

Abstract
Group II intron homing occurs primarily by a mechanism in which the intron RNA reverse splices into a DNA target site and is then reverse transcribed by the intron-encoded protein. The DNA target site is recognized by an RNP complex containing the intron-encoded protein and the excised intron RNA. Here, we analyzed DNA target-site requirements for the Lactococcus lactis Ll.LtrB group II intron in vitro and in vivo. Our results suggest a model similar to yeast mtDNA introns, in which the intron-encoded protein first recognizes a small number of nucleotide residues in double-stranded DNA and causes DNA unwinding, enabling the intron RNA to base-pair with the DNA for reverse splicing. Antisense-strand cleavage requires additional interactions between the protein and 3′ exon. Key nucleotide residues are recognized directly by the intron-encoded protein independent of sequence context, and there is a stringent requirement for fixed spacing between target site elements recognized by the protein and RNA components of the endonuclease. Experiments with DNA substrates containing GC-clamps or “bubbles” indicate a requirement for DNA unwinding in the 3′ exon but not the distal 5′ exon region. Finally, by applying the target-site recognition rules, we show that the L1.LtrB intron can be modified to insert at new sites in a plasmid-borne thyA gene in Escherichia coli. This strategy should be generally applicable to retargeting group II introns and to delivering foreign sequences to specific sites in heterologous genomes.
Keywords: DNA target site recognition, functional genomics, gene therapy, group II intron, intron mobility, reverse transcriptase
Mobile group II introns insert into specific DNA target sites by a remarkable mechanism in which the intron RNA reverse splices directly into double-stranded DNA and is then reverse transcribed by the intron-encoded reverse transcriptase (RT; Yang et al. 1996; Eskes et al. 1997; Matsuura et al. 1997; Cousineau et al. 1998). This mechanism is used ordinarily for a process termed retrohoming in which the intron undergoes duplication from an intron-containing to an intronless allele of the same gene. Retrohoming is highly efficient, occurring at frequencies approaching 100% in both organelles and bacteria (Lazowska et al. 1994; Moran et al. 1995; Cousineau et al. 1998, 2000). Attributes of this mechanism, discussed below, suggest that mobile group II introns might be adapted for targeted gene disruption and site-specific insertion of genetic information into DNA genomes.
Studies with the Saccharomyces cerevisiae mtDNA introns aI1 and aI2 and the Lactococcus lactis Ll.LtrB intron showed that retrohoming is dependent on three different activities of the intron-encoded protein: RT, maturase (promotion of RNA splicing), and site-specific DNA endonuclease (Moran et al. 1995; Zimmerly et al. 1995a,b; Matsuura et al. 1997; Cousineau et al. 1998). For splicing, the protein binds specifically to the intron in unspliced precursor RNA and promotes formation of the catalytically active RNA structure (Saldanha et al. 1999; Wank et al. 1999). After splicing, the protein remains associated with the excised intron RNA to form a DNA endonuclease/integrase, which recognizes the DNA target site and initiates mobility (Zimmerly et al. 1995a,b).
In the first step in retrohoming, the intron RNA uses its ribozyme activity to reverse splice into the top (sense) strand of the DNA target. Although this reaction is RNA catalyzed, it is also dependent on the maturase and DNA-binding activities of the intron-encoded protein (Zimmerly et al. 1995b). In the bacterial system, complete reverse splicing results in the insertion of linear intron RNA between the two DNA exons. After reverse splicing, a DNA endonuclease activity associated with the carboxy-terminal zinc finger-like region (Zn domain) of the intron-encoded protein cleaves the bottom (antisense) strand of the DNA target site after position +9 of the 3′ exon. Then, the protein uses the 3′ end of the cleaved antisense strand as a primer to synthesize a cDNA copy of the reverse-spliced intron RNA, and this cDNA is integrated by a RecA-independent repair mechanism (Cousineau et al. 1998).
Studies with the yeast aI1 and aI2 introns showed that both the RNA and protein components of the DNA endonuclease contribute to the recognition of the DNA target site (Guo et al. 1997; Yang et al. 1998). The DNA target site for aI2 extends from position −21 in the 5′ exon (E2) to +10 in the 3′ exon (E3). A 13-nucleotide region encompassing the intron–insertion site (E2−12 to E3+1) is recognized primarily by base-pairing with the intron RNA. This region includes three short, conserved sequence elements denoted intron-binding sites 1 and 2 (IBS1 and IBS2) and δ′ (E3+1), which base-pair with complementary sequences denoted exon binding sites 1 and 2 (EBS1 and EBS2) and δ, located at two different positions in domain I of the intron RNA (diagramed in Fig. 1A,B for the lactococcal intron). Base-pairing interactions between the same sequence elements are required for RNA splicing and also for reverse splicing of group II introns into RNA substrates (Michel and Ferat 1995).
Figure 1.

Target site and intron interactions. (A) DNA target site and EBS–IBS and δ–δ′ interactions. Partial sequence of the 72-bp DNA target site used for biochemical assays is shown. Uppercase letters indicate ltrB sequence, and lowercase letters indicate the 3′ EcoRI site added for cloning. The intron RNA's EBS1, EBS2, and δ sequences that potentially base-pair with the target DNA are shown at top. The positions of the intron–insertion site (top strand) and antisense-strand cleavage site (bottom strand) are indicated. (B) Schematic of the Ll.LtrB intron showing base-pairing interactions with flanking exon sequences (EBS1–IBS1, EBS2–IBS2, δ–δ′). (C) Synthesis of DNA substrates. 32P-Labeled DNA substrates were synthesized by annealing overlapping DNA oligonucleotides (straight lines) and then using Sequenase to fill in single-stranded regions in the presence of a 32P-labeled dNTP (wavy lines). (Top) The 72-bp DNA substrate with the arrows indicating the intron–insertion site (top strand) and antisense-strand cleavage site (bottom strand). (1) oligonucleotides used to introduce mutations into the distal 5′ exon and 3′ exon regions of the DNA target site; (2) oligonucleotides used to introduce IBS and δ′ mutations; (3) oligonucleotides used to introduce mutations between positions +1 and +5 in Fig. 6B. The bubble substrates were made by using the bottom strand oligonucleotide from pair 1 and the top strand oligonucleotide from pair 3 with appropriate mutations (Fig. 6).
The regions of the DNA target site flanking the IBS and δ′ sequences are recognized primarily by the protein subunit of the endonuclease (Guo et al. 1997; Yang et al. 1998). First, the protein recognizes a small number of fixed positions in the distal 5′ exon region of the DNA target site, and this leads to DNA unwinding, enabling the intron to base-pair to the IBS and δ′ sequences for reverse splicing. Antisense-strand cleavage is dependent on reverse splicing and also on additional interactions between the protein and 3′ exon region of the DNA target site.
Because a 13-nucleotide region of the DNA target site is recognized by base-pairing, it is, in principle, possible to design group II intron endonucleases to target specific 13-nucleotide DNA sequences for site-specific cleavage and insertion, taking into account the additional constraints of the fixed nucleotide residues recognized by the intron-encoded protein. Kinetic and thermodynamic analysis of EBS/IBS mismatches in RNA suggested that this system would have high specificity (Xiang et al. 1998; Qin and Pyle 1999). In the case of aI1, as little as a single nucleotide change in the IBS1 sequence strongly inhibited reverse splicing in vitro and abolished intron homing in vivo, and both could be restored by introducing the compensatory change in the EBS1 sequence of the intron RNA (Eskes et al. 1997).
The ability to design group II intron endonucleases to target specific DNA sequences could have widespread applications in genetic engineering and gene therapy. To study the splicing and mobility mechanisms in detail and to pursue these practical applications, we developed an efficient Escherichia coli expression system for the L. lactis Ll.LtrB intron (Matsuura et al. 1997). This expression system enables us to obtain large quantities of purified intron-encoded protein and RNP particles that carry out the splicing and mobility reactions (Saldanha et al. 1999). By using this expression system, we showed that it is possible to insert additional genetic markers into the intron RNA without impairing the mobility reactions (Matsuura et al. 1997), and such genetically marked introns were used to develop L. lactis and E. coli genetic systems in which the intron inserts into its target site in vivo (Cousineau et al. 1998). Thus, the lactococcal intron is the model system of choice for detailed studies of the DNA endonuclease activity, as well as for developing practical applications. Here, we performed in vitro and in vivo studies that elucidate rules for target-site recognition by the Ll.LtrB intron and show that it is possible to use these rules to retarget the Ll.LtrB intron to new sites in the E. coli thyA gene. These findings are the first demonstration of group II intron retargeting in vivo.
