

Abstract
It is difficult to investigate the mechanisms that mediate long-term changes in synapse function because synapses are small and deeply embedded inside brain tissue. Although recent fluorescence nanoscopy techniques afford improved resolution, they have so far been restricted to dissociated cells or tissue surfaces. However, to study synapses under realistic conditions, one must image several cell layers deep inside more-intact, three-dimensional preparations that exhibit strong light scattering, such as brain slices or brains in vivo. Using aberration-reducing optics, we demonstrate that it is possible to achieve stimulated emission depletion superresolution imaging deep inside scattering biological tissue. To illustrate the power of this novel (to our knowledge) approach, we resolved distinct distributions of actin inside dendrites and spines with a resolution of 60–80 nm in living organotypic brain slices at depths up to 120 μm. In addition, time-lapse stimulated emission depletion imaging revealed changes in actin-based structures inside spines and spine necks, and showed that these dynamics can be modulated by neuronal activity. Our approach greatly facilitates investigations of actin dynamics at the nanoscale within functionally intact brain tissue.
Introduction
Understanding the structural and molecular mechanisms that mediate synaptic plasticity is one of the central challenges for neurobiological research. Among the large number of proteins and molecular interactions that mediate changes at synapses, the structural protein actin stands out because of its critical role in regulating virtually all aspects of synapse structure and function, including the dynamics of spine morphology, the organization of the postsynaptic density, and the anchoring and trafficking of postsynaptic receptors (1–3).
Because of the small size of synapses, however, it has been next to impossible to resolve the dynamics of actin inside single synapses deeply embedded in functional synaptic networks. Hence, there is a clear need for methods that facilitate nanoscale imaging within physiological contexts such as brain slices. Currently, confocal and two-photon fluorescence microscopes are used for studying synapses in living brain tissue (4). But their resolution is limited by diffraction to ∼200–400 nm, yielding just a few meaningful pixels per synapse. As a result, these conventional techniques fail to properly resolve important subsynaptic structures such as spine necks or actin distributions therein, whose dimensions typically are well below 200 nm (5,6).
Whereas novel light microscopy techniques have recently overcome the diffraction barrier (see Hell (7) and Patterson et al. (8) for reviews), their application in neuroscience has been restricted to imaging dissociated cells or cells close to the surface of brain tissue (<10 μm). The achievable spatial resolution usually deteriorates quickly with imaging depth because of scattering, absorption, and aberrations induced by the sample (9). Commonly used labeling strategies for time-lapse imaging of actin dynamics are problematic as well, because they rely on overexpression of GFP variants fused to actin and thus are likely to perturb normal actin dynamics (10). We have addressed these challenges for imaging actin inside synapses embedded in three-dimensional tissue by developing an approach based on live-cell stimulated emission depletion (STED) fluorescence microscopy (11,12) using Lifeact as a label for actin (10).
STED microscopy has natural advantages over single-molecule superresolution methods (e.g., photo-activated localization microscopy and stochastic optical reconstruction microscopy) for imaging deep inside living tissue. By detecting molecular ensembles from predefined spatial coordinates, confocalized STED microscopy enables superior time resolution and overcomes the limitations that single-molecule movement and background impose on wide-field methods.
To improve penetration depth within the sample, we adapted STED microscopy for use with a high-numerical-aperture glycerol immersion objective lens equipped with a correction collar. This makes it possible to reduce spherical aberrations stemming from the mismatch in refractive index between the immersion medium of the lens and the brain tissue, which would otherwise degrade resolution below tissue surface.
To label actin, we used Lifeact, which is a 17-amino-acid-long actin-binding peptide derived from yeast (10,13,14). It is likely to be less invasive than methods based on overexpressing GFP-actin fusion constructs, and the signal is less prone to bleaching because it binds actin reversibly with micromolar affinity. Lifeact has been shown to be useful for visualizing actin dynamics in several experimental preparations, including dissociated neuronal cultures (10,13,14) and transgenic mice expressing the label (15).
In this study we show that our superresolution approach makes it possible to resolve, with 60–80 nm spatial resolution, distinct distributions of actin inside dendritic spines embedded up to 120 μm within a three-dimensional functional synaptic network. We illustrate the power of this approach by imaging the morphology and dynamics of actin-rich spines and spine necks, which cannot be properly resolved by diffraction-limited light microscopy. Time-lapse STED imaging reveals that spine necks are structurally heterogeneous and highly dynamic, and their widths can be modulated by neuronal activity.
