

Abstract
A central question in cancer therapy is how individual cells within a population of tumor cells respond to drugs designed to arrest their growth. However, the absolute growth of cells, their change in physical mass, whether cancerous or physiologic, is difficult to measure directly with traditional techniques. Here, we develop live cell interferometry for rapid, real-time quantification of cell mass in cells exposed to a changing environment. We used tunicamycin induction of the unfolded protein stress response in multiple myeloma cells to generate a mass response that was temporally profiled for hundreds of cells simultaneously. Within 2 h, the treated cells were growth suppressed compared to controls, with a few cells in both populations showing a robust increase (+15%) or little change (<5%) in mass accumulation. Overall, live cell interferometry provides a conceptual advance for assessing cell populations to identify, monitor, and measure single cell responses, such as to therapeutic drugs.
Introduction
Interference microscopy is an interesting biophysical approach to measure the spatial distribution of material inside cells and other transparent objects. We have previously shown that an adaptation of this technique, which we call live-cell interferometry (LCI), can sensitively detect and track the nanomechanical properties of hundreds of cells simultaneously (1). LCI can also be used to monitor the dynamic flow of the cytoplasm inside single cells as small indentions are made by highly magnetic probes on the surface of a cell (2). It was determined that an almost instantaneous redistribution of cell material resulted from indentation of the cell surface, which was beyond the detection limit of conventional optical microscopy. These results suggested that changes in cell mass could serve as a sensitive, real-time, and noninvasive marker of cellular fitness. If conducted in a highly parallel fashion, mass measurements could become an effective mechanism for profiling the differential response of cells in a population to internal or external stimuli.
How individual cells regulate their size is poorly understood, as is the relationship between cell mass and well-characterized biochemical pathways. Although quantitative mass measurements of single live cells began in the 1950s (3,4), only recently have newer approaches to increase the speed, precision, and practicality of cellular mass measurements become available. For example, a microelectromechanical systems-based approach has been developed to sensitively measure mass changes in a variety of nonadherent cell types flowing sequentially through a hollow microcantilever resonator (5,6). Our own studies with LCI, and the work of others using optical coherence-based microscopy (7,8), suggest a different and powerful approach for rapidly, simultaneously, measuring the masses of individual cells within large populations of cells.
The physical principal underlying LCI is as follows: The variation in phase imparted to coherent or semicoherent light propagating through a transparent cell body is linearly proportional to the material density of the cell (9–11). Interference microscopy can measure these changes in phase, for micron-sized objects, to a precision exceeding 1/1000 of a wavelength, or better than 0.5 nm for visible light. Cell mass can then be related to the measured phase retardation of each cell as (9) where m is the mass of the cell, α is a constant describing the relationship between phase shift and cell mass, ϕ is the measured fractional phase shift, λ is the illumination wavelength, and integration is performed across the entire cell area, A. Here, α = 1.8 × 10−3 m3kg−1, consistent with Ross (9) as an average value taking into account the usual contents of a cell. The exact value of α is not known, however, based on previous independent measurements, it is assumed that: 1), α remains constant across a wide range of concentrations and 2), α is not likely to vary > ∼5% due to changes in cellular content (9,11,12). Nevertheless, the specific value of α will not affect the accuracy of measurements of comparative growth rates (c.f. Figs. 2 and 3) and relative daughter cell masses after cell division (c.f. Fig. 5). Fig. 1 shows a schematic of the LCI, and typical optical thickness images of adherent and nonadherent cells.
Figure 2.
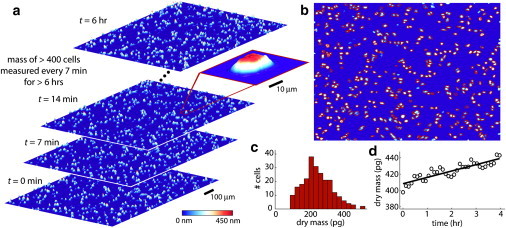
LCI enables high-throughput and longitudinal measurements of cell mass: Four sample images of H929 multiple myeloma cells (a) from the LCI show optical thickness profiles of cells over 6 h of monitoring. Color indicates the phase shift in nm, with dark blue indicating low thickness and white/red indicating high thickness. These sample images are composites of 25 successive charge-coupled device captures taken every 7 min. The inset shows a measurement of the phase shift across a single cell. Integrated phase shift across a cell is directly proportional to cell dry mass. (b) Hundreds of individual cells (outlined in red) are identified at unique positions in each frame and (c) the mass of each individual cell is determined, enabling high-throughput, population-level mass profiling over time. (d) The mass of individual cells is tracked longitudinally over time to examine single-cell growth dynamics. Measurements are shown as open symbols with a linear least squares best fit line. The measured growth rate in this case is 6.5 (se ± 0.72) pg/h. The variation about the linear trend, taken as the standard deviation of the residual error, is 5.0 pg or 1.17% of the median cell mass. The maximum peak-to-peak residual error is 11 pg at 102 min, or 2.61% of the median mass at that time point.
