

Abstract
Membrane inlet mass spectrometry (MIMS) uses diffusion across a permeable membrane to detect in solution uncharged molecules of small molecular weight. We point out here the application of MIMS to determine catalytic properties of decarboxylases using as an example catalysis by oxalate decarboxylase (OxDC) from Bacillus subtilis. The decarboxylase activity generates carbon dioxide and formate from the non-oxidative reaction, but is accompanied by a concomitant oxidase activity that consumes oxalate and oxygen and generates CO2 and hydrogen peroxide. The application of MIMS in measuring catalysis by OxDC involves the real-time and continuous detection of oxygen and product CO2 from the ion currents of their respective mass peaks. Steady-state catalytic constants for the decarboxylase activity obtained by measuring product CO2 using MIMS are comparable to those acquired by the traditional endpoint assay based on the coupled reaction with formate dehydrogenase, and measuring consumption of O2 using MIMS also estimates the oxidase activity. Use of isotope-labeled substrate (13C2-enriched oxalate) in MIMS provides a method to characterize the catalytic reaction in cell suspensions by detecting the mass peak for product 13CO2 (m/z 45), avoiding inaccuracies due to endogenous 12CO2.
Keywords: oxalate decarboxylase, mass spectrometry, carbon dioxide, stable isotope, enzyme assay
INTRODUCTION
The use of semi-permeable membranes in the mass spectrometric measurement of the concentration of small uncharged molecules in solution was first demonstrated by Hoch and Kok [1]. The method is based on the permeability of the membrane inlet to dissolved gases while being mostly impermeable to water and charged solutes. Membrane inlet mass spectrometry (MIMS) has been frequently used in the measurement of dissolved organic compounds [2] and in physiological studies [3–5]. The method has been used specifically to measure catalysis by the carbonic anhydrases using the exchange of 18O between CO2 and water [6,7], and in CO2 kinetics in suspensions of human red blood cells [8–10] and cyanobacteria [5]. The method is described in its various applications in several reviews [2,4,11]. However, MIMS has not been applied to the study of production of CO2 in catalysis by the decarboxylases. The aim of this work is to demonstrate the advantages of MIMS in the measurement of catalysis by decarboxylases using specifically oxalate decarboxylase (OxDC) from Bacillus subtilis.
Oxalate decarboxylases are found in a variety of fungi and bacteria [12–17]. They catalyze the aerobic degradation of oxalate into formate and CO2 in a unique non-oxidative mechanism which remains incompletely understood (eq 1) [14,16,18,19]
| (1) |
The current proposed mechanism suggests that dioxygen acts as a reaction cofactor which is not consumed in catalysis [20]. Oxalate oxidase, a different enzyme which structurally resembles oxalate decarboxylase, catalyzes the same oxalate substrate yet consumes the required dioxygen to form two equivalents of carbon dioxide and an equivalent of hydrogen peroxide (eq 2) [19, 21–23].
| (2) |
Oxalate decarboxylase from B. subtilis has been documented to possess a small inherent oxidase activity [20].
Current assays for oxalate decarboxylase activity are endpoint assays in which product formate is measured spectrophotometrically by coupling with formate dehydrogenase and NAD [24]. This does not provide a continuous progress curve for the catalytic reaction. The only documented real-time assay for oxalate decarboxylase involves the use of fourier transform infrared spectroscopy in which oxalate, formate, and carbonate are simultaneously detected [25]. This approach has been used to monitor both substrate and product but not other dissolved gases which may affect catalysis (e.g. NO, O2).
We demonstrate here the use of MIMS to measure the rate of accumulation of CO2 in solution in a direct, continuous, sensitive, and real-time assay of enzymatic activity of oxalate decarboxylase. This method permits simultaneous monitoring of other dissolved gases, such as oxygen which permits concomitant monitoring of the oxalate oxidase activity. Moreover, MIMS is easily adaptable to the use of stable isotope labeling, demonstrated here in the use of 13C-enriched oxalate. MIMS is demonstrated here to estimate steady-state constants for catalysis by B. subtilis oxalate decarboxylase, and to estimate the inherent oxidase activity accompanying the catalysis.