Materials and Methods
Organotypic hippocampal slice cultures
Hippocampal slices (350 μm thick) were prepared from postnatal day 5–7 wild-type C57BL/6 mice, embedded in a plasma clot on 0.14 mm thick glass coverslips, and incubated in a roller incubator at 35°C according to the method of Gähwiler et al. (16). The age of the slice cultures used in the experiments ranged from 12 to 42 days in vitro after the preparation. For the experiments, slice cultures were transferred to a heated chamber (32°C) and perpetually suffused with carbogenated (95% O2, 5% CO2) artificial cerebrospinal fluid (ACSF) at pH 7.4 containing (in mM) NaCl 126, KCl 2.5, CaCl2 2.5, MgCl2 1.3, glucose 30, NaH2PO4 1.25, and NaHCO3 26.
Recording solutions and experimental timeline
Slices were imaged for a baseline period (typically acquiring two time points) before they were subjected to the modified ACSF solutions. Image stacks were taken typically every 5–10 min. The ACSF that was designed to induce chemical long-term potentiation (LTP) contained (in mM) NaCl 99, KCl 5, MgCl2 0.1, CaCl2 5, glucose 24, TEA-Cl 25, NaH2PO4 1.25, and NaHCO3 26, and was carbogenated to maintain a pH of 7.4. This modified ACSF solution was washed in for a period of 7–9 min before it was washed out by the standard ACSF solution. The ACSF designed to block actin polymerization contained (in mM) NaCl 126, KCl 2.5, CaCl2 2.5, MgCl2 1.3, glucose 30, NaH2PO4 1.25, NaHCO3 26, and latrunculin B 0.01, and was carbogenated to maintain a pH of 7.4. For some experiments, we used a slightly different ACSF mixture with a greater concentration of latrunculin B, which contained (in mM) NaCl 126, KCl 2.5, CaCl2 2.5, MgCl2 1.3, glucose 30, HEPES 27, and latrunculin B 0.05; the pH was adjusted with NaOH to 7.4. This modified ACSF solution was washed in for a period of at least 10 min before it was washed out by the standard ACSF solution.
STED microscopy
We used a pulsed-laser diode (Toptica Photonics, Graefelfing, Germany) to deliver excitation pulses with a 490 nm wavelength, 100 ps duration, and average focal power of 0.7–2.8 μW. The pulses for the STED beam were delivered by a Ti:Sapphire laser (MaiTai; Spectra-Physics, Darmstadt, Germany) operating at 80 MHz and emitting at λ = 795 nm; the pulses were converted to λ = 595 nm by an optical parametric oscillator (APE, Berlin, Germany). The pulses of originally 200 fs duration were stretched to ≈300 ps by dispersion in a 25-cm-long glass rod (SF6) and then in a 50-m-long polarization-preserving fiber (OZ Optics, Ottowa, Canada). The maximal focal power of the STED beam was 33.6 mW. To create the STED focal doughnut, a polymeric phase plate (RPC Photonics, Rochester, NY) was introduced into the path of the expanded STED beam, imprinting a helical phase ramp of exp(iϕ), with 0 < ϕ < 2π, onto the wavefront. The STED and excitation pulses were synchronized via external triggering of the laser diode, and the delay was adjusted with an in-house-built electronic delay generator. Both beams were overlapped with a custom-made dichroic mirror and focused into the 1.3 NA objective lens (PL APO, CORR CS, 63×, glycerol; Leica, Wetzlar, Germany). The fluorescence was collected by the same lens, separated by another dichroic mirror, filtered with a 535/50 band-pass, and imaged onto a multimode optical fiber connected to an avalanche photodiode (PerkinElmer, Waltham, MA). The diameter of the fiber corresponded to 1.47 of the back-projected Airy disc, and thus the fiber served as a confocal pinhole. Images were recorded with resonant mirror scanning (15 kHz, SC-30; EOPC, Glendale, NY) along the x axis and piezo-stage scanning (P-733; Physik Instrumente, Karlsruhe, Germany) along the y axis.
The correction collar of the objective lens was designed for coverslips of 0.14–0.18 mm thickness. A glycerol/water (80:20) mixture (type G, ne23 = 1.45; Leica Microsystems, Wetzlar, Germany) was used as an immersion medium for depths < 50 μm, and a refractive index liquid (nD25 = 1.46, series A; Cargille, Cedar Grove, NJ) was used for depths > 50 μm. The correction collar was adjusted for each measurement, and the setting that maximized the fluorescence signal was chosen.
Image acquisition and analysis
Images were recorded line-wise with 18 ms recording time of a line, which is an average over 153 individual lines of the 15 kHz resonant mirror, with a pixel size of 30 nm over a field of view up to 20 μm × 20 μm. The average frame rate was 11 s/frame. Image stacks extended between 1.5 μm to 3.5 μm along the z axis (0.3 μm < Δz < 0.5 μm). All images shown were smoothed by a median filter (2 pixels), but all quantitative analyses were performed on untreated images only. Raw data images were analyzed with the use of ImageJ. First, two-dimensional maximum intensity projections of three-dimensional image stacks were created. Then, values of FWHM were determined from 3-pixel-wide line intensity profiles of the narrowest part of spine necks. The spine head signal intensity was averaged over a small area around the brightest spot inside the spine head and then normalized relative to the average brightness of the dendrite shaft. The spine neck length (defined as the curvilinear distance between the edge of the dendrite shaft to the tip of the spine head) was measured directly.