Figure 3.
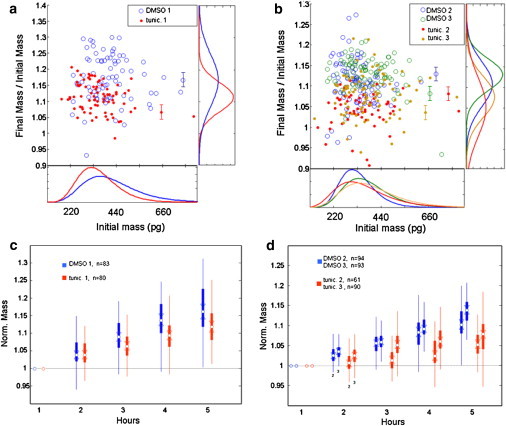
Drug response of H929 multiple myeloma cells profiled by single-cell mass accumulation: Results of LCI longitudinal mass measurements on populations of H929 multiple myeloma cells, comparing the mass accumulation of DMSO-treated controls with TM-treated (10 μg/ml) cells. Data are taken over 5 h as described in Methods. The treated cells grow more slowly than do the controls. Experiments Nos. 2 and 3 were conducted at 32°C vs. 37°C for No. 1, which accounts for the slightly lower overall growth rates observed. The scatter plots (a and b) depict the growth of individual cells at 5 h versus their initial mass (normalized by initial mass). Error bars represent ± 2% CV, our estimate of the measurement error (see Methods). Error bars apply to all data, but are omitted for the majority of points in the plot for clarity. In the box plots of normalized mass versus time (c and d), circles indicate the sample median, and triangles indicate the 95% confidence interval for the median. Solid boxes indicate limits of the 25 and 75 percentiles, and whiskers represent two standard deviations from the mean.
Figure 5.
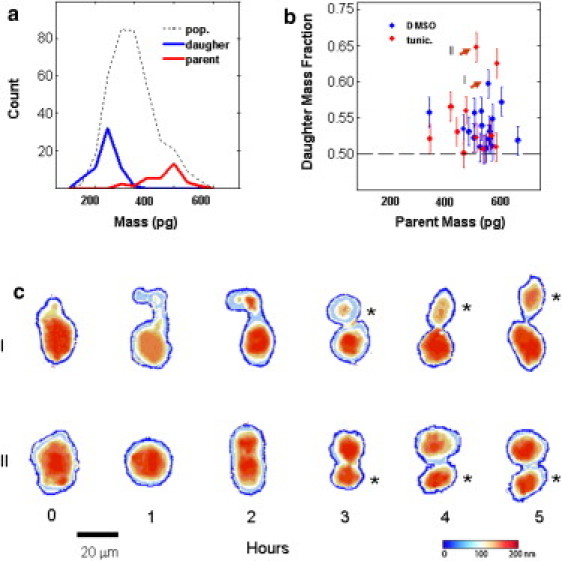
Mass dynamics of cell division: Twenty-eight division events were recorded from the treated and untreated populations in all experiments (from a total of ∼600 cells). (a) We determined the mass range of dividing cells by observing individual divisions and measuring the mass of the parent and daughter cells directly. Panel (a) compares the mass distribution of all cells measured (treated and untreated; dashed line) with the masses of those cells that divided during the experiment (red before the division, blue after division). (b) Surprisingly, a number of cell divisions were highly asymmetric, with ∼55% or more, of the total parent cell mass remaining in the larger of the two daughter cells. (c) Two examples of highly asymmetric division are shown over the 5-h time course. The smaller of the daughter cells in these divisions (indicated by an asterisk) contained 35% and 40%, respectively, of the parent cell mass. These division events are indicated by red-solid circles in (b). Error bars represent ±2% CV, our estimate of the measurement error (see Methods).
Figure 1.