MATERIALS AND METHODS
Materials
Unless otherwise stated, all chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA). Protein concentrations were determined using the Coomassie Protein Plus Kit (Pierce, Rockford, IL) with calibration curves generated utilizing bovine serum albumin as standard. DNA sequencing services were done by the core facility of the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida. Solutions of carbon-13 oxalate (13C2-labeled oxalic acid; 99% 13C; Cambridge Isotope Laboratories - Andover, MA) were prepared by adjusting pH to 4.2 with minimum amounts of KOH.
Oxalate decarboxylase (OxDC)
The OxDC:pET-32a plasmid construct for the polyhistidine-tagged wild-type B. subtilis OxDC was acquired from the research group of Dr. Stephen Bornemann of the John Innes Centre in Norwich, UK. This plasmid was cloned into BL-21/DE3 E. coli cells in which the polyhistidine-tagged wild-type OxDC was expressed and later purified using previously established methods [26, 27], except that grown cells were lysed via sonication. Pooled elution fractions from Ni-NTA agarose (Quiagen) affinity chromatography were eluted through a 100-mL G-25 Sephadex Desalting column equilibrated with 50 mM Tris-Cl (pH 8.5) and 0.5 M NaCl storage buffer.
Enzyme samples were treated with Chelex-100 resin (BioRad) for at least 1 hour with gentle swirling to remove trace metals. Final samples were buffer exchanged into Chelex-treated storage buffer and further concentrated to within 6 – 10 mg/mL using YM-30 Centriprep concentrators (Millipore). Purity was assessed from the 44 kDa protein band on a 12% resolving SDS-PAGE gel, from protein fractions taken at different stages of the purification.
Metal incorporation of purified protein (2.25 mgs) was quantified in 1% nitric acid protein solutions [28] by the ICP-MS [29] services of the University of Wisconsin Soil and Plant Analysis Laboratory. Our samples of OxDC contained 1.4 Mn/monomer. Each monomer contains two manganese binding sites, one of which is catalytic [15–17]. The distribution of metal ions between these two sites is not known. In the kinetic results reported here, the concentration of enzyme has been set equal to the protein content of solution.
Membrane Inlet Mass Spectrometry (MIMS)
MIMS uses a membrane permeable to dissolved gases as an inlet to a mass spectrometer (Figure 1). The inlet probe to the mass spectrometer comprised a 1 cm length of silicon rubber tube (1.5 mm i.d. and 2.0 mm o.d., Silastic a Dow Corning product), which was sealed by a glass bead at one end and interfaced to an Extrel EXM-200 quadrupole mass spectrometer [7]. The inlet probe was immersed in a 2 mL reaction solution contained in a gas-tight quartz cuvette (1 cm pathlength). The reaction vessel was jacketed at a temperature of 25 °C, and sealed with injection septa and Teflon screws for the introduction of samples and inert gas. This apparatus is previously described [30].
Figure 1.
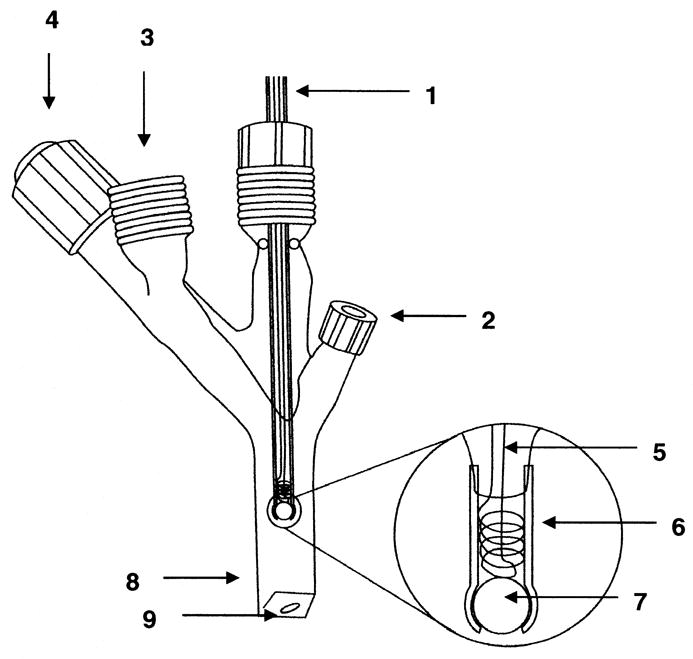
The membrane inlet inserted in an air-tight cell for mass spectrometric measurements: 1) tubing leads to mass spectrometer; 2) sample introduction port with septum; 3) threaded glass port for connecting to vacuum or for introduction of inert gas; 4) vacuum needle valve stopcock; 5) wire helix for support of the Silastic tubing; 6) Silastic tubing of length 1 cm (1.5 mm i.d. and 2.0 mm o.d.); 7) glass bead to seal the Silastic tubing; 8) glass optical cuvette; 9) magnetic stirring bar.