Transfection and labeling
For transfection, we injected a modified Semliki Forest virus containing the pSCA-Lifeact-EYFP vector construct with a CMV promoter into the CA1 area of the slice cultures using a patch pipette connected to a pressure generator (Picospritzer; Parker, Pine Brook, NJ). The cultures were then incubated overnight for at least 16 h and imaged the following day.
Results
We imaged the dynamic distribution of actin in dendritic spines of living neurons in hippocampal slice cultures using the synthetic peptide Lifeact and STED microscopy. To that end, we built a STED setup based on an inverted confocal microscope, using pulsed laser sources in the visible range for the excitation and STED beams, a resonant beam scanning system, and a recording chamber equipped with heating and a perfusion system to maintain organotypic brain slices (Fig. 1 a). To reduce aberrations associated with imaging lipid-enriched brain tissue, we used a glycerol-immersion lens with a correction collar, which resulted in greatly improved penetration depths of up to 120 μm.
Figure 1.
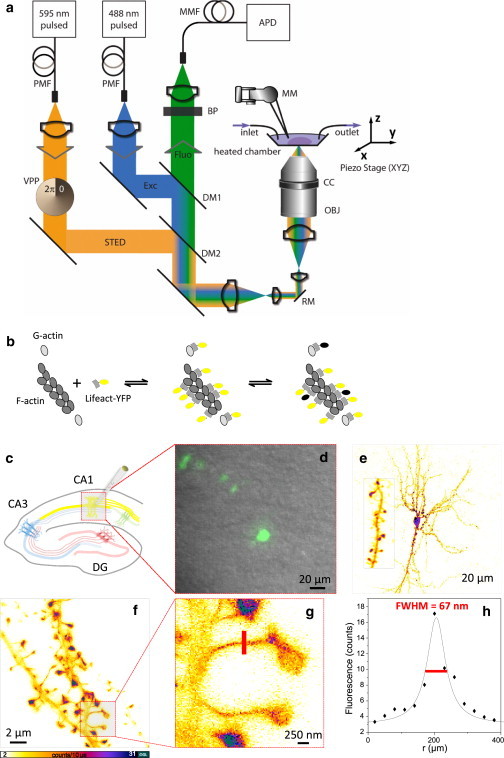
STED microscopy of hippocampal neurons labeled with Lifeact-YFP. (a) Schematic of the experimental setup. A red-shifted STED beam is superimposed on an excitation beam, thus enabling subdiffraction imaging. The imaged hippocampal slice is maintained in a heated chamber (32°C) and perfused with ACSF. PMF, polarization maintaining optical fiber; MMF, multimode optical fiber; VPP, vortex phase plate; DM, dichroic mirror; RM, resonating mirror; BP, bandpass filter; APD, avalanche photodiode; OBJ, glycerol objective lens; CC, correction collar; MM, micromanipulator. (b) Schematic of Lifeact-YFP binding to actin; low binding affinity leads to constant replenishment of potentially bleached YFP. (c) Schematic of the hippocampal formation of a mouse; CA1 pyramidal neurons were labeled with Lifeact-YFP. (d) Overlay of epifluorescence and transmitted light images of CA1 pyramidal neurons. (e) Two-photon image of a Lifeact-infected CA1 pyramidal neuron (4× zoom into a stretch of dendrite). (f) STED image of dendritic structures of CA1 pyramidal neurons. (g) High magnification of the boxed region in f. (h) Profile of pixel intensity across the neck of a spine following the marked line in g, showing an FWHM of the Lorentzian fit of 67 nm.
We expressed the actin label in CA1 pyramidal neurons of hippocampal slice cultures by locally injecting viral particles carrying the Lifeact-YFP construct (Fig. 1, b and c). We checked the transfection of Lifeact-YFP in hippocampal neurons by wide-field fluorescence microscopy (Fig. 1 d). Two-photon images clearly showed that the label was concentrated more highly in dendritic spines than in the parent dendrite (Fig. 1 e). This was expected, because spines are enriched in actin (17). The two-photon images, however, did not reveal any actin substructures within individual spines, which is not surprising given the 300–400 nm spatial resolution of a two-photon microscope.