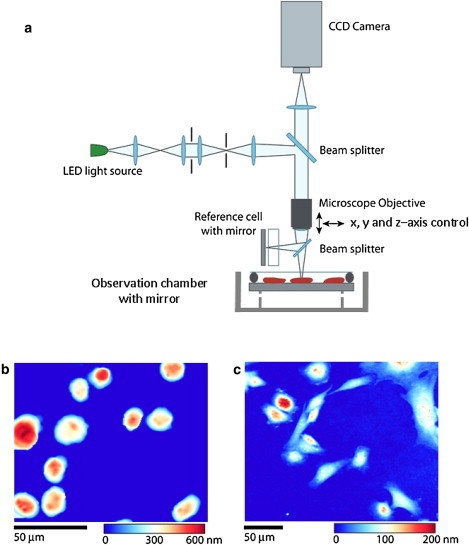
Live cell interferometer (LCI). The LCI (a) is a Michelson-type interference microscope that compares the optical thickness of a reference cell to the optical thickness of samples placed in the observation chamber. Suspended in the observation chamber is a mirrored substrate, allowing the LCI to make measurements of optical thickness on transparent cells. The relative position of the microscope objective and observation chamber is controlled by computer and translatable in three-dimensions allowing for rapid, automated image acquisition. Throughout data collection, cells in the observation chamber are maintained in standard cell culture conditions (e.g., pH 7.4, 37°C, 5% CO2). The LCI is capable of measuring the mass of both adherent and nonadherent cells. Frame (b) shows several nonadherent H929 cells attached to the observation chamber substrate after coating the substrate with Poly-L-Lysine solution, whereas frame (c) shows adherent female Indian Muntjac (9) cells cultured directly on the substrate. The color maps show optical thickness measurements with blue being a low optical thickness relative to background and red being a high optical thickness.
Because it is a wide-field imaging technique, LCI provides simultaneous mass measurements of hundreds of cells (Fig. 2). Throughout the data collection, cells can be maintained in standard culture dishes in physiological conditions (e.g., pH 7.4, 37°C, 5% CO2) enabling periodic, longitudinal measurements for 6 h or longer (Fig. 2 a). With an automated image processing algorithm, hundreds of cells can be identified and mass profiled in each image in rapid succession (Fig. 2 b). In these conditions, the single-cell mass measurements are highly repeatable (<3% CV (coefficient of variation); see Methods: measurement errors). At each time point, therefore, the population-wide distribution of cell mass can be determined (Fig. 2 c). Furthermore, individual cells can be tracked over long periods of time to yield growth rate curves (nonaqueous cell mass changes), as in Fig. 2 d.
Materials and Methods
Interferometer
The live cell interferometer has been described in detail previously (1). Briefly, the system is an optical microscope, based on a modified Veeco NT9300 optical profiler, with a 20X 0.28NA Michelson interference objective that allows for the observation of not only lateral features with typical optical resolution (1.16 μm for the 20× objective) but also height dimensions of reflective objects below the scale of 1 nm. The Michelson interferometer is composed of a beam splitter, reference mirror, and compensating fluid cell to adjust for optical path differences induced by fluid surrounding the specimen. The phase shifting interferometry (PSI) (14) method was used to capture phase images of the cell bodies in situ. During measurement, a piezoelectric translator decreases the light path a small amount causing a phase shift between the test and reference beams. The system records the irradiance of the resulting interference pattern at many different phase shifts and then converts the irradiance to phase wavefront data by integrating the irradiance data using a PSI algorithm. As currently implemented, the autofocus and PSI measurement cycle takes 12 s. The PSI measurement itself takes 1–2 s, and is limited by the camera frame rate (60 fps). In our present experiments, one set of 25 images, containing 400–1000 cells, was captured every 7 min. Each set of 25 images contained hundreds of cells, with data from the first five images presented here, and therefore each run includes ∼80 cells. All cells within each of the selected images were measured.
Data analysis
The software native to the Bruker NT9300 (Bruker, Tucson, AZ) allows automated optical thickness measurements of cells selected manually from the phase image. The optical thickness is converted to mass as described in the text, using the conversion constant, α = 1.8 × 10−3 m3kg−1, consistent with Ross (9). The boundary of each cell was automatically selected by an algorithm that partitions objects from the background using a threshold determined from the histogram of pixel heights (15). Conversion of the raw phase image into optical thickness uses a series of well-established phase unwrapping routines (16). Occasionally, this conversion from phase to optical thickness is incorrect by a factor of negative one wavelength (530 nm), which causes contiguous regions with the cell to have an apparent optical thickness one wavelength less than the true value. This error is easily detected as a nonphysical discontinuity in optical thickness, and corrected by adding back one wavelength of optical thickness to the affected pixels. This process is not currently fully automated.
