeling of the interaction of myelin basic protein (MBP) with calmodulin (CaM)-diversity and conformational adaptability of MBP CaM-targets. J. Struct. Biol. 2004;148:353–369. doi: 10.1016/j.jsb.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 59.Libich D.S., Hill C.M.D., Harauz G. Myelin basic protein has multiple calmodulin-binding sites. Biochem. Biophys. Res. Commun. 2003;308:313–319. doi: 10.1016/s0006-291x(03)01380-9. [DOI] [PubMed] [Google Scholar]
- 60.Duplicated reference deleted in proof.
- 61.Roth G.A., Gonzalez M.D., Cumar F.A. Myelin basic protein domains involved in the interaction with actin. Neurochem. Int. 1993;23:459–465. doi: 10.1016/0197-0186(93)90130-w. [DOI] [PubMed] [Google Scholar]
- 62.Baryłko B., Dobrowolski Z. Ca2+-calmodulin-dependent regulation of F-actin-myelin basic protein interaction. Eur. J. Cell Biol. 1984;35:327–335. [PubMed] [Google Scholar]
- 63.Dobrowolski Z., Osińska H., Baryłko B. Ca2+-calmodulin-dependent polymerization of actin by myelin basic protein. Eur. J. Cell Biol. 1986;42:17–26. [PubMed] [Google Scholar]
- 64.Hartwig J.H., Thelen M., Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–622. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 65.Harauz G., Ishiyama N., Bates I.R. Analogous structural motifs in myelin basic protein and in MARCKS. Mol. Cell. Biochem. 2000;209:155–163. doi: 10.1023/a:1007176216360. [DOI] [PubMed] [Google Scholar]
- 66.Liao M.C., Hoos M.D., Van Nostrand W.E. N-terminal domain of myelin basic protein inhibits amyloid β-protein fibril assembly. J. Biol. Chem. 2010;285:35590–35598. doi: 10.1074/jbc.M110.169599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuxreiter M., Simon I., Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004;338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 68.Tompa P., Szász C., Buday L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 69.Fuxreiter M., Tompa P., Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 70.Mészáros B., Tompa P., Dosztányi Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 2007;372:549–561. doi: 10.1016/j.jmb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 71.Tompa P., Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 72.Bates I.R., Boggs J.M., Harauz G. Membrane-anchoring and charge effects in the interaction of myelin basic protein with lipid bilayers studied by site-directed spin labeling. J. Biol. Chem. 2003;278:29041–29047. doi: 10.1074/jbc.M302766200. [DOI] [PubMed] [Google Scholar]
- 73.Ahmed M.A.M., Bamm V.V., Ladizhansky V. The BG21 isoform of Golli myelin basic protein is intrinsically disordered with a highly flexible amino-terminal domain. Biochemistry. 2007;46:9700–9712. doi: 10.1021/bi700632x. [DOI] [PubMed] [Google Scholar]
- 74.Cavanagh J., Fairbrother W.J., Skelton N.J. Academic Press, Boston, MA and Elsevier Science; San Diego, CA: 1996. Protein NMR Spectroscopy: Principles and Practice. [Google Scholar]
- 75.Clubb R.T., Thanabal V., Wagner G. A constant-time 3-dimensional triple-resonance pulse scheme to correlate intraresidue H-1(N), N-15, and C-13 chemical-shifts in N-15-C-13-labeled proteins. J. Magn. Reson. 1992;97:213–217. [Google Scholar]
- 76.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 77.Grzesiek S., Bax A. Improved 3D triple-resonance NMR techniques applied to a 31-kDa protein. J. Magn. Reson. 1992;96:432–440. [Google Scholar]
- 78.Grzesiek S., Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 79.Kay L.E., Keifer P., Saarinen T. Pure absorption gradient-enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992;114:10663–10665. [Google Scholar]
- 80.Kay L.E., Xu G.Y., Yamazaki T. Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson. A. 1994;109:129–133. [Google Scholar]
- 81.Keller R. Cantina-Verlag; Goldau, Switzerland: 2007. The Computer-Aided Resonance Assignment Tutorial. [Google Scholar]
- 82.Kouyama T., Mihashi K. Fluorimetry study of n-(1-pyrenyl)iodoacetamide-labeled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- 83.Marion D., Ikura M., Bax A. Rapid recording of 2D NMR spectra without phase cycling—application to the study of hydrogen-exchange in proteins. J. Magn. Reson. 1989;85:393–399. [Google Scholar]
- 84.Mossessova E., Lima C.D. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 85.Muhandiram D.R., Kay L.E. Gradient-enhanced triple-resonance 3-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. B. 1994;103:203–216. [Google Scholar]
- 86.Palmer A.G., Cavanagh J., Rance M. Sensitivity improvement in proton-detected 2-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 1991;93:151–170. [Google Scholar]
- 87.Pardee J.D., Spudich J.A. Purification of muscle actin. Methods Cell Biol. 1982;24:271–289. doi: 10.1016/s0091-679x(08)60661-5. [DOI] [PubMed] [Google Scholar]
- 88.Porumb T., Crivici A., Ikura M. Calcium binding and conformational properties of calmodulin complexed with peptides derived from myristoylated alanine-rich C kinase substrate (MARCKS) and MARCKS-related protein (MRP) Eur. Biophys. J. 1997;25:239–247. doi: 10.1007/s002490050036. [DOI] [PubMed] [Google Scholar]
- 89.Schägger H., von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 90.Schleucher J., Sattler M., Griesinger C. Coherence selection by gradients without signal attenuation—application to the 3-dimensional HNCO experiment. Angew. Chem. Int. Ed. Engl. 1993;32:1489–1491. [Google Scholar]
- 91.Schleucher J., Schwendinger M., Griesinger C. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR. 1994;4:301–306. doi: 10.1007/BF00175254. [DOI] [PubMed] [Google Scholar]
- 92.Schwarzinger S., Kroon G.J., Dyson H.J. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 93.Wang Y., Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wittekind M., Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α-carbon and β-carbon resonances in proteins. J. Magn. Reson. B. 1993;101:201–205. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.