We then imaged transfected neurons using STED microscopy. Besides confirming the enrichment of the actin label in spines, the STED images revealed details in the subdendritic and subspine distributions of Lifeact-YFP (Fig. 1, f and g), highlighting the improvement in spatial resolution in the focal plane of STED microscopy over diffraction-limited approaches (Fig. 1 h). The sparse labeling of neurons resulted in background-free images that were virtually devoid of autofluorescence from the surrounding tissue. We observed marked spatial variations in the Lifeact signal, presumably reflecting concentration gradients in binding sites for Lifeact associated with F-actin structures. Of interest, the general morphology of the dendrites was also fairly well outlined in the images, presumably because unbound Lifeact diffuses freely inside the cytosol.
Of importance, transfected neurons and their dendritic spines appeared entirely normal in terms of density and shape compared with neurons from Thy-1YFP transgenic mice (12), suggesting that neither the infection nor the label led to any deleterious effects on neuronal viability and morphology within 24 h of transfection. This is consistent with the absence of any signs of cytotoxicity, abnormal morphology, or growth defects after Lifeact transfection in a variety of other cellular preparations, as reported previously (10). In addition, we were able to repeatedly acquire superresolved stacks of images in time-lapse experiments over several hours without blebbing or other signs of photodynamic damage to the neurons.
To confirm that Lifeact labels actin inside dendritic spines, we combined time-lapse imaging with pharmacological experiments using latrunculin B, an actin filament modulator that sequesters actin monomers and promotes depolymerization of actin filaments. If Lifeact indeed labels actin-based structures, the Lifeact signal within spines should be sensitive to the drug. In addition, drug application should also lead to changes in spine shape, because actin inhibitors have been shown to disrupt the morphological integrity of spines (18). Indeed, after bath application of latrunculin B (∼30 min, >10 μM) we observed that the Lifeact signal inside the spines began to disperse and the spines became smaller (see Fig. S1 a in the Supporting Material). We quantified these effects by measuring the spine length (defined as the curvilinear distance between the edge of the dendrite shaft to the tip of the spine head) and spine head signal intensity before and after drug application, as compared with untreated control conditions (Fig. S1 b). Taken together, the drug effects indicate that Lifeact indeed labels actin-based structures, which normally sustain spine morphology.
To estimate the spatial resolution of STED microscopy that can be achieved beyond the immediate surface of the sample (i.e., several cell layers deep), we imaged Lifeact-positive dendrites and spines at different depths below the surface of brain slices. To reduce spherical aberrations, which degrade image quality and effectively limit spatial resolution, we used a glycerol objective lens with a correction collar. We optimized image quality by first recording small confocal images near the intended location for different settings of the correction collar (see Fig. S2). After optimally adjusting the correction collar, we were able to acquire STED images of Lifeact-labeled spines up to 90 μm below the sample surface without compromising image quality or spatial resolution relative to images taken close to the surface (Fig. 2 a). Images could be taken in depths up to 120 μm, but at depths beyond 90–100 μm, the correction collar reached its limit and could no longer be optimally adjusted. To quantitatively confirm the observed resolution, we determined the full width at half-maximum (FWHM) of Lifeact-labeled spine necks, systematically varying the imaging depth and visually selecting thin structures in the images. To guard against noise, we used 3-pixel-wide line profiles. To obtain a sound resolution estimate, we averaged the five lowest FWHM values at each level of imaging depth and plotted the mean value as a function of depth (using data bins ∼20 μm wide; Fig. 2 b). Even at 90 μm depth inside the sample, we consistently achieved a STED microscopy resolution of 60 nm, compared with a resolution of ∼190 nm for the confocal case. At depths beyond 90–100 μm, both STED and confocal resolution started to deteriorate as the limit of the correction collar was reached. Also, the images were noticeably dimmer at depths beyond 90 μm. Despite this, we observed resolutions of 220 nm and 80 nm for confocal and STED imaging, respectively, at depths of 90–120 μm.
Figure 2.
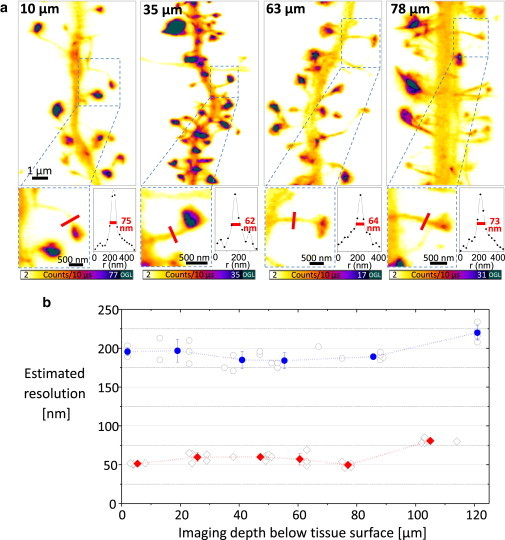
Glycerol objective lens with correction collar improves resolution of STED imaging deep inside brain slices. (a) STED images of dendrites belonging to hippocampal neurons recorded at various depths (from left to right: 10, 35, 63, and 78 μm) below the tissue surface. Tissue surface (z = 0 μm) was defined as the position of the first visible labeled structure. (b) Estimated spatial resolution at various depths inside brain tissue. Displayed are the thinnest spine neck diameters (FWHMs) as a function of imaging depth for both confocal (open circles) and STED imaging (open diamonds).The data bins are 20 μm wide, except for the superficial (0–10 μm) and deepest (90–120 μm) layers. The spatial resolution is estimated for each depth interval by averaging the five smallest FWHM values in that data bin. This leads to an average resolution of 190 nm for confocal microscopy (blue) and 60 nm for STED microscopy (red) at depths up to 90 μm. With the correction collar reaching its limit, the resolution starts to deteriorate at depths > 90 μm.