Quantification of measurement errors
The accuracy of interference microscopy for cell mass measurements is firmly established in the electromagnetic theory (17,18), and by a variety of reference techniques that include ultracentrifugation (3,4,10–12,19–21), refractometry of protein solutions, hydrogels, and transparent films (22–24), x-ray densitometry (25), and electron microcopy (26–30). To characterize the accuracy and stability of our LCI system, we conducted several benchmark experiments, the detail for which is given in the Supporting Material, see Fig. S2, Fig. S3, Fig. S4, and Fig. S5. The lower limit of CV for LCI mass measurements, which is a function of the temporal stability of the interferometric optical path (1.2 Å; Fig. S2 a), was determined to be ∼0.35%. Similar CVs were determined for serial measurements of partially melted polystyrene beads, which simulated cells (CV < 0.4%; Fig. S2 b), and for short repeated measurements of actual live cells (CV < 1%; Fig. S3). We measured populations of 6 μm diameter polystyrene spheres (Fig. S4 a) normally used as calibration standards in flow cytometry (Flow Check, Polysciences), and for which a population mean volume and standard deviation are provided by the manufacturer; the population mass CV determined by LCI (6.8%) was considerably smaller than that determined by the manufacturer (15%). We also measured a population of red blood cells (RBCs) freshly obtained from a 15-week-old female C57BL/6 mouse (Fig. S4, b and c). Mouse RBCs serve as an informative independent standard because there exits an established range of values for average cell mass (determined by photochemical and other methods). Our LCI-determined value of mean RBC cell mass, 19.4 pg, are in excellent agreement with the range of published values at 15–21 pg (9–12,31). Finally, for comparison we measured the masses of populations of a variety of mammalian cell types (Fig. S4 b and Fig. S5). These are plotted together with the mouse RBC and polystyrene sphere data in the Supporting Material and Fig. 3 b. To estimate the scale of measurement variation in multihour live cell experiments, all single-cell mass versus time data, (representing ∼480 cells) were fitted to a simple exponential growth model (mass(t) = m0∗Ct, where the constant C is close to unity) and residual error calculated as the percent difference between the trend and the actual data at each time point (Fig. S6 a). The residuals are symmetrically distributed about zero (Fig. S6 b) and the range between the 25% and 75% quartile varies from 0.0126 (c2) to 0.027 (c3); the mean interquartile range was 0.02. Taken together, these results indicate a lower bound of measurement repeatability on the order of 0.5–1.0% and an outer bound of 2.0–3.0%. The main difference between short- and long-term measurements of live cells is the shape change that occurs over the scale of hours. This can cause added variation in the integrated optical thickness from: 1), small errors in partitioning the cell boundaries; 2), optical averaging of closely spaced fringes present at the edge of rounded cells; and 3), a potential change in the value of α, the mass-to-optical thickness constant, although previous work suggests this error would be relatively small (3). It is established that 1), α is unaffected by changes in concentration, even up to the limit of crystallized protein solutions (9); 2), α reflects the mass interacting with light at a specific location (9–12,31) and is, therefore, not affected by how much area the cell occupies within the field of view as it grows; and 3), the value of α remains close to 0.0018 over a wide range of materials found in cells (32).
Cell lines and tissue culture
H929 human multiple myeloma cells were maintained at 37°C in 5% CO2 in RPMI 1640 growth media supplemented with 10% defined fetal bovine serum (HyClone) and antibiotics. The observation chamber was 4.5 cm in diameter and 1.5 cm deep with a 2 × 2 cm silicon substrate placed on top of a plastic shelf such that the silicon was near the top of the fluid surface. The imaging cell was completed by a piece of optical glass (BK7 glass, Quartz Plus, Brookline, NH) separated from the silicon surface by resting on top of three 600 μm stainless steel beads (Salem Specialty Ball Company, Canton, CT) to create a uniform thickness sample chamber. Media bubbled with 5% CO2 air was continuously flowed through the incubation chamber using a peristaltic perfusion pump at a rate of 0.5 ml/min. The 530 nm wavelength LED illumination (Luxeon Star LED, Brantford, Ontario) incident on the sample chamber had a power of 15 μW spread over a 1.2 mm diameter illumination spot. We have measured cell responses to external stimuli for as long as 7 h in this configuration, and observed unperturbed cultures for up to 12 h, although the upper limit of experiment duration has not been determined.
Drug treatment, cell cycle analysis, and nucleic acid isolation
H929 cells were seeded in 6-well culture plates at a density of 1 × 106 cells/well. Before plating cells in the LCI's observation chamber, either 1 μl of Tunicamycin (TM) (T7765; Sigma-Aldrich) in dimethyl sulfoxide (DMSO), or DMSO alone were added to the media at a concentration of 10 mg/ml, DMSO/media (1:1000 dilution). Mass measurements commenced 1 h after the cells were plated in the observation chamber to allow the experimental system to stabilize, i.e., culture acclimation, temperature stabilization, etc. For cell cycle analysis, cells from each time point were collected and incubated with a hypotonic DNA- staining buffer containing propidium iodide and later analyzed by flow cytometry. RNA for each time point was extracted using the Trizol reagent (Invitrogen).