Results
Effect of single-nucleotide substitutions in the distal 5′ and 3′ exon regions of the DNA target site on reverse splicing
Nucleotide sequence requirements in the Ll.LtrB target site were analyzed by using a series of mutant DNA substrates having single-nucleotide substitutions at each position between −26 and +10 from the intron–insertion site. In initial experiments, reverse-splicing reactions were performed with RNP particle preparations from E. coli strains expressing pLI1, which contains the full-length Ll.LtrB intron and flanking exons. In this construct, the intron-encoded protein LtrA is translated in E. coli from its own Shine–Dalgarno sequence within the intron. It then splices the Ll.LtrB intron and remains associated with the excised intron in RNP particles having DNA endonuclease activity (Matsuura et al. 1997). For reverse-splicing assays, the RNP particles were incubated with small (72-bp), 32P-labeled DNA substrates containing the target site, and the products were analyzed in a 1% agarose gel (Figs. 1A and 2). The gels show a mixture of products resulting from the two steps of reverse splicing. The first step (“partial” reverse splicing) results in the linkage of intron lariat RNA to the 3′ exon, and the second step (“full” reverse splicing) results in the insertion of linear intron RNA between the two DNA exons (see schematics to right of gel in Fig. 2B). In addition, a substantial proportion of the intron RNA expressed in E. coli is nicked in domain IV by endogenous nucleases but nevertheless remains catalytically active (Matsuura et al. 1997). As a result of these nicks, the reverse-spliced products appear in denaturing agarose gels as a series of radiolabeled bands of >l kb containing nicked RNA and extending up to ∼2.5 kb for products containing the full-length intron RNA.
Figure 2.
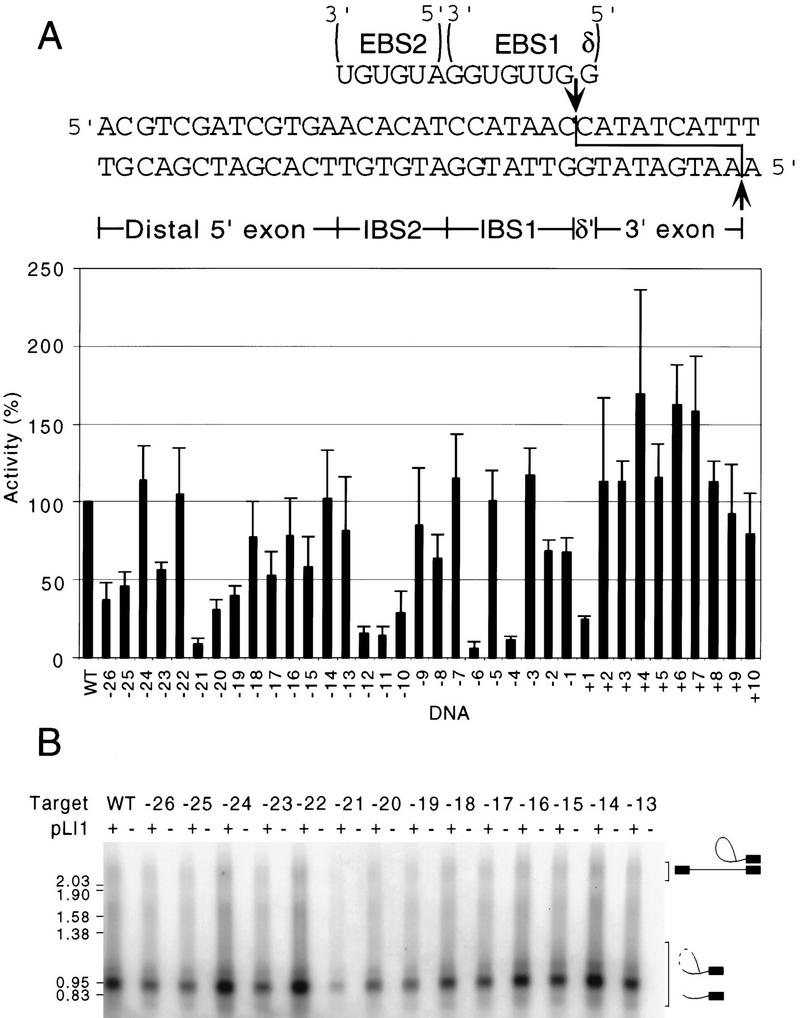
Effect of single-nucleotide substitutions in the DNA target site on reverse splicing. Reverse-splicing assays were performed by incubating pLI1 RNP particles with 32P-labeled 72-bp DNA substrates having single-nucleotide substitutions. Products were analyzed on a 1% agarose gel to detect the intron RNA reverse spliced into the DNA target site. For the distal 5′ exon and 3′ exon regions (positions −26 to −14 and +2 to +10, respectively), mutant DNA substrates have equimolar mixture of three non-wild-type nucleotides at each position. For the IBS and δ′ sequences (−13 to +1), mutant DNA substrates have purines replaced with an equimolar mixture of the two pyrimidines and pyrimidines replaced with an equimolar mixture of the two purines. Position −13 at the boundary between the distal 5′ exon region and IBS2 was tested with both sets of substrates with similar results; shown are data for the substrate having an equimolar mixture of A and G residues at this position. (A) Bar graph summarizing reverse-splicing activity of the mutant DNA target sites relative to the wild-type target site, assayed in parallel. The data are the average of at least three independent experiments, with the s.d. indicated by the thin lines. The wild-type DNA target site sequence is shown at top, with the distal 5′ exon, IBS2, IBS1, δ′, and 3′ exon regions delineated. The intron insertion site in the top strand and the antisense-strand cleavage site in the bottom strand are indicated by arrows. (B) Gel showing representative reverse-splicing assays with DNA substrates having mutations in the distal 5′ exon region of the DNA target site. Lanes labeled + or − indicate DNA substrates incubated with or without RNP particles. Schematics at right show structures of fully and partially reverse spliced products (█, DNA; thin lines, RNA) and products containing nicked or degraded intron RNA (broken lines). The major ∼1-kb band contains 3′ exon DNA attached to a 3′ segment of the intron (see Matsuura et al. 1997). Numbers at left indicate sizes (kb) and positions of molecular mass markers.
For the distal 5′ exon and 3′ exon regions, which are probably recognized by the protein subunit of the endonuclease, we prepared a series of labeled DNA substrates in which equal proportions of the three non-wild-type nucleotides were substituted at each position. Figure 2A summarizes data for mutations at each position of the DNA target, with a representative gel for positions −26 to −14 shown below (Fig. 2B). In the distal 5′ exon region, the most critical position was −21, where the single-nucleotide substitution reduced reverse-splicing activity to ∼9% of that with the wild-type target site. The substitutions at positions −26, −25, −23, −20, −19, −17, and −15 reduced reverse splicing to 31%–58% of the wild-type level, whereas substitutions at other positions had less effect (70%–114% of the wild-type level). Single-nucleotide substitutions in the 3′ exon between positions +2 and +10 did not inhibit reverse splicing significantly, as expected from results with the yeast mtDNA introns (Guo et al. 1997; Yang et al. 1998). In other experiments, increasing the length of the 5′ exon to 40 nucleotides or changing the sequence from −25 to −40 to its complement had no effect on the reverse-splicing efficiency, and a DNA substrate in which segments +2 to +19, +21 to +24, and +30 to +32 were changed to their complements gave ∼50% of the wild-type substrate reverse-splicing activity (not shown). These findings indicate that there are no strong sequence requirements in these regions and that as long as the same GC content is maintained, they have relatively little effect on the efficiency of reverse splicing.