Experiments were initiated by the addition of enzyme or substrate, and masses were recorded continuously using an Extrel EXM-200 mass spectrometer at an electron impact ionization of 70 eV using an emission current close to 1 mA. Source pressures were approximately 1 × 10−6 torr. The reaction vessel was washed between experiments with a solution of 2.5 M KOH and 2.5 M EDTA, and thoroughly rinsed with deionized water in order to prevent any carry over of residual enzyme or unreacted material.
Coupled Enzyme Assays
Oxalate decarboxylase activity was measured through an endpoint assay coupled with NAD-requiring formate dehydrogenase enzyme [26,28]. Assay reaction mixtures were composed of 50 mM sodium acetate buffered at pH 4.2, 0.2% Triton X-100, 0.5 mM o-phenylenediamine, and 4 – 125 mM potassium oxalate. Reactions were initiated with the addition of 1.4 μM wild-type enzyme in a total reaction volume of 100 μL incubated at ambient temperature of 23 –25 °C. The reaction was quenched after a predetermined amount of time with the addition of 1M sodium hydroxide (10 μL) and the amount of formate product was quantified by a coupled assay reaction using 50 mM potassium phosphate buffered at pH 7.8, 1.4 mM NAD+, and 0.4–1.0 U/mg of formate dehydrogenase at a final volume of 1 mL. The amount of NADH was measured by absorbance of assay mixtures at 340 nm after overnight incubation at 37 °C. Corresponding production of formate was quantified against a calibration curve generated from the relative absorbances A340 of pre-quenched OxDC assay mixtures containing known amounts of formate [28].
RESULTS
In demonstrating the application of MIMS to kinetic studies of decarboxylases, we measured the decarboxylation of 13C-labeled oxalate to yield 13CO2 and formate catalyzed by OxDC from B. subtilis. Dissolved gases were detected by immersing the membrane inlet into the air-equilibrated reaction solution and monitoring different masses using the ion current at peak heights. Reactions were carried out in an air-tight vessel such as shown in Figure 1. The catalyzed reaction was initiated by addition of enzyme at time 0 to solutions containing substrate (Figure 2). The progress curve for the accumulation of product CO2 was observed through the m/z 45 peak (13CO2). The data in Figure 2 represent a single experiment; however, the results were very reproducible even when measured on separate days (Supplementary Material Figure 1).
Figure 2.

Ion current (arbitrary scale) in the production of 13CO2 from 13C2-oxalate catalyzed by OxDC. Dissolved gases were monitored from the ion currents at their respective peak heights: (red) 13CO2 at m/z 45; (green) O2 at m/z 32; and (cyan) N2 at m/z 28. The solution contained 50 mM potassium 13C2-oxalate, 0.2% Triton X-100, 50 mM sodium acetate buffer at pH 4.2 and 25°C. Reaction was initiated at time 0 by the addition of (polyhistidine-tagged) wild-type OxDC to a final concentration of 1.4 μM in a total reaction volume of 2 mL.
Dinitrogen, also shown in Figure 2, originated from the air equilibration of this sample and is not generated or consumed in the catalysis; hence, its peak at m/z 28 is a control for the stability of the method. A very slight decrease in the m/z 28 peak over the course of the experiment (Figure 2) is a measure of the rate of loss of N2 from solution by passage across the membrane inlet into the mass spectrometer or into the headspace. This was negligible compared with changes in 13CO2 levels.