Reverse transcription-polymerase chain reaction and quantitative RT-PCR
CDNA was synthesized from 3 μg of total RNA with oligo(dT) primers using the Superscript III first strand cDNA synthesis kit (Invitrogen). RT-PCR for XBP1 spliced and unspliced isoforms were performed using Platinum Taq (Invitrogen) at an annealing temperature of 58°C for 25 cycles. Quantitative RT-PCR for CHOP (DDIT3) mRNA was performed using the SYBR green real-time PCR kit (Diagenode) and an Applied Biosystems (Foster City, CA) 7700 sequence detector as described (33). Samples were analyzed for 36b4 expression as a normalization control. Primer sequences are available on request.
Results and Discussion
Mass accumulation dynamics have not been previously reported on a cell-by-cell basis over long time scales (several hours) for an entire population of ∼100 cells measured simultaneously. To test the hypothesis that LCI mass profiling can rapidly determine a response to external cell stimuli, such as a drug response, we exposed H929 multiple myeloma cells to the drug TM, a protein glycosylation inhibitor (34), and compared the growth profiles of TM-treated to untreated control cells by measuring mass continuously over 5 h.
We determined that the initial distribution of H929 cell masses is approximately log-normal, with a range of 200–700 pg. The majority of cells had mass >200 pg and <400 pg, although a small fraction (3–6%) are much larger than average, with masses above 500 pg. Both the treated and untreated populations exhibited growth, but the mass accumulation rate was much lower in the treated cells (Fig. 3). The growth profiles of both populations are clearly heterogeneous (Fig. 3, a and b), and in both, a minority of cells exhibited either a vigorous increase in mass (+15% growth), or little to no mass accumulation (<5% growth). The suppression of growth of the treated population appears within 2 h, and is readily apparent by the fourth hour (Fig. 3, c and d). Thus, whole population detection and quantification of cell drug responses were attained within several hours of treatment. The variation in growth rates within the treated and untreated groups (Fig. 3, c and d) at 5 h approached the magnitude of the variation between treated and untreated cultures at the same time point. These experiments were conducted on separate days, with distinct subcultures taken from a master stock. Therefore, they are biological not technical replicates, and the difference in behavior likely reflects biological variation. We used technical replicates on controlled samples to estimate the measurement error to be <3% CV (see Methods). Nonetheless, we note that the differences in normalized final mass (final/initial) between each treated sample and each untreated sample are statistically significant with p < 0.05 (Fig. 3, c and d). This supports the conclusion that the LCI is capable of detecting differences in growth rates between treated and untreated populations of cells.
At the single cell level, the growth rate of individual cells is largely independent of cell mass, within experimental error, for both treated and untreated cells (see Fig. S1). An exception is treated population Tm1, which showed a statistically significant linear trend toward slower growth in its larger cell subpopulation. The reason for this difference is unclear. Interestingly, the spread in growth rates within any particular mass fraction cannot be explained entirely by measurement error, suggesting a biological origin of this variation as well. This variation, taken as the norm of the residuals of a linear least squares fit to the growth versus mass data, ranges from 3.1–5.8% (Fig. S6), whereas we estimate the mass measurement error is <3% CV (see discussion of errors in Methods).
To link the kinetics of mass accumulation with biochemical signaling, we profiled molecular markers with PCR and conducted a cell cycle analysis on the treated population. The divergence in growth rates between the treated and untreated populations occurs synchronously with the up-regulation of transcription factors CHOP and the spliced form of XBP1 (XBP1-s), in the treated population (Fig. 4, a and b). CHOP and XBP1-s activate a host of genes responsible for mitigating the effects of protein misfolding in the endoplasmic reticulum, through increased production of molecular chaperones to aid protein folding, and accelerated degradation of misfolded proteins (the so-called unfolded protein response, UPR), and the endoplasmic reticulum-associated protein degradation pathway (35). This is consistent with the known mechanism of TM action (34). Both the UPR and endoplasmic reticulum-associated protein degradation molecular pathways are emerging targets for therapeutic intervention in a wide range of diseases, including multiple myeloma.
Figure 4.