Next, we determined whether it was possible to resolve spatially restricted distributions of actin within spines and dendrites. Indeed, in many neurons we could discern actin structures, some of which extended from the dendrite to the spine head, that resembled actin bundles observed in spines by electron microscopy (5). We detected different types of geometries, ranging from straight to coiled actin structures (Fig. 3). In some cases, the actin bundles seemed to define the morphology of the spines (Fig. 3, a–c), whereas in others they clearly did not, because they were contained well within the cytosolic volume of the neurons (Fig. 3 d). Furthermore, we observed a variety of different Lifeact distributions inside spine necks and heads, including actin bundles running in parallel inside spine necks (Fig. 3 f), discrete hot spots in spine necks (Fig. 3 g) and heads (Fig. 3, h and i), and ring-like structures around the circumference of spine heads (Fig. 3 j). By acquiring stacks of images by time-lapse microscopy, we were able to observe a variety of spatial rearrangements of actin inside spines occurring rapidly within minutes, including changes in the geometry of actin structures (Fig. 3, k–m) or in the location of actin hotspots (Fig. 3, n–p) inside spines (also see Movie S1).
Figure 3.
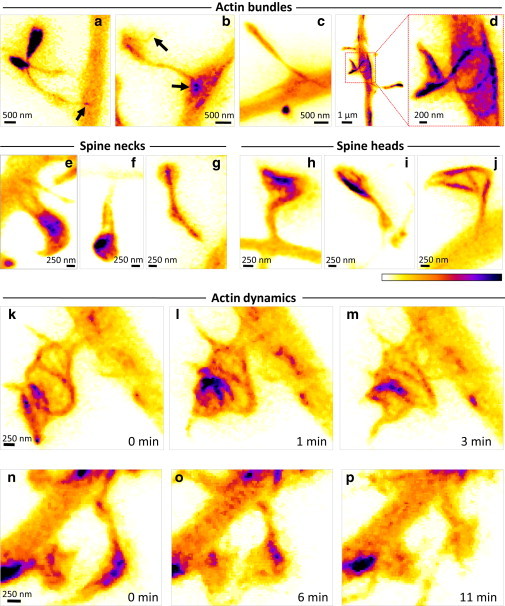
STED microscopy of Lifeact-YFP reveals dynamic organization of actin inside synapses. (a–d) Superresolved live-cell images of actin distributions inside dendritic spines reveal curvilinear bundles linking spine heads to parent dendrites. (e–j) Examples of actin-based substructures that can be seen inside spine necks (e–g) and spine heads (h–j). (k–p) Examples of rapid reorganization of actin distribution inside dendritic spines.
To further explore the power of this novel (to our knowledge) approach, we examined dynamic changes in actin-based spine morphology. Focusing on spine necks with diameters < 250 nm (i.e., below the diffraction limit), we analyzed spine neck dynamics under two different experimental conditions (Fig. 4, a and b).
Figure 4.