A small component of total catalysis by OxDC is an oxidase that consumes oxygen and yields two molecules of CO2 in each catalytic cycle (eq 2). Consumption of oxygen in Figure 2 is observed by the rate of decrease in the m/z 32 peak which is greater than the rate of decrease in the control peak for m/z 28. A further control was performed in a separate experiment in which dioxygen was introduced into the reaction buffer containing oxalate but in the absence of OxDC. The observed rate of signal decay for m/z 32 was negligible (data not shown). Hence the MIMS method has the capability to measure the oxidase function of OxDC in the same experiment as the decarboxylase activity.
Calibration
The membrane inlet mass spectrometer was calibrated by rapid injection into the reaction vessel of solutions of known CO2 concentration. The most accurate procedure was to prepare solutions of K2CO3 at pH 10.2 and inject known volumes into the reaction vessel containing a concentrated solution of acetic acid (pH ~ 2). At this pH, 99.9% of all carbonate species exist as CO2. The ion current at m/z 44 was recorded when it reached a maximum. A plot of ion current versus CO2 concentration was linear (Figure 3). We tested detectability of CO2 by extending the calibration to 5 μM concentration of CO2 (Supplementary Material Figure 2). With the current apparatus, our lower limit of detection was 10 μM with a standard deviation for three experiments at 22%. The instrument was similarly calibrated for O2 by recording the average ion currents at m/z 32 in solutions of different O2 concentration prepared by dilution of air-saturated reaction buffer at 25°C ([O2] is 256 μM) [31]. The resulting plot of ion current versus O2 concentration was linear (not shown). The baseline signal corresponding to 0 μM O2 was verified using degassed reaction buffer containing 1 mg glucose oxidase which was deoxygenated by the addition of 7 mM glucose [20, 32].
Figure 3.
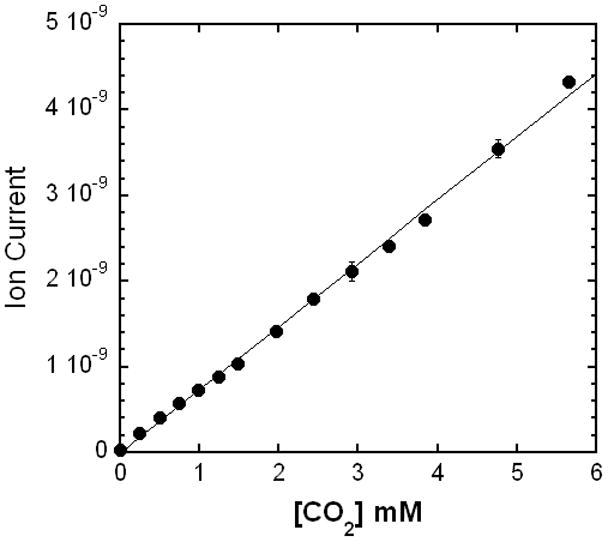
Ion current (arbitrary scale) at m/z 44 using the membrane inlet mass spectrometer observed using solutions of known CO2 content. Solutions containing CO2 were prepared from step additions resulting in 0.25 mM increments of K2CO3 into concentrated acetic acid (pH 2) and 25 °C. Peak heights were recorded when equilibrium was reached. The average of three repetitions with standard deviations was 3% at 4.8 mM CO2, 8% at 3.0 mM CO2, and 10% at 0.4 mM CO2. The solid line is a least-squares fit with a correlation coefficient of 0.999.
Response time
The response time of the apparatus was measured by rapid injection into the reaction vessel of a solution containing CO2 and measuring the time to reach a plateau of ion current at m/z 44. The final concentration of CO2 was approximately 2 mM and the time to reach plateau was adequately fit to a first-order process with a half-time of 4 seconds.