Mutations in the IBS/δ′ region
For the IBS/δ′ region of the DNA target site (positions −13 to +1), which is recognized by the intron RNA, we performed reverse-splicing assays with mutant DNA substrates in which positions having an A or G residue were changed to a mixture of pyrimidines, whereas positions having a C or T residue were changed to a mixture of purines, thereby eliminating the possibility of wobble base pairs. As summarized in Figure 2A, the strongest effects were observed for substitutions at positions −12, −11, −10, −6, −4, and +1, where the activity was reduced to 6%–29% of the wild-type substrate. Mutations at positions −13, −9, −8, −2, and −1 had moderate effects (64%–85% of wild-type activity), whereas mutations at −7, −5, and −3 had little if any effect (101%–117% of wild-type activity). A DNA substrate in which positions −7, −5, and −3 were changed simultaneously to their complement gave ∼70% of wild-type reverse-splicing activity, consistent with the relatively small effects of the single-nucleotide substitutions at these positions (not shown). These findings indicate that residues in the IBS and δ′ regions do not contribute equally to DNA target-site recognition and that some unpaired nucleotides are tolerated.
Direct recognition of nucleotide residues in the distal 5′ exon region of the DNA target site
In general, the critical nucleotide residues in the distal 5′ exon region of the DNA target site could be recognized directly by the protein component of the endonuclease or the mutations at these positions could inhibit recognition indirectly by affecting the structure of the DNA target site. To assess whether the critical nucleotide residues are recognized directly, we started with a mutant DNA substrate in which 10 contiguous positions from −22 to −13 were changed simultaneously to a mixture of the three non-wild-type nucleotide residues (Fig. 3). This target (N) gave ∼4% of the wild-type level of reverse splicing, which was found by additional experiments to reflect about equal utilization of the normal insertion site and cryptic sites on the bottom strand (not shown). Starting with this substrate, restoration of the wild-type nucleotide residues at two (−21, −20), three (−21, −20, −19), four (−21, −20, −19, −17), or five (−21, −20, −19, −17, and −15) of the more critical positions resulted in a progressive increase in the amount of reverse splicing (9%, 14%, 30%, and 57% of wild-type activity for N-2, N-3, N-4, and N-5, respectively). A control (N-3GTG), in which three of the less critical positions (−17 to −15) were restored, did not significantly increase the amount of reverse splicing. These findings demonstrate that the critical nucleotide residues are recognized by the endonuclease in a variety of sequence contexts, implying direct interaction.
Figure 3.
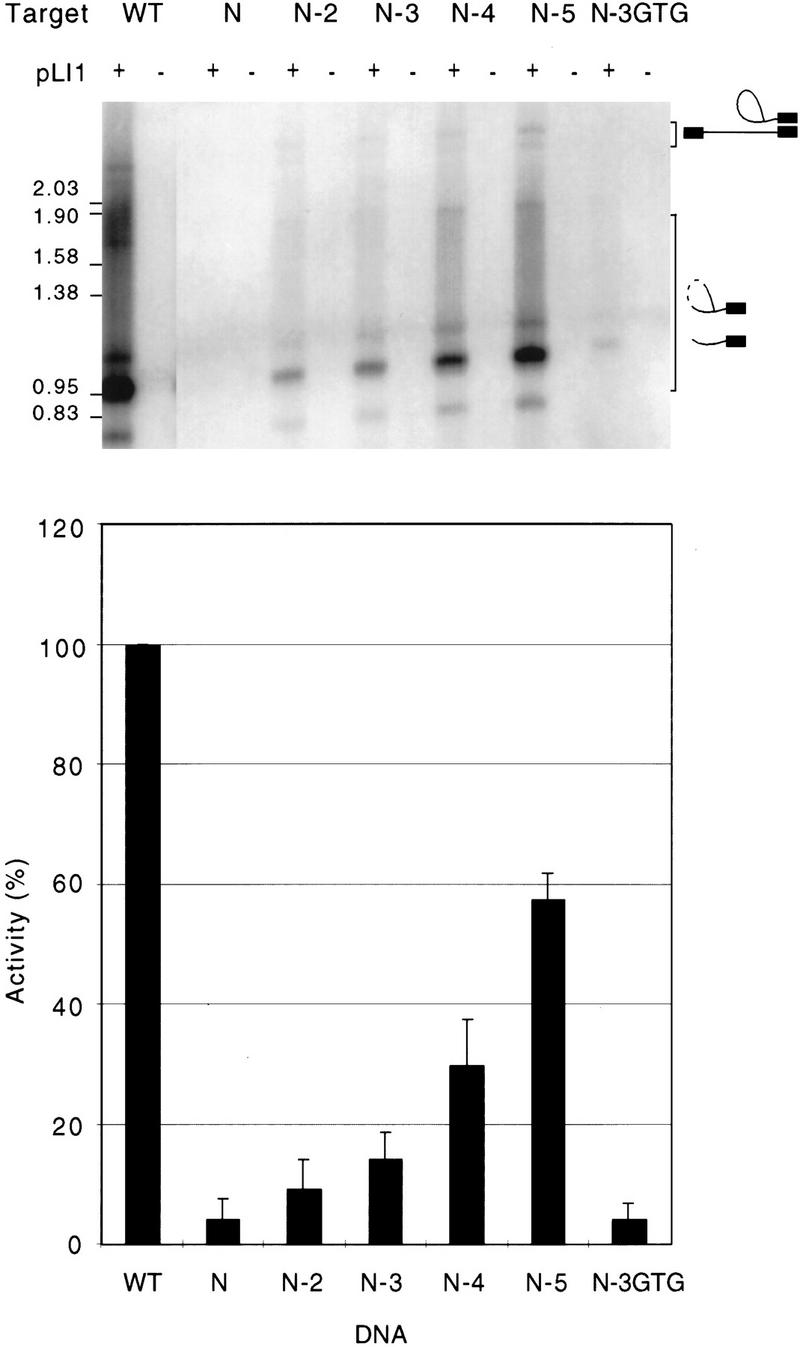
Direct recognition of nucleotide residues in the distal 5′ exon region of the DNA target site. Reverse-splicing assays were performed as in Fig. 2 with mutant DNA substrates having multiple substitutions. Target site N has equimolar mixtures of the three non-wild-type nucleotide residues at each position from −13 to −22. The N-2, N-3, N-4, N-5, and N-3GTG targets have wild-type nucleotide residues restored at the following positions of the N-target site: (N-2) −21 and −20; (N-3) −21, −20, and −19; (N-4) −21, −20, −19, and −17; (N-5) −21, −20, −19, −17, and −15; (N-3GTG) −17, −16, and −15.
Identification of critical positions recognized by base-pairing with the intron RNA
In the IBS/δ′ region, we showed previously that inhibition of reverse splicing by a C-6G mutation at position −6 in IBS1 could be restored by a compensatory change at the corresponding position of EBS1 of the intron RNA, indicating that this position is recognized by base-pairing (Matsuura et al. 1997). Here, we performed analogous experiments to investigate whether the other critical positions in IBS1 and IBS2 are also recognized by base-pairing. To facilitate modification of the intron RNA, these experiments were performed out with RNP particles reconstituted with purified LtrA protein and in vitro-synthesized lariat RNA obtained by self-splicing of in vitro transcripts containing a ΔORF derivative of the intron. In experiments with reconstituted RNP particles, the fully and partially reverse-spliced products are detected as a doublet of radiolabeled bands of ∼1 kb containing the intact ΔORF intron covalently linked or inserted into the DNA target site.
Figure 4A shows an experiment in which we tested all possible combinations of nucleotide residues at position −4 in IBS1 and the corresponding EBS1 position of the intron RNA. For the wild-type intron and DNA target site, this position is a G:T pair taken as 100% activity. All combinations that could form a Watson–Crick or wobble pair gave relatively high levels of reverse splicing, indicating that position −4 is recognized by base-pairing (>136% of wild-type for C:G, U:A, and A:T and 49% and 65% of wild-type for U:G and G:C, respectively). In addition, relatively strong signals were also observed for U:T and C:T (86% and 50% of wild-type, respectively), pyrimidine–pyrimidine mismatches that retain the wild-type T residue at IBS1-4. All other nonpaired combinations gave ≤27% of the wild-type activity, with the lowest activity found for the mismatches G:A, A:A, and A:C (<15% wild type). These results indicate that IBS1-4 is recognized primarily by base-pairing with the intron RNA, but there is some leeway for pyrimidine–pyrimidine mismatches that retain the wild-type T residue at this position.
Figure 4.