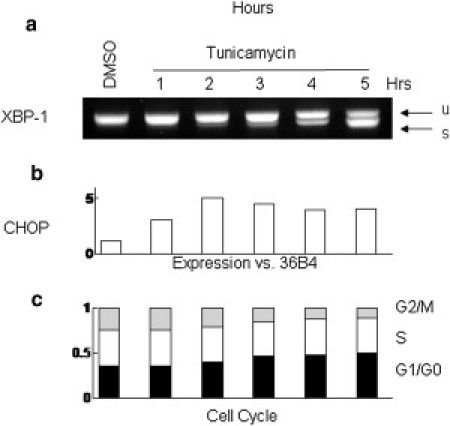
Molecular profile of H929 response to TM. The divergence in growth rates between the treated and untreated populations occurs synchronously with the up-regulation of the transcription factor CHOP (a) and the alternative splicing of transcription factor XBP1 (b) in the treated population. CHOP and XBP1-s activate a host of genes responsible for mitigating the effects of protein misfolding in the endoplasmic reticulum. This is consistent with the known mechanism of TM action, an inhibitor of protein glycosylation. (c) Cell cycle data show a rapid reduction in the G2/M phase population and a corresponding increase in the G1/G0 population, consistent with cell cycle arrest. This shift becomes pronounced after 3 h of treatment, leaving 50% of cells in G1/G0 by the end of 5 h of treatment.
XBP1 is a context-dependent positive or negative regulator of cell growth and differentiation in multiple myeloma cells (34). The molecular dynamics of its bipolar transcriptional potential is not well understood. In the context of our experiments, induction of XBP1 mRNA splicing is associated with slowing mass accumulation, but not cell shrinkage or apoptosis. This time-resolved, nondestructive measurement of cell mass greatly helps interpretations of conflicting pro- and antiproliferate molecular signals, assayed through traditional techniques, including immunohistochemistry or quantitative PCR. Cell cycle data show a rapid reduction in the G2/M phase population and a corresponding increase in the G1/G0 population, consistent with cell cycle arrest (Fig. 4 c). This shift becomes pronounced after 3 h of TM exposure, leaving 50% of the cells in G1/G0 by the end of 5 h of treatment. This is also consistent with observations that activation of the UPR pathway leads to cell cycle arrest (35,36).
We determined the mass range of dividing cells by observing individual divisions and measuring the mass of the parent and daughter cells directly. Twenty-eight cell divisions were observed across all experiments, out of a total of ∼600 cells. The number of divisions was skewed in favor of the untreated population 18:11. This is consistent with the observed higher growth rates in that population. The mass at which a cell divides was tightly regulated, and similar in both treated and untreated populations (Fig. 5 a). The median mass at division was 515 pg (±75 pg), with the two resulting daughter cells each having a median mass of 250 pg (±40pg). This results enable us to infer via mass values that individual cells in the population are likely to be in early-, mid- and late-phases of the cell cycle. Although the mass fraction for the daughter cells was ∼50/50 in most instances, a minority of cell divisions were highly asymmetric, with the smaller of the two daughter cells retaining <45% of the parent's cell mass (Fig. 5 b). Mass maps of two cells undergoing asymmetric cell division are shown in Fig. 5 c.
There are clear advantages of LCI over other established and emerging methods for single-cell mass measurements. Unlike hollow cantilever microelectromechanical mass measurement devices (5,6), which require nonadherent cells, LCI is equally compatible with adherent or nonadherent cells (Fig. 6). The ability to work with adherent cells is absolutely critical for probing the relationship between mass accumulation/distribution and cell-substrate interactions, and for assessing epithelial or stromal cell types, which form the bulk of human malignancies. LCI is also an excellent approach for linking mass profiling with a whole class of cell migration, motility, and tissue invasiveness assays commonly used in drug discovery. The interferometric microscope permits full optical access to the specimen, meaning high resolution light micrographs and fluorescent images are easily obtained. This enables the combined use of mass profiling and the extensive armamentarium of fluorescent reporter assays used in cell biology, for simultaneous assessments. Furthermore, LCI demonstrates tracking and quantification of individual cell masses throughout, and following, cell division. To our knowledge, this will directly enable, for the first time, broad spectrum profiling of mass partitioning in stem cells, for example.
Figure 6.
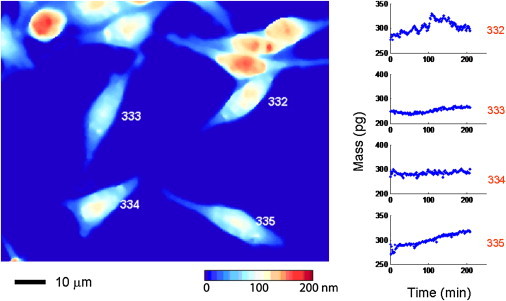
Live cell interferometer measures the mass of adherent cells: Frame shows several mouse fibroblast cells cultured directly on a polished silicon substrate. The color map indicates optical thickness measurements with blue being a low optical thickness relative to background and red being a high optical thickness. To the left are mass measurements of the four cells, as indicated, taken every 2 s for 200 min. The smaller optical thickness of the adherent cells versus nonadherent cells is easily measured by the LCI.