Catalysis by oxalate decarboxylase
Oxalate containing two 13C labels (99% 13C) was used as substrate to distinguish catalytically generated 13CO2 (m/z 45) from pre-existing 12CO2 (m/z 44) in reaction samples. Progress curves were measured to show catalytically generated 13CO2 at various initial concentrations of oxalate; the initial segments of such curves are shown in Figure 4. The slow phase in the beginning 8 – 12 sec is the response time. The initial velocities of the catalytic production of CO2 were determined from the linear slopes of Figure 4 at times 5% to 10% of approximate time to equilibrium. The rate of the uncatalyzed reaction is negligible as is the loss of CO2 into the instrument and headspace. The initial rates of catalysis were adequately fit to a simple Michaelis-Menten curve (Figure 5) with catalytic constants given in Table 1. These constants are in reasonable agreement with those determined by coupled assay using NAD-requiring formate dehydrogenase (Table 1). The Michaelis-Menten plot for the formate dehydrogenase assay is shown in Supplementary Figure 3. From a concomitant decrease in the m/z 32 peak, such as shown in Figure 2, we measured a rate of O2 consumption in catalysis of the oxidation reaction. Comparing with increases in m/z 45, we determined that 0.3% to 0.5% of the rate of overall catalysis by OxDC at saturating substrate (50 mM oxalate) and air equilibration is due to the inherent catalysis of the oxidase reaction (eq 2). Under similar conditions Tanner et al. estimated this value to be 0.2% [20]. This is too small a rate to affect significantly the constants for the decarboxylase activity given in Table 1.
Figure 4.
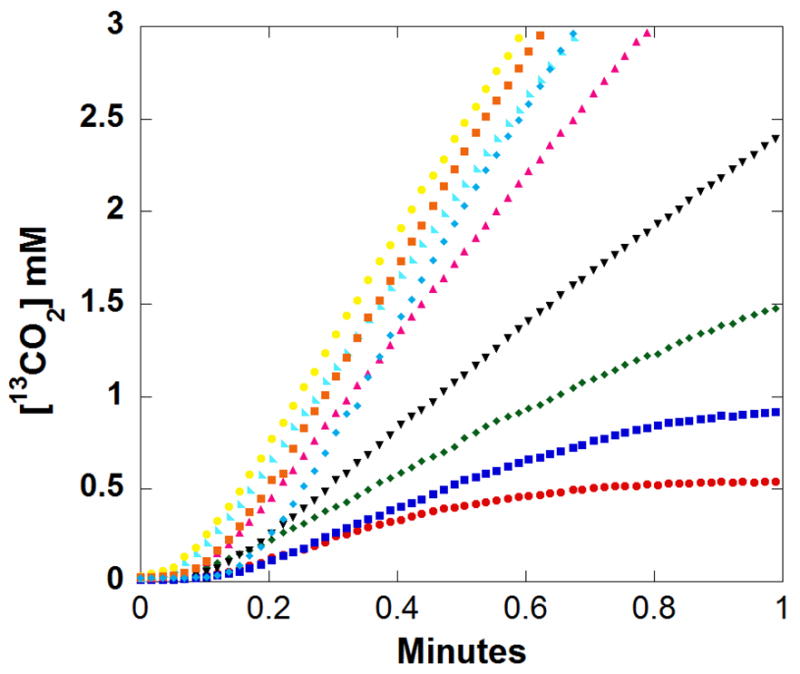
The accumulation of 13CO2 in solution resulting from the catalysis by OxDC. Initial concentrations of 13C2-labeled (99%) oxalate were: (red) 0.5 mM; (dark blue) 1 mM; (green) 2 mM; (black) 4 mM; (purple) 8 mM; (cyan) 16 mM; (yellow) 30 mM; (orange) 50 mM; and (light blue) 80 mM. Each curve for different concentrations of oxalate represents a single experiment. Other components of the solutions were as described in Figure 2.
Figure 5.

The initial rates of the appearance of 13CO2 in solutions resulting from catalysis of the decarboxylation of oxalate by OxDC. Each data point represents an initial velocity measured from the data of Figure 4. The solid line is a fit to the Michaelis-Menten equation with constants for catalysis given in Table 1.
Table 1.