Testing the IBS1–EBS1 and δ–δ′ pairings. (A) Reverse-splicing assays with reconstituted RNP particles (50 nm) and wild-type or mutant DNA targets (10 nm) to test all 16 possible combinations of nucleotide residues at IBS1-4 and EBS1-4. (B) Reverse-splicing assays to test base-pairing at positions −12, −11, −10, and +1. The bar graphs below the gels summarize the reverse-splicing activity of the mutant DNA target site/intron RNA combinations relative to the wild-type combination assayed in parallel. The data are the average of at least three independent experiments, with the s.d. indicated by the thin lines. Wild-type nucleotide residues at the relevant positions are boxed. The schematic (bottom) shows the potential base pairs in the wild-type EBS–IBS and δ–δ′ interactions, with boxed base pairs indicating those confirmed here or by Matsuura et al. (1997).
For the other critical positions (−10, −11, −12, and +1), we tested single-nucleotide substitutions with and without compensatory changes in the intron RNA (Fig. 4B). The mutations at each position result in <25% reverse-splicing activity with the wild-type intron, and in each case the activity could be increased by the compensatory change in the intron RNA. The compensatory changes at positions −11 and +1 completely restored the reverse-splicing activity, whereas the compensatory changes at positions −10 and −12 only partially restored activity (40% and 65% wild-type activity, respectively). Similar partial restoration of activity was observed for compensatory changes at the 5′ end of IBS2 in the aI2 intron (Guo et al. 1997) and could reflect that substitutions in this region of EBS2 are deleterious for ribozyme activity or that these positions are recognized in part by the intron-encoded protein, perhaps on the DNA strand opposite that involved in base-pairing.
Considered together, these findings indicate that the critical positions +1, −4, −10, −11, and −12, along with −6 identified previously, are recognized by base-pairing with the intron RNA. It is likely that other positions in IBS1 and IBS2 are also recognized by base-pairing, but mismatches at these positions appear to be tolerated better for reverse splicing in vitro (see Fig. 2).
Identification of critical nucleotide residues required for antisense-strand cleavage
To identify positions required for antisense-strand cleavage, DNA endonuclease assays were performed with pLI1 RNP particles and a series of DNA substrates having nucleotide substitutions in both the 5′- and 3′-flanking regions. In all cases, the wild-type nucleotide was changed to an equimolar mixture of the other three nucleotides, except for the C at the δ′ position (+1), which was changed to mixture of purines. The results are shown in Figure 5, A and B.
Figure 5.

Antisense-strand cleavage assays. DNA endonuclease assays were performed using wild-type and mutant DNA substrates labeled at the 5′ end of the antisense strand. (A,B) Data for positions −26 to −13 and +1 to +10, respectively. The mutant DNA substrates were synthesized with equimolar mixtures of the three non-wild-type nucleotide residues, except for the C at position +1, which was changed to an equimolar mixture of A and G. Lanes labeled + or − indicate DNA substrates incubated with or without pLI1 RNP particles. The cleavage products were analyzed in denaturing 10% polyacrylamide gels, which were dried and quantitated with a PhosphorImager. The 5′-end-labeled antisense-strand cleavage product has a length of 34 nucleotides (see Fig. 1C). The bar graphs below the gels summarize antisense-strand cleavage activity of the mutant DNA target sites relative to the wild-type target site. The data are the average of at least three independent experiments, with the s.d. indicated by the thin lines.
As expected if reverse splicing is required for antisense-strand cleavage, all single-nucleotide substitutions in the 5′ exon that inhibited reverse splicing had parallel effects on antisense-strand cleavage (cf. Fig. 5A with Fig. 2A). In the 3′ exon, where additional interactions with the protein are required for antisense-strand cleavage, the strongest effect was observed for the mutation at position +5, which essentially abolished antisense-strand cleavage activity. The substitutions at +1 and +7 also had relatively strong inhibitory effects (<20% wild-type activity), whereas substitutions at +2, +3, +4, +6, and +10 had less effect (45%–78% wild-type activity), and substitutions at +8 and +9 had slightly increased activity. The strong reduction seen with the +1 mutation could be reversed completely by introducing the compensatory change in the δ position of the intron RNA (not shown), indicating that the inhibition of antisense-strand cleavage is due to impairment of the δ–δ′ interaction, not to protein recognition of this position. Additional experiments with reconstituted RNP particles showed that the mutation at +5, which strongly inhibits antisense-strand cleavage, resulted in an increased ratio of full to partial reverse splicing (not shown). This was also the case for 3′ exon mutations that inhibit antisense-strand cleavage by the yeast aI2 intron and probably reflects that the second step of reverse splicing and antisense-strand cleavage are partially competing reactions (Guo et al. 1997).
Effect of GC content and “bubbles” in different regions of the DNA target site
Base-pairing between the DNA target site and intron RNA requires strand displacement, which could be influenced by the GC content of the DNA target site. We tested the effect of increasing the GC content in both the 5′ and 3′ exon regions of the DNA target site by substituting G:C for A:T base pairs at positions not essential for reverse splicing based on the data of Figure 2. For the 5′ exon, the GC1 substrate has G:C substituted for A:T pairs at −26 and −23, resulting in a target site with nine contiguous G:C pairs (−21 to −29), and the GC2 substrate has G:C pairs substituted at positions −16 and −14, resulting in five contiguous G:C pairs (−14 to −18). Figure 6A shows that the reverse-splicing activity with the GC1 target was affected only moderately, whereas activity with the GC2 target was relatively unaffected (∼40% and 94% of the wild-type target, respectively). The inhibition by the GC1 substitutions could simply reflect the nucleotide substitutions at position −26 and −23, which alone reduced reverse-splicing activity to about the same level (see Fig. 2A). These relatively small effects indicate that high GC content in the upstream flanking region does not strongly affect reverse-splicing activity.
Figure 6.

Introduction of GC-clamps and bubbles in the 5′ and 3′ regions of the DNA target site and changes in spacing of recognition elements. (A,B) Reverse-splicing assays with DNA target sites having GC-clamps or bubbles at different positions. The GC1 and GC2 DNA targets have G:C base pairs substituted at positions −23 and −26 or positions −16 and −14, respectively; GC3T, GC3B, and GC3TB have complementary GC-rich sequences (top 5′-TGCG-3′) substituted at positions +2 to +5 in the top, bottom, or both strands; and T+3G and T+5G have G:C base pairs substituted at positions +3 and +5, respectively. (C) Reverse-splicing assays with DNA target sites having altered spacing between different recognition elements. −14/+A and −14/+3A have one or three A residues inserted at position −14; −14/ΔA has position A-14 deleted; +2/+A has an A residue inserted at position +2; and −8/+T has a T residue inserted at position −8. The bar graphs summarize the reverse-splicing activity of the mutant DNA target sites relative to the wild-type target site assayed in parallel. The data are the average of at least three independent experiments, with the s.d. indicated by thin lines. The schematic at bottom shows the modifications in the DNA target sites.
We noticed that the sequence immediately downstream of the intron–insertion site in E2 is AT rich and had reason to suspect that this might be an important parameter for reverse splicing, because tests with several DNA target sites that otherwise obeyed rules for flanking sequence and IBS interactions failed to give significant reverse splicing (not shown). To test this possibility, we used a DNA target site (GC3TB) in which positions +3 to +5 were changed to G:C or C:G base pairs. Figure 6B shows that this target site was used poorly in reverse-splicing assays (<10% of the wild-type target). In contrast, targets that contain the same GC-rich sequence in either the top or bottom strands (GC3T and GC3B, respectively), leading to a single-stranded bubble at these positions, were very good substrates (65%–82% of wild type). Single G:C substitutions at positions +3 (T+3G) or +5 (T+5G) only moderately decreased activity (40% wild type) compared with the target site having multiple G:C substitutions. We conclude that increasing the GC content immediately downstream of the EBS/IBS pairing significantly inhibits reverse splicing, presumably because it impedes the strand displacement required for protein binding or base-pairing of the intron RNA.
DNA endonuclease activity requires the correct spacing between sequence elements
The above experiments along with those for the yeast aI1 and aI2 introns distinguish three distinct elements of the DNA target site: the distal 5′ exon region (positions −25 to −14), the IBS/δ′ region (−13 to +1), and the 3′ exon (+2 to +10). A series of constructs was designed to test whether a fixed spacing is required between these regions (Fig. 6C). Mutations that alter the spacing between the distal 5′ exon region and IBS sequences (−14/+A, −14/+3A, and −14/ΔA) strongly inhibited reverse splicing (<10% wild-type activity). A mutation that alters the spacing between IBS1 and IBS2 (−8/+T) inhibited reverse splicing by more than sixfold, whereas a one-nucleotide displacement of the 3′ exon sequences (+2/+A) had no effect on reverse splicing but virtually abolished antisense-strand cleavage (Fig. 6C; data not shown). These findings indicate a relatively stringent requirement for fixed spacing between the different regions of the DNA target site.