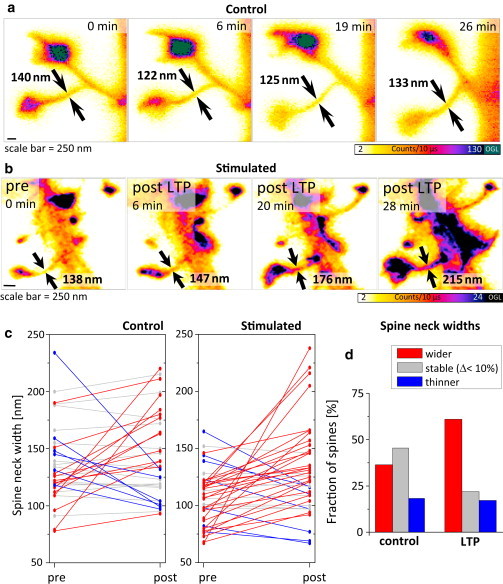
STED imaging of postsynaptic morphological plasticity after chemical LTP stimulation. (a and b) Zoom-in of dendritic spines observed under control conditions (a), as well as before and at subsequent times after chemical LTP stimulation (b). Spine neck diameters are indicated in each frame by arrows. (c) Changes in spine neck diameter after pure ACSF perfusion (control) or chemical LTP treatment. Spine neck growth is shown in red, shrinking in blue, and changes of <10% in gray (paired two-sided t-test, α-error = 0.05; control: p = 0.17, n = 34; LTP: p = 0.00096, n = 41). We checked for normal distribution by examining the residuals and performing a Lilliefors (modified Kolmogorov-Smirnov) test. We assessed the homogeneity of variances by using box plots and performing a Levene test of homogeneity. (d) Ratio of dendritic spines showing a measurable change in spine neck diameter after a certain amount of time (10–30 min) after pure ACSF perfusion (control, n = 74, left) or chemical LTP treatment (n = 64, right).
Under control conditions, the spine necks displayed bidirectional changes in diameter that were substantial in magnitude (mean of absolute value of % changes: <|Δ|> = 24 ± 5%, mean ± standard error (SE)) but did not significantly change the mean value across a population of spine necks between two time points (p = 0.17, paired t-test, n = 34; Fig. 4 a). Next, we used a chemical LTP protocol to induce morphological and functional changes in synapses (19). After bath application of the potentiation medium for 5 min, the spine necks became wider overall (on average by 28%, from 107 ± 4 nm to 131 ± 6 nm, mean ± SE; p < 0.001, paired t-test, n = 41; Fig. 4, b and c). This was reflected in both the larger magnitude of changes and the increased number of spine necks that showed widening (Fig. 4 d). The effect became apparent a few minutes after the stimulation and lasted for >30 min. All of the changes we report here occurred on a much longer timescale than the frame acquisition time (11 s). Motion artifacts, such as a displacement of consecutive lines, were never observed. Taken together, the data obtained by our new approach indicate that spine necks are flexible, dynamic structures that become wider after synaptic plasticity-inducing stimulation.
Discussion
Our study represents a significant advance toward achieving superresolution live-cell imaging deep inside brain tissue. By combining aberration-corrected STED microscopy with Lifeact for labeling actin, we were able to image actin inside synapses up to 120 μm below the tissue surface inside living brain slices with a spatial resolution of 60–80 nm.
The recent development of fluorescence nanoscopy techniques holds great promise for studies of the inner workings of cells and their subcompartments. By offering a spatial resolution well beyond the diffraction limit, these techniques facilitate investigations, which have been largely out of reach for dynamic light microscopy studies. Of importance, they retain the essential benefits of fluorescence microscopy, such as the ability to specifically label and perform live-cell imaging of virtually any cellular structure or protein of interest, and the ease of obtaining relatively large data sets as compared with electron microscopy. On the downside, the depth penetration inside living tissue for these novel approaches has been seriously limited, typically restricting the application of nanoscale imaging to thin samples, such as dissociated cell cultures. This is unfortunate because many important investigations in cellular neuroscience require the use of three-dimensional, thick-tissue preparations, such as acute brain slices or even intact brains, and hence require a method that can image at least a few cell layers below the tissue surface.
As with any other light microscopy technique, the tissue penetration of STED microscopy is limited by scattering and aberrations. Spherical aberrations are generally caused by a mismatch of refractive indices between the immersion medium of the objective lens and the tissue sample (9). To reduce such aberrations, we adapted an inverted STED microscope for use with a glycerol-immersion objective, which was designed for samples embedded in a medium with a refractive index of 1.451 (e.g., glycerol 80%/water 20%). We selected a glycerol objective over water- or oil-immersion objective lenses for several reasons. First, we wanted to better match the refractive index of the brain tissue to the immersion medium of the objective lens. Although the refractive index of our hippocampal brain slices was not known precisely, we made a rough estimate based on the refractive index of various tissue types (e.g., brain, muscle, and liver, ranging from 1.37–1.41) and the refractive index of adipose tissue (1.45) (20–22). Thus, in the case of lipid-enriched brain tissue, the refractive index should be closer to that of glycerol (1.45) than that of water (1.33). Second, the glycerol objective lens had a higher numerical aperture than its water counterpart, and the objective's correction collar provided another degree of freedom to compensate for the remaining refractive index mismatch. The increase in depth penetration we report here makes it much easier to find regions of interest within the sample, and, of more importance, should allow the use of acute brain slices or even intact brains as preparations without sacrificing the subdiffraction resolution enhancement of STED.