Steady-state constants for the decarboxylation of oxalate catalyzed by polyhistidine-tagged OxDC measured by membrane inlet mass spectrometry and by the formate dehydrogenase coupled assay. Reaction mixtures contained an initial concentration of 50 mM oxalate. Other buffer conditions were as described in Figure 2. Uncertainties are standard errors in the fit to the Michaelis-Menten expression.
| Assay | Km (mM) | kcat (s−1) | kcat/Km (mM−1s−1) |
|---|---|---|---|
| MIMS | 4.0 ± 0.5 | 71 ± 2 | 18 ± 1 |
| Dehydrogenase- coupled Endpoint Assay | 3.9 ± 0.4 | 44 ± 1 | 11 ± 1 |
DISCUSSION
We have shown the applicability of membrane inlet mass spectrometry (MIMS) as applied to catalysis by a decarboxylase. MIMS technology has not been used previously, to our knowledge, to examine decarboxylases. Using MIMS to measure product CO2 in catalysis by OxDC from B. subtilis, we have obtained steady-state constants in reasonable agreement with those by an endpoint assay using formate dehydrogenase (Table 1). The values of kcat and kcat/Km were somewhat larger when measured by MIMS, perhaps related to the advantages of MIMS in being able to provide a sensitive, continuous, and real-time measure of CO2 in solution. Many dissolved gases can be observed in one experiment, shown here to estimate concomitantly both the oxidase and decarboxylase activities of OxDC. From the data of Figure 2, and similar repeated experiments, we estimate that approximately 0.3% to 0.5% of the overall rate of catalytic degradation of oxalate is due to the oxidase pathway of catalysis by OxDC rather than the decarboxylase pathway.
MIMS can be applied to cell suspensions allowing the method to be extended to the study of whole cell physiology. Moreover, stable isotope labeling of substrate can be done with this method to clarify the catalytic mechanism and improve accuracy, for example by use of 13C-enriched oxalate while observing the accumulation of 13CO2. This avoids complications arising from pre-existing 12CO2 contaminants and offers a considerable advantage, especially in experiments evaluating catalysis in a cellular environment.
The design of the inlet is quite flexible and the geometry of the membrane inlet determines to a large extent its sensitivity. The design of the inlet used here (Figure 1) is quite similar to that used previously [2,30]; however, membrane inlets designed as flow cells through silicon rubber tubes are also used to achieve more rapid response times [11]. Other membrane materials have been used, such as Teflon and polyethylene, which allows further adaptations [1].
The MIMS technology offers an advantage compared with the use of the Clark electrode for oxygen in the particular application of measuring oxidase activity of oxalate decarboxylase. In our experience, the use of the Clark electrode for this purpose was accompanied by overestimates of O2 levels due to the presence of high concentrations of CO2. This observation may be associated with the possible buildup of insoluble Ag2CO3 on the oxidized silver electrode resulting in a disrupted electron flow and inaccurate estimates of concentrations of O2. Examining enzymatic reactions in which considerable amounts of CO2 are generated could lead to an artifactual signal using the oxygen electrode.
In conclusion, MIMS provides a direct, continuous, sensitive, and real time assay to monitor decarboxylase activity, yielding results comparable to those acquired from traditional indirect endpoint assays. Importantly, MIMS provides the added feature of simultaneously monitoring other gaseous species providing, for example, a concomitant measure of oxidase activity of OxDC. MIMS measures isotopically labeled products providing a wide flexibility in monitoring effects on reaction catalysis such as addition of inhibitors and changes in reaction conditions. This renders MIMS a versatile tool in studying enzymic catalysis in CO2 generating reactions.