In vivo assays with mutant DNA target sites
The biochemical experiments identified key nucleotide residues in the Ll.LtrB DNA target site where mutations inhibit reverse splicing and/or antisense-strand cleavage. To determine the effects of inhibiting these reactions in vivo, we measured retrohoming into selected mutant DNA target sites in E. coli, using the two-plasmid genetic assay developed by Cousineau et al. (1998). In this assay, the AmpR donor plasmid, pLI1td+KR′, contains ltrB exons flanking a modified Ll.LtrB intron in which a KanR marker and the group I td intron have been inserted. This construct, referred to as a “twintron,” is expressed from a T7 promoter (pT7; Fig. 7A). As shown previously, loss of the td intron via RNA splicing provides a minimum estimate of the level of retrohoming (Cousineau et al. 1998). Compatible CamR recipient plasmids contain a 50-bp segment corresponding to wild-type or mutant homing sites (ligated E1–E2 sequence, extending from −25 to +25 from the intron–insertion site). After induction of T7 polymerase expression with IPTG, to activate pT7, inheritance of the KanR-marked group II intron by the receipient was detected by selecting for CamRKanR homing products. These recombinants were distinguished from cotransformants by their AmpS phenotype. The level of retrohoming among homing products was estimated by group I intron loss at >80% (Cousineau et al. 1998).
Figure 7.
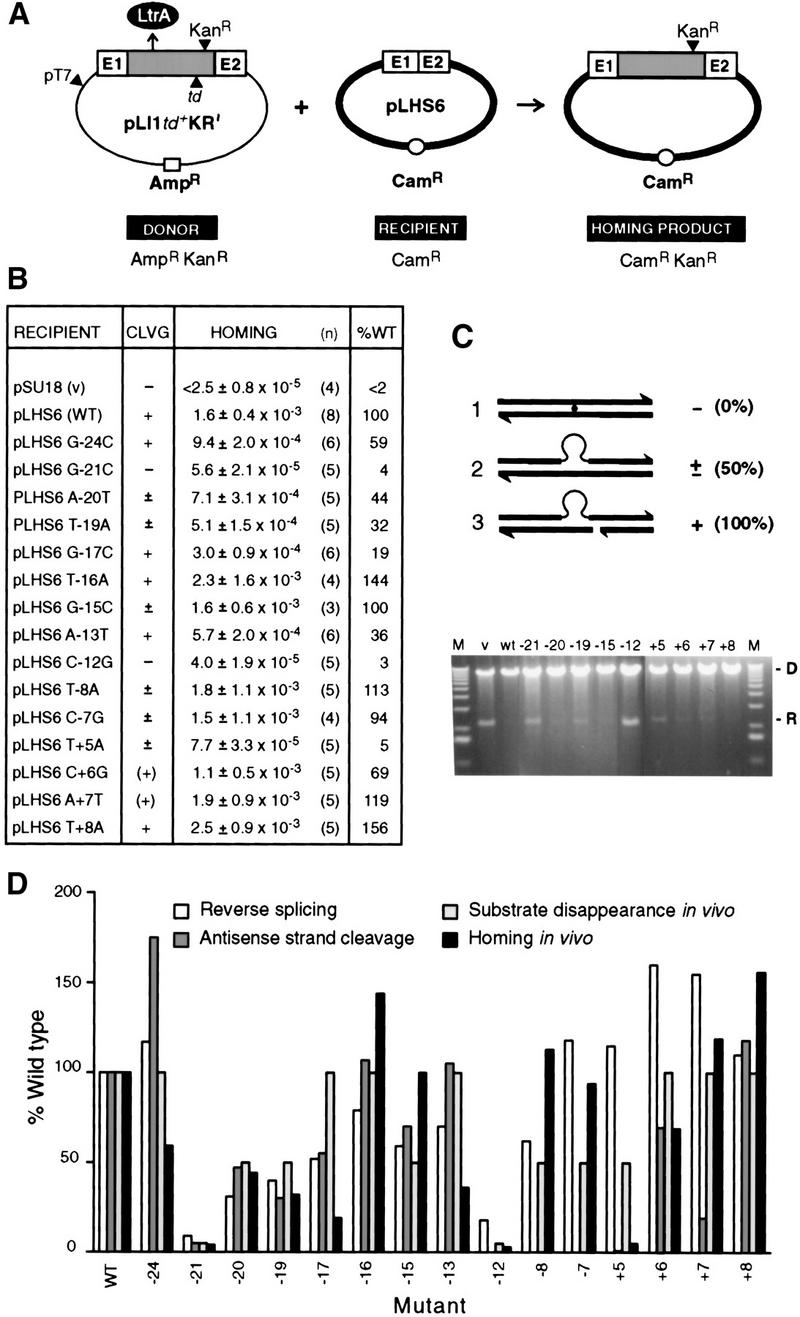
In vivo properties of mutant target sites. (A) Genetic retrohoming assay. Crosses between the AmpRKanR donor (pLI1td+KR′) and CamR recipients (pLHS6 derivatives) allowed selection for CamRKanR homing products. (Shaded box) Ll.LtrB intron; (LtrA) product of intron ORF; (td) group I intron; (E1 and E2) exons. (B) Summary of assay data. Homing frequency is defined as the number of CamRKanR homing products per CamR recipient. (n) Number of trials; (WT) wild-type target site. Cleavage (CLVG), assayed by substrate disappearance, is represented as −, ±, and + as defined in C. Parentheses for the +6 and +7 mutants indicate some variability among experiments. (C) Target-site cleavage assay. Donor (D) and recipient (R) DNAs were extracted from aliquots of cultures 3 hr after IPTG induction, linearized, and separated in a 1% agarose gel. Representative data show persistence of substrate, [(−); −21, −12], partial disappearance of substrate [(±); −20, −19, −15, +5], and virtually complete disappearance of substrate [(+); +6, +7, +8]. Schematics at top depict uncleaved (1), an example of partially cleaved (2), and completely cleaved (3) target. (V) Vector; (M) molecular weight markers. (D) Comparison of in vivo and in vitro data. Reverse splicing and antisense-strand cleavage in vitro from Figs. 2 and 5 are compared with substrate disappearance and homing in vivo for indicated mutant target sites. The data are represented in the histogram as a percentage of the wild-type target site. Substrate disappearance is arbitrarily represented as 100% for +, 50% for ±, and 5% for −. Antisense-strand cleavage assays were not performed for the −12, −8, and −7 substrates, and those positions of the histogram have been left blank.
As summarized in Figure 7B, changes to the complementary nucleotide in the 50-bp homing site resulted in a precipitous drop in homing levels, to ≤5% of wild type at positions −21, −12, and +5. Among the positions tested, the −21 and −12 residues are precisely the same ones where mutations most strongly inhibited reverse splicing in vitro, whereas the +5 residue is the one that most strongly inhibited antisense-strand cleavage. The mutations at other positions had less dramatic effects. Changes at positions −24, −20, −19, −17, −15, −13, −7, and +6 resulted in homing levels of 19%–100% of the wild-type, whereas changes at positions −16, −8, +7, and +8 caused an elevation in homing, to 113%–156% of wild type. The elevation in homing with the +7 mutation was surprising because mutations at this position inhibited antisense-strand cleavage by 80% (Figure 6B). This discrepancy could reflect either that the residual antisense-strand cleavage was not limiting for homing or that a mixture of all three nucleotide substitutions was assayed in vitro whereas only a single substitution was assayed in vivo. After switching the orientation of the 50-bp wild-type homing site in the recipient, homing was still at ∼50% (not shown), indicating that residues outside of this 50-bp region are not required for targeting in vivo.