LCI is high-throughput and allows longitudinal measurements of the same cells over time; it is also massively parallel, enabling hundreds of longitudinal measurements simultaneously and reducing interexperiment error due to varying conditions. However, occasionally the conversion from phase to optical thickness is incorrect by a factor of negative one wavelength (530 nm), due to the ambiguity in phase shifts >2π. This situation causes contiguous regions with the cell to have an apparent optical thickness one wavelength less than the true value. This error is easily detected as a nonphysical discontinuity in optical thickness, and corrected by adding back one wavelength of optical thickness to the affected pixels. This correction process is not currently fully automated, although a substantial body of work addressing this issue exists in the literature (16).
We have measured cell responses to external stimuli for as long as 7 h, and observed unperturbed cultures up to 12 h. In principle, measurements can continue for much longer durations because the cells remain viable for days under tightly controlled culture conditions. One limitation, common to LCI and alternative approaches (5,7,8), is the time required for the system to stabilize after cells are introduced into the observation chamber, or after media with a different temperature or density is introduced. In the present experiments, we have conservatively allowed 1 h of settling time, although if required this settling time could be reduced by at least a factor of two.
In summary, high throughput LCI mass profiling is a sensitive and precise mechanism for quantifying single-cell, population-based responses to environmental perturbations, such as medically relevant drug responses.
Acknowledgments
We thank K. Niazi and S. Rabizadeh (Abraxis Bioscience and California NanoSystems Institute, University of California, Los Angeles) for helpful discussions.
This project was supported by National Institute of General Medical Sciences (NIGMS) (R21GM074509), a University of California Discovery/Abraxis Bioscience Biotechnology Award (Bio07-10663) and by the National Institutes of Health Roadmap for Medical Research Nanomedicine Initiative (PN2EY018228).
Contributor Information
Jason Reed, Email: jreed@cnsi.ucla.edu.
Michael A. Teitell, Email: mteitell@ucla.edu.
Supporting Material
References
- 1.Reed J., Frank M., Gimzewski J.K. High throughput cell nanomechanics with mechanical imaging interferometry. Nanotechnology. 2008;19:235101. doi: 10.1088/0957-4484/19/23/235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reed J., Troke J.J., Gimzewski J.K. Live cell interferometry reveals cellular dynamism during force propagation. ACS Nano. 2008;2:841–846. doi: 10.1021/nn700303f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barer R., Joseph S. Refractometry of living cells. Part 1. Basic principles. Q. J. Microsc. Sci. 1954;95:399–423. [Google Scholar]
- 4.Davies H.G., Wilkins M.H.F., Lacour L.F. The use of the interference microscope to determine dry mass in living cells and as a quantitative cytochemical method. Q. J. Microsc. Sci. 1954;95:271–304. [Google Scholar]
- 5.Godin M., Delgado F.F., Manalis S.R. Using buoyant mass to measure the growth of single cells. Nat. Methods. 2010;7:387–390. doi: 10.1038/nmeth.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryan A.K., Goranov A., Manalis S.R. Measurement of mass, density, and volume during the cell cycle of yeast. Proc. Natl. Acad. Sci. USA. 2010;107:999–1004. doi: 10.1073/pnas.0901851107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park Y.K., Diez-Silva M., Suresh S. Refractive index maps and membrane dynamics of human red blood cells parasitized by Plasmodium falciparum. Proc. Natl. Acad. Sci. USA. 2008;105:13730–13735. doi: 10.1073/pnas.0806100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popescu G., Park Y., Badizadegan K. Optical imaging of cell mass and growth dynamics. Am. J. Physiol. Cell Physiol. 2008;295:C538–C544. doi: 10.1152/ajpcell.00121.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross K.F.A. Edward Arnold; London, UK: 1967. Phase Contrast and Interference Microscopy of Cell Biologists. [Google Scholar]
- 10.Barer R. Interference microscopy and mass determination. Nature. 1952;169:366–367. doi: 10.1038/169366b0. [DOI] [PubMed] [Google Scholar]
- 11.Davies H.G., Wilkins M.H.F. Interference microscopy and mass determination. Nature. 1952;169:541. doi: 10.1038/169541a0. [DOI] [PubMed] [Google Scholar]
- 12.Barer R., Ross K.F.A., Tkaczyk S. Refractometry of living cells. Nature. 1953;171:720–724. doi: 10.1038/171720a0. [DOI] [PubMed] [Google Scholar]
- 13.Reference deleted in proof.