Supplementary Material
Acknowledgments
We thank Stephen Bornemann for a plasmid construct of the polyhisitidine-tagged wild-type B. subtilis OxDC. This work was supported by a grant from the NIH GM25154 and NIH DK061666.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoch G, Kok B. A mass spectrometer inlet system for sampling gases dissolved in liquid phases. Arch Biochem Biophys. 1963;101:160–170. doi: 10.1016/0003-9861(63)90546-0. [DOI] [PubMed] [Google Scholar]
- 2.Brodbelt JS, Cooks RG, Tou JC, Kallos GJ, Dryzga MD. In vivo mass spectrometric determination of organic compounds in blood with a membrane probe. Anal Chem. 1987;59:454–458. doi: 10.1021/ac00130a017. [DOI] [PubMed] [Google Scholar]
- 3.Gerster R. Kinetics of Oxygen Exchange between Gaseous C18O2 and Water. International Journal of Applied Radiation and Isotopes. 1971;22:339. [Google Scholar]
- 4.Lauritsen FR, Lloyd D. Direct-Detection of Volatile Metabolites Produced by Microorganisms - Membrane Inlet Mass-Spectrometry. Mass Spectrometry for the Characterization of Microorganisms. 1994;541:91–106. [Google Scholar]
- 5.Tu C, Spiller H, Wynns GC, Silverman DN. Carbonic Anhydrase and the Uptake of Inorganic Carbon by Synechococcus sp. (UTEX-2380) Plant Physiol. 1987;85:72–77. doi: 10.1104/pp.85.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher SZ, Tu C, Bhatt D, Govindasamy L, Agbandje-McKenna M, McKenna R, Silverman DN. Speeding up proton transfer in a fast enzyme: kinetic and crystallographic studies on the effect of hydrophobic amino acid substitutions in the active site of human carbonic anhydrase II. Biochemistry. 2007;46:3803–3813. doi: 10.1021/bi602620k. [DOI] [PubMed] [Google Scholar]
- 7.Silverman DN. Carbonic anhydrase: oxygen-18 exchange catalyzed by an enzyme with rate-contributing proton-transfer steps. Methods Enzymol. 1982;87:732–752. doi: 10.1016/s0076-6879(82)87037-7. [DOI] [PubMed] [Google Scholar]
- 8.Endeward V, Gros G. Extra- and intracellular unstirred layer effects in measurements of CO2 diffusion across membranes--a novel approach applied to the mass spectrometric 18O technique for red blood cells. J Physiol. 2009;587:1153–1167. doi: 10.1113/jphysiol.2008.165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itada N, Forster RE. Carbonic anhydrase activity in intact red blood cells measured with 18O exchange. J Biol Chem. 1977;252:3881–3890. [PubMed] [Google Scholar]
- 10.Tu C, Wynns GC, McMurray RE, Silverman DN. CO2 kinetics in red cell suspensions measured by 18O exchange. J Biol Chem. 1978;253:8178–8184. [PubMed] [Google Scholar]
- 11.Lewis RS, Deen WM, Tannenbaum SR, Wishnok JS. Membrane mass spectrometer inlet for quantitation of nitric oxide. Biol Mass Spectrom. 1993;22:45–52. doi: 10.1002/bms.1200220106. [DOI] [PubMed] [Google Scholar]
- 12.Kathiara M, Wood DA, Evans CS. Detection and partial characterization of oxalate decarboxylase from Agaricus bisporus. Mycological Research. 2000;104:345–350. [Google Scholar]
- 13.Mehta A, Datta A. Oxalate Decarboxylase from Collybia-Velutipes - Purification, Characterization, and Cdna Cloning. J Biol Chem. 1991;266:23548–23553. [PubMed] [Google Scholar]
- 14.Shimazono H. Oxalic Acid Decarboxylase, a New Enzyme from the Mycelium of Wood Destroying Fungi. J Biochem. 1955;42:321–340. [Google Scholar]
- 15.Shimazono H, Hayaishi O. Enzymatic decarboxylation of oxalic acid. J Biol Chem. 1957;227:151–159. [PubMed] [Google Scholar]
- 16.Emiliani E, Bekes P. Enzymatic Oxalate Decarboxylation in Aspergillus Niger. Arch Biochem Biophys. 1964;105:488–493. doi: 10.1016/0003-9861(64)90040-2. [DOI] [PubMed] [Google Scholar]