In parallel with the homing assays, we also examined the persistence of the recipient on agarose gels, an indication of whether the mutations inhibited cleavage by the Ll.LtrB RNP particles (Cousineau et al. 1998) (Fig. 7C). For the wild-type intron, the recipient plasmid was almost completely degraded after 3 hr of induction. In contrast, the mutations at positions −21 and −12, which inhibited homing completely, prevented degradation of the recipient plasmid (−). Mutations at −20, −19, −15, −8, −7, and +5 partially inhibited cleavage (±), while mutations at other positions had little or no effect on cleavage (+). Together, these results show generally good correspondence between the in vivo and in vitro effects of the mutations (compared in Fig. 7D). In addition, the stringent requirement for the wild-type nucleotide at +5 for retrohoming demonstrates that antisense-strand cleavage in addition to reverse splicing is required in vivo.
Retargeting the Ll.LtrB intron to insert into the E. coli thyA gene
Because at least a 14-nucleotide region of the DNA target site is recognized by base-pairing with the intron RNA, we can, in principle, design derivatives of the Ll.LtrB intron targeted for specific 14-nucleotide DNA sequences, taking into account the additional fixed positions recognized by the protein subunit of the endonuclease. As an initial demonstration of the feasibility of this approach, we designed derivatives of the Ll.LtrB intron targeted to the E. coli thyA gene. The Ll.LtrB target site has only two fixed positions stringently recognized by the protein: G-21, which is required for reverse splicing, and T+5, which is required for antisense-strand cleavage (see above). The thyA coding region (795 nucleotides) was first scanned for matches to these positions by using the search sequence 5′-G(N)24T-3′. The collection of 75 potential target sites was then inspected for GC content, discarding those that have >60% G + C residues between positions −21 and +5, and the remainder were evaluated for matches to other positions that contribute to target-site recognition by LtrA (positions −23, −20, −19, −17, −15, +7). Six target sites were chosen for testing, and three of these sites (denoted thyA1, thyA2, and thyA3) that gave detectable mobility in vivo are shown in Figure 8.
Figure 8.



Retargeting of the Ll.LtrB intron to the thyA gene. (A) Sequences of thyA target sites and potential base-pairing interactions with the retargeted introns. The wild-type ltrB target site and base-pairing interactions with the wild-type Ll.LtrB intron are shown at top. Nucleotides in the thyA target sites that match those in the wild-type ltrB site are shaded. Dots above G-21 and T+5 correspond to critical residues representing the two fixed positions in the search for new target sites. The arrow indicates the intron–insertion site. (B) Mobility assay with retargeted introns. The retargeted introns were expressed from pLIthyA derivatives of the AmpR donor plasmid pLI1KR′ and have a KanR resistance marker in domain IV. The CamR-recipient plasmid pSUthyA contains the E. coli thyA coding sequence in place of the ltrB target site in pSU18. The locations of the thyA1, thyA2, and thyA3 target sites in the thyA gene are indicated by arrows. Insertion of the retargeted intron into the thyA target site was detected by a CamRKanRAmpS phenotype and confirmed by PCR analysis and restriction digests. (C) DNA sequence analysis of intron–insertion sites in mobility products. Intron–exon boundaries were amplified by PCR, using the forward and reverse sequencing primers in conjunction with primers near the 5′ and 3′ ends of the intron, and the PCR products were sequenced. The thyA3 site is in the top strand, whereas the thyA1 and thyA2 sites are in the bottom strand. Uppercase and lowercase letters indicate exon and intron sequences, respectively; arrows indicate intron–exon junctions.
For each thyA site, retargeted Ll.LtrB introns were constructed by modifying the EBS2, EBS1, and δ sequences to base-pair to positions −13 to +1 (Fig. 8A). To test whether the modified introns would recognize their intended DNA target sites in vivo, we used the two-plasmid genetic assay. The donor introns were derivatives of the full-length Ll.LtrB intron in pLI1KR′, with the retargeted EBS2, EBS1, and δ sequences, along with complementary changes of the 5′ exon's IBS sequences for efficient splicing. The δ–δ′ interaction is not required for RNA splicing in vitro or in vivo (G. Mohr, X. Cui, and A.M. Lambowitz, unpubl.). The modified introns carry a KanR marker in domain IV, and mobility was followed into a CamR-recipient plasmid (pSUthyA) in which the E. coli thyA gene was inserted in place of the normal ltrB target site (Fig. 8B). Although the mobility frequencies were significantly decreased compared with the wild-type intron (10−5 to 10−6 compared with 10−3 to 10−4), we readily identified mobility products in which all three introns had inserted precisely into their intended target sites (confirmed by sequencing in six cases for thyA1, eight for thyA2, and seven for thyA3; Fig. 8C). Furthermore, in none of 54 mobility products analyzed by PCR did the retargeted intron insert at any location in the thyA plasmid other than its intended target site (not shown). These results demonstrate that it is possible to use the targeting rules determined by biochemical analysis to redirect homing of the Ll.LtrB intron to new target sites in vivo.
Discussion
Our results indicate that the mode of DNA target-site recognition for the L. lactis Ll.LtrB intron is similar to that of the yeast mtDNA introns, with the IBS and δ′ sequences of the DNA target site recognized primarily by base-pairing with the intron RNA and the flanking regions recognized primarily by the intron-encoded protein. As for the yeast mitochondrial introns, mutations beyond δ′ in the 3′ exon region inhibit antisense-strand cleavage but not reverse splicing. Furthermore, substitutions at distal positions of EBS2 (E1−10 and E1−12) are not fully restored by compensatory base changes, possibly reflecting that this region is recognized in part by the intron-encoded protein, perhaps on the strand opposite that used for base-pairing (cf. Guo et al. 1997). Additionally, by using the facile E. coli genetic assay available for the lactococcal intron, we demonstrate that DNA target-site mutations that inhibit reverse splicing or antisense-strand cleavage in vitro strongly inhibit retrohoming in vivo. These findings validate the in vitro analysis of target-site recognition and indicate that both reverse splicing and antisense-strand cleavage are required for efficient mobility.
Despite the overall similarity in target-site recognition by the yeast and bacterial introns, there are nevertheless several differences. Thus, the 5′ boundary of the DNA target site for the lactococcal intron extends farther than that for the yeast introns (−23 to −26 compared with −21), and the critical positions recognized by the intron-encoded protein differ in both the distal 5′ exon and 3′ exon regions. Although these differences could in principle reflect indirect readout of DNA structure, the nucleotide residues putatively recognized by the LtrA protein were sufficient to restore reverse-splicing activity when substituted into an otherwise randomized upstream sequence. Thus, the differences in target specificity probably reflect direct recognition of different nucleotide residues by the intron-encoded protein. The situation is similar to that for group I intron homing endonucleases, where different enzymes of the same family have adapted to recognize different nucleotide residues in their DNA target sites (Lambowitz and Belfort 1993).
The detailed analysis of the Ll.LtrB target site carried out here reveals several additional features of group II intron target-site recognition. First, we show that the spacing between the different sequence elements is relatively stringent. Small deletions or insertions that changed the spacing between the distal 5′ exon region and the IBS sequences or between IBS1 and IBS2 strongly inhibited reverse splicing. Likewise, a one-nucleotide displacement of the 3′ exon elements did not affect reverse splicing but abolished antisense-strand cleavage. These findings suggest that there is relatively limited flexibility in the spacing of different recognition elements in the RNP particle. The requirement for fixed spacing between IBS1 and IBS2 in the double-stranded DNA substrate differs from that in RNA substrates, where studies with the yeast aI5γ intron showed that IBS1 and IBS2 could be separated by a flexible linker of at least 10 nucleotides without affecting binding (Qin and Pyle 1999).
Second, we find that individual positions in IBS1 and IBS2 differ greatly in their tolerance for mutations that disrupt base-pairing. Thus, assays with mutant DNA target sites showed that mismatches at positions E1−4, E1−6, E1−10, E1−11, and E1−12 strongly inhibit reverse splicing, whereas mismatches at other positions (e.g., E1−3, E1−5, E1−7, E1−9) have relatively little effect. For the G:T pair at position −4, analysis of all possible nucleotide combinations showed the greatest tolerance for pyrimidine–pyrimidine mismatches that retain the wild-type T residue in the target site and the greatest intolerance for purine–purine mismatches, which are expected to be most disruptive for helices. The positions where mutations most strongly affect reverse splicing into the DNA target site appear to be clustered toward the 5′ ends of IBS2 (−12 to −10) and IBS1 (−6 and −4). Three of the most critical positions are G:C pairs, one is G:T, and the other is U:A. These results for the RNP reaction again differ from those for the RNA-only reaction of the aI5γ intron, where mismatches in the center of the IBS/EBS helices had greater effects than mismatches at the ends (Xiang et al. 1998). The difference here could reflect the possibility that some nucleotides in the IBS region of the DNA target site are recognized in part by the intron-encoded protein, perhaps on the DNA strand opposite that which participates in base-pairing.