- 14.Schmit J., Creath K. Window function influence on phase error in phase-shifting algorithms. Appl. Opt. 1996;35:5642–5649. doi: 10.1364/AO.35.005642. [DOI] [PubMed] [Google Scholar]
- 15.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979;9:62–66. [Google Scholar]
- 16.Ghiglia D., Pritt M. John Wiley & Sons; Hoboken, NJ: 1998. Two-Dimensional Phase Unwrapping: Theory, Algorithms, and Software. [Google Scholar]
- 17.Adair G.S., Ogston A.G., Johnston J.P. Osmotic pressures and sedimentation velocity of Gastrophilus methaemoglobin. Biochem. J. 1946;40:867–869. doi: 10.1042/bj0400867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedersen K.O. Ultracentrifugal and electrophoretic studies on the milk proteins: the lactoglobulin of Palmer. Biochem. J. 1936;30:961–970. doi: 10.1042/bj0300961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adair G.S., Robinson M.E. The specific refraction increments of serum-albumin and serum-globulin. Biochem. J. 1930;24:993–1011. doi: 10.1042/bj0240993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies H.G. On microscope interferometry and the specific refraction increment of a crystalline protein. J. Histochem. Cytochem. 1958;6:393. [Google Scholar]
- 21.Barer R., Joseph S. Refractometry of living cells. 3. Technical and optical methods. Q. J. Microsc. Sci. 1955;96:423–447. [Google Scholar]
- 22.Grampp W., Hallen O., Rosengren B. Mass determination by interference microscopy and x-ray microscopy. A comparative study. Exp. Cell Res. 1960;19:437–442. doi: 10.1016/0014-4827(60)90053-7. [DOI] [PubMed] [Google Scholar]
- 23.Gamble C.N., Glick D. Determination of the total dry mass of human erythrocytes by interference microscopy and x-ray microradiography. J. Histochem. Cytochem. 1960;8:332–333. doi: 10.1083/jcb.8.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ottoson R., Kahn K., Glick D. Studies in histochemistry. 48. Dry mass of mast cells measured by interference microscopy and x-ray absorption. Exp. Cell Res. 1958;14:567–574. doi: 10.1016/0014-4827(58)90161-7. [DOI] [PubMed] [Google Scholar]
- 25.Ruch F., Bahr G.F. Dry mass determination by interference microscopy; agreement with quantitative electron microscopy. Exp. Cell Res. 1970;60:470. doi: 10.1016/0014-4827(70)90547-1. [DOI] [PubMed] [Google Scholar]
- 26.Magnani M., Rossi L., Fornaini G. Effect of age on some properties of mice erythrocytes. Mech. Ageing Dev. 1988;42:37–47. doi: 10.1016/0047-6374(88)90061-9. [DOI] [PubMed] [Google Scholar]
- 27.Myśliwski A., Lass P. Increase of size and dry mass of mouse erythrocytes depending on age of donors. Mech. Ageing Dev. 1985;29:107–110. doi: 10.1016/0047-6374(85)90010-7. [DOI] [PubMed] [Google Scholar]
- 28.Nie Z., Cui F., Yu A.L. High-speed mass analysis of whole erythrocytes by charge-detection quadrupole ion trap mass spectrometry. Anal. Chem. 2007;79:7401–7407. doi: 10.1021/ac071207e. [DOI] [PubMed] [Google Scholar]
- 29.Vaysse J., Vassy R., Pilardeau P. Does red blood cell size correlate with red blood cell age in mouse? Mech. Ageing Dev. 1988;44:265–276. doi: 10.1016/0047-6374(88)90027-9. [DOI] [PubMed] [Google Scholar]
- 30.Wirth-Dzieciolowska E., Karaszewska J., Gajewska M. Selected peripheral blood cell parameters in twelve inbred strains of laboratory mice. Anim. Sci. Pap. Rep. 2009;27:69–77. [Google Scholar]
- 31.Barer R. Refractometry and interferometry of living cells. J. Opt. Soc. Am. 1957;47:545–556. doi: 10.1364/josa.47.000545. [DOI] [PubMed] [Google Scholar]
- 32.Dawson D.W., Hong J.S., Teitell M.A. Global DNA methylation profiling reveals silencing of a secreted form of Epha7 in mouse and human germinal center B-cell lymphomas. Oncogene. 2007;26:4243–4252. doi: 10.1038/sj.onc.1210211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J.Y., Koi M., Feinberg A.P. Simple purification of human chromosomes to homogeneity using muntjac hybrid cells. Nat. Genet. 1994;7:29–33. doi: 10.1038/ng0594-29. [DOI] [PubMed] [Google Scholar]
- 34.Shaffer A.L., Shapiro-Shelef M., Staudt L.M. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 35.Brewer J.W., Diehl J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA. 2000;97:12625–12630. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brewer J.W., Hendershot L.M., Diehl J.A. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc. Natl. Acad. Sci. USA. 1999;96:8505–8510. doi: 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.