- 17.Tanner A, Bornemann S. Bacillus subtilis YvrK is an acid-induced oxalate decarboxylase. J Bact. 2000;182:5271–5273. doi: 10.1128/jb.182.18.5271-5273.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinhardt LA, Svedruzic D, Chang CH, Cleland WW, Richards NG. Heavy atom isotope effects on the reaction catalyzed by the oxalate decarboxylase from Bacillus subtilis. J Am Chem Soc. 2003;125:1244–1252. doi: 10.1021/ja0286977. [DOI] [PubMed] [Google Scholar]
- 19.Svedruzic D, Jonsson S, Toyota CG, Reinhardt LA, Ricagno S, Lindqvist Y, Richards NGJ. The enzymes of oxalate metabolism: unexpected structures and mechanisms. Arch Biochem Biophys. 2005;433:176–192. doi: 10.1016/j.abb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Tanner A, Bowater L, Fairhurst SA, Bornemann S. Oxalate decarboxylase requires manganese and dioxygen for activity - Overexpression and characterization of Bacillus subtilis YvrK and YoaN. J Biol Chem. 2001;276:43627–43634. doi: 10.1074/jbc.M107202200. [DOI] [PubMed] [Google Scholar]
- 21.Chiriboga J. Purification and properties of oxalic acid oxidase. Arch Biochem Biophys. 1966;116:516–523. doi: 10.1016/0003-9861(66)90057-9. [DOI] [PubMed] [Google Scholar]
- 22.Kotsira VP, Clonis YD. Oxalate oxidase from barley roots: purification to homogeneity and study of some molecular, catalytic, and binding properties. Arch Biochem Biophys. 1997;340:239–249. doi: 10.1006/abbi.1997.9896. [DOI] [PubMed] [Google Scholar]
- 23.Lane BG. Oxalate, germin, and the extracellular matrix of higher plants. FASEB J. 1994;8:294–301. doi: 10.1096/fasebj.8.3.8143935. [DOI] [PubMed] [Google Scholar]
- 24.Magro P, Marciano P, Dilenna P. Enzymatic Oxalate Decarboxylation in Isolates of Sclerotinia-Sclerotiorum. Fems Microbiology Letters. 1988;49:49–52. [Google Scholar]
- 25.Muthusamy M, Burrell MR, Thorneley RN, Bornemann S. Real-time monitoring of the oxalate decarboxylase reaction and probing hydron exchange in the product, formate, using fourier transform infrared spectroscopy. Biochemistry. 2006;45:10667–10673. doi: 10.1021/bi060460q. [DOI] [PubMed] [Google Scholar]
- 26.Burrell MR, Just VJ, Bowater L, Fairhurst SA, Requena L, Lawson DM, Bornemann S. Oxalate decarboxylase and oxalate oxidase activities can be interchanged with a specificity switch of up to 282,000 by mutating an active site lid. Biochemistry. 2007;46:12327–12336. doi: 10.1021/bi700947s. [DOI] [PubMed] [Google Scholar]
- 27.Just VJ, Burrell MR, Bowater L, McRobbie I, Stevenson CE, Lawson DM, Bornemann S. The identity of the active site of oxalate decarboxylase and the importance of the stability of active-site lid conformations. Biochem J. 2007;407:397–406. doi: 10.1042/BJ20070708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moomaw EW, Angerhofer A, Moussatche P, Ozarowski A, Garcia-Rubio I, Richards NG. Metal dependence of oxalate decarboxylase activity. Biochemistry. 2009;48:6116–6125. doi: 10.1021/bi801856k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olivares JA. Inductively-coupled plasma mass spectrmetry. Methods Enzymol. 1988;158:205–232. doi: 10.1016/0076-6879(88)58057-6. [DOI] [PubMed] [Google Scholar]
- 30.Tu C, Swenson ER, Silverman DN. Membrane inlet for mass spectrometric measurement of nitric oxide. Free Radic Biol Med. 2007;43:1453–1457. doi: 10.1016/j.freeradbiomed.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 31.Delieu T, Walker DA. Improved Cathode for Measurement of Photosynthetic Oxygen Evolution by Isolated Chloroplasts. New Phytologist. 1972;71:201–225. [Google Scholar]
- 32.Patil PV, Ballou DP. The use of protocatechuate dioxygenase for maintaining anaerobic conditions in biochemical experiments. Analyt Bioch. 2000;286:187–192. doi: 10.1006/abio.2000.4802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.