It should be possible to push depth penetration and resolution even further by using additional adaptive optical elements to correct higher-order aberrations. For deeper (>100 μm) penetration into the tissue, however, resolution will be increasingly limited by scattering and absorption of the incident photons (23) because the resolution of STED microscopy depends on the peak-to-valley contrast of the focal STED doughnut. Increasing losses in doughnut contrast caused by scattering and absorption will reduce the resolution accordingly. The most common approach for minimizing the detrimental effects of scattering involves the use of two-photon excitation techniques. In a recent study, Ding et al. (24) augmented a two-photon excitation microscope with a single-photon STED beam and recorded images at depths up to 100 μm below the surface of brain slices. They reported a lateral resolution of >240 nm, which, however, is still not sufficient to resolve the fine morphological details of spines and their actin organization.
We have shown that in combination with the actin label Lifeact, STED microscopy can resolve the dynamics of actin within individual dendritic spines of hippocampal neurons embedded in a functional synaptic network. The Lifeact label is particularly well suited for time-lapse imaging by virtue of its low binding affinity for actin, which allows it to rapidly turn over on the actin-binding site, rendering the fluorescence signal less sensitive to bleaching. In addition, the use of an exogenous peptide for labeling endogenous actin avoids problems associated with overexpressing the protein of interest. Moreover, the unbound form of Lifeact produced a useful background signal that outlined the morphology of the dendrites.
During the experiments, we carefully checked for possible photodynamic damage inflicted on the neurons by the STED beam. Usually we were able to select imaging parameters (e.g., laser power and dwell time) that permitted extended time-lapse imaging of the living nervous tissue. The fact that we did not see any signs of excessive heating of the tissue, such as dendrite blebbing, is consistent with a low absorption cross section for 595 nm light, which is the dominant parameter for heating. In fact, in white brain matter, the absorption of light at 595 nm is comparable to the absorption at 900 nm used in two-photon microscopy (25).
Using this approach, we were able to resolve distinct distributions of actin inside dendrites and spines. The actin bundles observed inside spine necks may represent long-distance cytoskeletal links that flexibly anchor spine heads to dendritic shafts (5), and the actin hotspots within spine heads and necks may mark dense scaffolds of branched actin associated with postsynaptic densities and exo-/endocytotic pits at perisynaptic zones (26,27).
Furthermore, the potential of this new approach is demonstrated by the fact that we were able to observe changes in actin distribution and spine morphology that are invisible to traditional approaches. Time-lapse imaging revealed that the width of spine necks is dynamic and changes in response to the induction of chemical LTP. Although previous studies have shown increases in spine volume after the induction of LTP using two-photon approaches (28,29), no direct measurements of these dynamic changes at the level of the spine neck have been reported to date. It remains to be determined whether these changes are due to a specific change in actin accumulation inside spine necks and/or they reflect a genuine increase in spine neck diameter. In any case, our experiments lend support to the view that spine necks act as activity-dependent valves to dynamically regulate the degree of functional coupling between spine synapses and their dendrites, which has been the subject of controversy (30–33).
The novel (to our knowledge) method we have presented here should greatly facilitate experiments on spine morphology and actin dynamics inside synapses in brain slice preparations within a host of physiological contexts.
Acknowledgments
We thank Roland Wedlich-Söldner (Max Planck Institute of Biochemistry) for the Lifeact-YFP construct, Claudia Huber (Max Planck Institute of Neurobiology) for the Lifeact virus, and Frank Voss (Max Planck Institute of Neurobiology) and Tanja Gilat (Max Planck Institute for Biophysical Chemistry) for slice culture work. We thank Sebastian Berning (Max Planck Institute for Biophysical Chemistry) and Johann Engelhardt (German Cancer Research Center) for valuable discussions, and Jan Tonnesen (University of Bordeaux) and Thomas Mrsic-Flogel (University College London) for valuable comments on the manuscript.
Contributor Information
Stefan W. Hell, Email: shell@gwdg.de.