Third, experiments in which we introduced GC-clamps or bubbles at different positions of the DNA target site indicate that DNA unwinding in the 3′ exon is required for efficient reverse splicing. A requirement for DNA unwinding was anticipated so that the intron RNA could base-pair with the IBS and δ′ sequences for reverse splicing. However, it was expected that DNA unwinding would initiate in the distal 5′ exon region, because results with the yeast aI2 intron showed that the interaction of the endonuclease with this region is required for reverse splicing into double-stranded but not single-stranded DNA substrates (Guo et al. 1997). The yeast and lactococcal introns may differ in that access to the IBS and δ′ sequences is from the 5′ exon in the yeast case and from the 3′ exon in the bacterial case. Alternatively, in both cases, the distal 5′ exon region may be required for the initial DNA endonuclease interaction, which then leads to DNA unwinding in the 3′ exon, as suggested by our results for the lactococcal intron.
The target-site analysis here and for the yeast aI1 and aI2 introns shows that mutations at many positions inhibit, but do not abolish, the reaction. Similar observations have been made for I-TevI, the mobility endonuclease encoded by the group I td intron. I-TevI has relaxed specificity for individual nucleotides within its 40-bp homing site, although the spacing of residues within the contacted regions of the DNA can be critical (Bryk et al. 1993, 1995). Flexible specificity, in addition to a lengthy asymmetric recognition site, are properties of many group I intron endonucleases of different families (Mueller et al. 1993; Argast et al. 1998). The flexible recognition specificity of group II intron endonucleases suggests that there may be redundant interactions in a direct read-out of nucleotides in the DNA and/or that the structure of the target site also makes some contribution to recognition specificity in an indirect read-out mode. This flexibility may allow for the movement of the group II introns to new locations having suboptimal target sites. Our in vivo analysis suggests that the wild-type Ll.LtrB target site itself may be suboptimal, because mutations at a number of positions (−16, −8, +7, and +8) actually increased the homing frequency (Fig. 7). In vivo, the transposition of group II introns to ectopic sites that resemble the normal homing site occurs at very low frequency (reviewed in Lambowitz et al. 1999), presumably reflecting that the flexible recognition specificity is balanced by the long target site.
Finally, we show that the target-site recognition rules elucidated here can be used to retarget the Ll.LtrB intron to integrate into new sites in the E. coli thyA gene. The retargeted introns insert accurately into their new target sites in vivo. However, their efficiency was decreased significantly compared with retrohoming of the wild-type intron (10−5 to 10−6 compared with 10−3 to 10−4), and only three of the six retargeted introns tested gave detectable insertions. These problems could result from several factors including the cumulative effects of multiple nucleotide substitutions, thermodynamic constraints for DNA unwinding and/or the EBS/IBS interactions, and adverse effects of some nucleotide substitutions on the ribozyme activity of the intron RNA. As these factors become better understood, the ability to design group II introns to target specific DNA sequences could have widespread applications in genetic engineering and gene therapy.
Materials and methods
E. coli strains and growth conditions
E. coli strain BLR(DE3) lon ompT recA::Tn10(TcR) was used for expression of the Ll.LtrB intron from pLI1 (Matsuura et al. 1997), and BL21(DE3) lon ompT (Novagene, Madison, WI) was used for expression of LtrA protein from pImp-1P (Saldanha et al. 1999). HMS174(DE3) recA and BL21(DE3) were used for in vivo mobility assays, and DH5α and DH10B were used for cloning. Strains were grown in LB with Bacto-agar (1.5%) used for solid media (Sambrook et al. 1989). Antibiotics were added at the following concentrations: 100 μg/ml ampicillin, 25 μg/ml chloramphenicol, and 40 μg/ml kanamycin.
Construction of mutant introns
For biochemical experiments, introns with modified EBS and δ sequences were synthesized from DNA templates generated by PCR of pLI2-ΔORF DNA (Cousineau et al. 1998). The PCR was performed with Pfu DNA polymerase (Stratagene, La Jolla, CA), using multiple sets of primers without cloning. To introduce a single-nucleotide change into the EBS or δ sequences, three different PCR reactions were performed with overlapping primers to amplify the 5′ end of the intron, the EBS/δ region, and the 3′ end of the intron, and for multiple changes, a fourth PCR reaction was performed for the 5′ exon to introduce compensatory changes into the IBS sequences for efficient RNA splicing. In the next step, approximately equal amounts of the three or four independently amplified segments were mixed and used as templates for PCR with the outer primers T7lacTEMPLATE (5′GGGAATTCTAATACGACTCACTATAGGGCCCACGTCGATCGTGA) and CB44 (5′-CGGGATCCAGTATAAAGATTCGTAGAA), to yield the modified intron and flanking exons positioned downstream of the phage T7 promoter. Transcription of gel-purified PCR products with T7 RNA polymerase yields Ll.LtrB RNAs containing a 32-nucleotide 5′ exon, the 902-nucleotide intron, and a 41-nucleotide 3′ exon.
For in vivo experiments, introns targeted for the E. coli thyA gene were cloned into pLI1KR′ (Cousineau et al. 1998). First, the required modifications in the intron's EBS and δ sequences and compensatory changes in the IBS sequences of the 5′ exon were generated by PCR, as described above, except that the final PCR step was performed with primers PETNAR (5′-TGGCGCCGGTGATGCCGGCCACGATG) and LtrBsrGb (5′-GATTGTACAAATGTGGTGATAAC), which are just upstream and downstream of BglII and BsrGI sites, respectively. The PCR product was digested with BglII and BsrGI, and the resulting 555-bp fragment was cloned in place of the corresponding BsrGI–BglII fragment of pLI1KR′ (Cousineau et al. 1998). The PCR-amplified region was sequenced completely to ensure that no adventitious nucleotide changes had been introduced. In all cases, the modified introns are identical to the parent construct, except for the changes specified.
DNA target sites
The standard DNA substrate for biochemical assays was a 72-bp double-stranded DNA that contains the Ll.LtrB intron insertion site, with 29 bp of 5′ exon, 35 bp of 3′ exon, and an EcoRI site downstream of the ltrB 3′ exon (see Fig. 1A). For experiments using pLI1 RNP particles, DNA substrates having mutations in different regions were synthesized by annealing overlapping oligonucleotides (7 pmoles each; see Fig. 1C, schematic) and filling in with Sequenase in a reaction mix containing 333 μm each dATP, dGTP, and dCTP, 20 μm dTTP, and 40 or 50 μCi [α-32P]dTTP (3000 Ci/mmole; NEN, Boston, MA). DNA target sites labeled at the 5′ ends of the sense or antisense strands were made similarly by using 5′-end-labeled DNA oligonucleotides with a reaction mix containing 333 μm each dNTP. The labeled DNA target sites were purified in nondenaturing 6% or 8% polyacrylamide gels, extracted twice with phenol–chloroform–isoamyl alcohol (25:24:1; phenol–CIA), and ethanol-precipitated.
For experiments using reconstituted RNP particles, larger amounts of DNA substrates were made by PCR using Taq DNA polymerase (GIBCO BRL, Gaithersburg, MD) with 0.05 μg of pLHS DNA (Matsuura et al. 1997) as template and 0.1 μg (∼300 nm) of oligonucleotides depicted in Figure 1C as primers. For internally labeled DNA substrates, the PCR was performed in reaction mix containing 20 μCi [α-32P]dTTP and 0.125 mm each dNTP for 25 cycles (94°C, 55°C, and 72°C for 30 sec each). DNA target sites labeled at the 5′ ends of the sense or antisense strands were made similarly by using 5′-end-labeled primers. The DNA target sites were extracted with phenol–CIA, centrifuged through a Sephadex G50 spin column to remove unincorporated nucleotides, ethanol-precipitated, and purified on a 4% agarose gel.