U. Valentin Nägerl, Email: valentin.nagerl@u-bordeaux2.fr.
Supporting Material
References
- 1.Halpain S. Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci. 2000;23:141–146. doi: 10.1016/s0166-2236(00)01576-9. [DOI] [PubMed] [Google Scholar]
- 2.Renner M., Specht C.G., Triller A. Molecular dynamics of postsynaptic receptors and scaffold proteins. Curr. Opin. Neurobiol. 2008;18:532–540. doi: 10.1016/j.conb.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Sheng M., Hoogenraad C.C. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 4.Svoboda K., Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50:823–839. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 5.Korobova F., Svitkina T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell. 2010;21:165–176. doi: 10.1091/mbc.E09-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris K.M., Kater S.B. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 7.Hell S.W. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 8.Patterson G., Davidson M., Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu. Rev. Phys. Chem. 2010;61:345–367. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hell S., Reiner G., Stelzer E.H. Aberrations in confocal fluorescence microscopy induced by mismatches in refractive index. J. Microsc. 1993;169:391–405. [Google Scholar]
- 10.Riedl J., Crevenna A.H., Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat. Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nägerl U.V., Bonhoeffer T. Imaging living synapses at the nanoscale by STED microscopy. J. Neurosci. 2010;30:9341–9346. doi: 10.1523/JNEUROSCI.0990-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nägerl U.V., Willig K.I., Bonhoeffer T. Live-cell imaging of dendritic spines by STED microscopy. Proc. Natl. Acad. Sci. USA. 2008;105:18982–18987. doi: 10.1073/pnas.0810028105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Era A., Tominaga M., Ueda T. Application of Lifeact reveals F-actin dynamics in Arabidopsis thaliana and the liverwort, Marchantia polymorpha. Plant Cell Physiol. 2009;50:1041–1048. doi: 10.1093/pcp/pcp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vidali L., Rounds C.M., Bezanilla M. Lifeact-mEGFP reveals a dynamic apical F-actin network in tip growing plant cells. PLoS ONE. 2009;4:e5744. doi: 10.1371/journal.pone.0005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riedl J., Flynn K.C., Wedlich-Söldner R. Lifeact mice for studying F-actin dynamics. Nat. Methods. 2010;7:168–169. doi: 10.1038/nmeth0310-168. [DOI] [PubMed] [Google Scholar]
- 16.Gähwiler B.H., Capogna M., Thompson S.M. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 1997;20:471–477. doi: 10.1016/s0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- 17.Hotulainen P., Hoogenraad C.C. Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allison D.W., Gelfand V.I., Craig A.M. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J. Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hosokawa T., Rusakov D.A., Fine A. Repeated confocal imaging of individual dendritic spines in the living hippocampal slice: evidence for changes in length and orientation associated with chemically induced LTP. J. Neurosci. 1995;15:5560–5573. doi: 10.1523/JNEUROSCI.15-08-05560.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolin F.P., Preuss L.E., Ference R.J. Refractive index of some mammalian tissues using a fiber optic cladding method. Appl. Opt. 1989;28:2297–2303. doi: 10.1364/AO.28.002297. [DOI] [PubMed] [Google Scholar]
- 21.Dirckx J.J., Kuypers L.C., Decraemer W.F. Refractive index of tissue measured with confocal microscopy. J. Biomed. Opt. 2005;10:44014. doi: 10.1117/1.1993487. [DOI] [PubMed] [Google Scholar]
- 22.Lue N., Bewersdorf J., Popescu G. Tissue refractometry using Hilbert phase microscopy. Opt. Lett. 2007;32:3522–3524. doi: 10.1364/ol.32.003522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmchen F., Denk W. Deep tissue two-photon microscopy. Nat. Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- 24.Ding J.B., Takasaki K.T., Sabatini B.L. Supraresolution imaging in brain slices using stimulated-emission depletion two-photon laser scanning microscopy. Neuron. 2009;63:429–437. doi: 10.1016/j.neuron.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaroslavsky A.N., Schulze P.C., Schwarzmaier H.J. Optical properties of selected native and coagulated human brain tissues in vitro in the visible and near infrared spectral range. Phys. Med. Biol. 2002;47:2059–2073. doi: 10.1088/0031-9155/47/12/305. [DOI] [PubMed] [Google Scholar]
- 26.Frost N.A., Shroff H., Blanpied T.A. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron. 2010;67:86–99. doi: 10.1016/j.neuron.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rácz B., Blanpied T.A., Weinberg R.J. Lateral organization of endocytic machinery in dendritic spines. Nat. Neurosci. 2004;7:917–918. doi: 10.1038/nn1303. [DOI] [PubMed] [Google Scholar]
- 28.Lang C., Barco A., Zakharenko S.S. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc. Natl. Acad. Sci. USA. 2004;101:16665–16670. doi: 10.1073/pnas.0407581101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuzaki M., Honkura N., Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Araya R., Jiang J., Yuste R. The spine neck filters membrane potentials. Proc. Natl. Acad. Sci. USA. 2006;103:17961–17966. doi: 10.1073/pnas.0608755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bloodgood B.L., Giessel A.J., Sabatini B.L. Biphasic synaptic Ca influx arising from compartmentalized electrical signals in dendritic spines. PLoS Biol. 2009;7:e1000190. doi: 10.1371/journal.pbio.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch C., Zador A. The function of dendritic spines: devices subserving biochemical rather than electrical compartmentalization. J. Neurosci. 1993;13:413–422. doi: 10.1523/JNEUROSCI.13-02-00413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svoboda K., Tank D.W., Denk W. Direct measurement of coupling between dendritic spines and shafts. Science. 1996;272:716–719. doi: 10.1126/science.272.5262.716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.