The mutant target sites used for in vivo analysis (Fig. 7) were made in the same way as pLHS6 (Cousineau et al. 1998) with 25 bp of exon 1 and 25 bp of exon 2. Overlapping complementary oligonucleotides, one of which contained the desired mutation, were kinased, annealed, filled in with Klenow polymerase, and cloned into the SmaI site of pSU18. The resulting series of plasmids were designated with the mutational change (e.g., T+5A) following pLHS6. These recipients were used for homing assays with the donor pLI1td+KR′ (Cousineau et al. 1998) in host cell BL21(DE3) (Fig. 7).
pSUthyA, the recipient plasmid used for mobility assays with the retargeted introns, contains the entire thyA coding sequence (795 nucleotides) cloned in place of the ltrB target site in pSU18. The thyA coding sequence was amplified by PCR of E. coli HMS174 chromosomal DNA using primers that introduce terminal SphI and EcoRI sites and then cloned between the corresponding sites of pSU18.
Reverse splicing and DNA endonuclease assays with RNP particles
RNP particles having DNA endonuclease activity were isolated from E. coli expressing plasmid pLI1, which contains the full-length Ll.LtrB intron and flanking exons (Matsuura et al. 1997), or reconstituted with 50 nm each of purified LtrA protein and self-spliced Ll.LtrB RNA containing wild-type or modified introns (Saldanha et al. 1999). Prior to reconstitution, the RNA was renatured by heating to 50°C in 10 mm KCl, 5 mm MgCl2, 50 mm Tris-HCl (pH 7.5), and 5 mm DTT and then slowly cooled to 37°C.
Reverse splicing and DNA endonuclease assays were performed by incubating the RNP particles (0.025 of OD260) with 32P-labeled DNA substrate (1.5 nm; ∼150,000 cpm) in 10 μl of 50 mm Tris-HCl (pH 7.5), 10 mm KCl, 10 mm MgCl2, and 1 mm DTT at 37°C (Matsuura et al. 1997). Reactions with the E. coli pLI1 RNP particles were incubated for 20 min, and those with the reconstituted RNP particles were incubated for 5 min to remain in the linear range. For reverse-splicing assays, the products were denatured with glyoxal and analyzed in a 1% agarose gel, and for DNA endonuclease assays, the products were analyzed in denaturing 6% or 8% polyacrylamide gels. Gels were loaded with equal counts per minute and checked for equal recovery of uncleaved target DNA (not shown). The gels were dried and quantitated with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
In vivo homing and cleavage experiments
The donor and recipient plasmids were cotransformed into BL21(DE3), and cotransformants were grown overnight at 37°C in media containing the appropriate antibiotics. Cultures were diluted 1:100 into 5 ml of medium and grown to an OD600 of 0.2. One-half of the culture was then induced by adding IPTG at 2 mm and grown for an additional 3 hr. Plasmid DNA was extracted, and one-third was digested with SapI, PpuMI, and AvaII, which cut only the donor plasmid, to allow selection of homing products. This DNA was electroporated into DH5α, and cells were plated on selective media. The homing frequency was calculated as the number of homing products (CamR, KanR) per recipient (CamR). To monitor in vivo cleavage, which is correlated with disappearance of the recipient plasmid, two-thirds of the plasmid DNA was cut with SapI and SacII, which linearize the donor and recipient, respectively (Cousineau et al. 1998). The DNA was then separated in a 1% agarose gel. The in vivo homing and cleavage assays were performed at least three times.
To assay the ability to retarget the Ll.LtrB intron, derivatives of pLI1KR′ containing Ll.LtrB introns targeted to the E. coli thyA gene (pLIthyA) were cotransformed into HMS174(DE3) with target plasmid pSUthyA containing the thyA coding sequence in pSU18 (see above). The pool of plasmid DNAs extracted from these cells was digested with ApaLI, EcoO109, PpuMI, and PshAI, which cleave only the donor plasmid, then electroporated into DH10B and plated on selective medium as above. CamRKanR colonies were restreaked on LB with chloramphenicol and kanamycin and on LB with chloramphenicol, kanamycin, and ampicillin to verify the absence of the AmpR donor plasmid. Plasmid DNAs were isolated from AmpS colonies and analyzed by restriction digestion, PCR, and sequencing to confirm the correct insertion of the retargeted intron.
Acknowledgments
We thank Ms. Judy Edwards for preparation of LtrA protein and Mr. Robert Coon for technical assistance. This work was supported by National Institutes of Health grants to A.M.L. (GM37949) and M.B. (GM39422 and GM44844).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lambowitz@mail.utexas.edu; FAX (512) 232-3420.
References
- Argast GM, Stephens KM, Emond MJ, Monnat RJ., Jr I-PpoI and I-CreI homing site sequence degeneracy determined by random mutagenesis and sequential in vitro enrichment. J Mol Biol. 1998;280:345–353. doi: 10.1006/jmbi.1998.1886. [DOI] [PubMed] [Google Scholar]
- Bryk M, Quirk SM, Mueller JE, Loizos N, Lawrence C, Belfort M. The td intron endonuclease I-TevI makes extensive sequence-tolerant contacts across the minor groove of its DNA target. EMBO J. 1993;12:2141–2149. doi: 10.1002/j.1460-2075.1993.tb05862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk M, Belisle M, Mueller JE, Belfort M. Selection of a remote cleavage site by I-TevI, the td intron-encoded endonuclease. J Mol Biol. 1995;247:197–210. doi: 10.1006/jmbi.1994.0133. [DOI] [PubMed] [Google Scholar]
- Cousineau, B., S. Lawrence, D. Smith, and M. Belfort. 2000. Retrotranspositon of a bacterial group II intron. Nature in press. [DOI] [PubMed]
- Cousineau B, Smith D, Lawrence-Cavanagh S, Mueller JE, Yang J, Mills D, Manias D, Dunny G, Lambowitz AM, Belfort M. Retrohoming of a bacterial group II intron: Mobility via complete reverse splicing, independent of homologous DNA recombination. Cell. 1998;94:451–462. doi: 10.1016/s0092-8674(00)81586-x. [DOI] [PubMed] [Google Scholar]
- Eskes R, Yang J, Lambowitz AM, Perlman PS. Mobility of yeast mitochondrial group II introns: Engineering a new site specificity and retrohoming via full reverse splicing. Cell. 1997;88:865–874. doi: 10.1016/s0092-8674(00)81932-7. [DOI] [PubMed] [Google Scholar]
- Guo H, Zimmerly S, Perlman PS, Lambowitz AM. Group II intron endonucleases use both RNA and protein subunits for recognition of specific sequences in double-stranded DNA. EMBO J. 1997;16:6835–6848. doi: 10.1093/emboj/16.22.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, Belfort M. Introns as mobile genetic elements. Annu Rev Biochem. 1993;62:587–622. doi: 10.1146/annurev.bi.62.070193.003103. [DOI] [PubMed] [Google Scholar]
- Lambowitz AM, Caprara MG, Zimmerly S, Perlman PS. Group I and group II ribozymes as RNPs: Clues to the past and guides to the future. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA world. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. pp. 451–485. [Google Scholar]
- Lazowska J, Meunier B, Macadre C. Homing of a group II intron in yeast mitochondrial DNA is accompanied by unidirectional co-conversion of upstream-located markers. EMBO J. 1994;13:4963–4972. doi: 10.1002/j.1460-2075.1994.tb06823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura M, Saldanha R, Ma H, Wank H, Yang J, Mohr G, Cavanagh S, Dunny GM, Belfort M, Lambowitz AM. A bacterial group II intron encoding reverse transcriptase, maturase, and DNA endonuclease activities: Biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes & Dev. 1997;11:2910–2924. doi: 10.1101/gad.11.21.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Ferat JL. Structure and activities of group II introns. Annu Rev Biochem. 1995;64:435–461. doi: 10.1146/annurev.bi.64.070195.002251. [DOI] [PubMed] [Google Scholar]
- Moran JV, Zimmerly S, Eskes R, Kennell JC, Lambowitz AM, Butow RA, Perlman PS. Mobile group II introns of yeast mitochondrial DNA are novel site-specific retroelements. Mol Cell Biol. 1995;15:2828–2838. doi: 10.1128/mcb.15.5.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller JE, Bryk M, Loizos N, Belfort M. Homing endonucleases. In: Linn SM, Lloyd RS, Roberts RJ, editors. Nucleases. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 111–143. [Google Scholar]
- Qin PZ, Pyle AM. Antagonistic substrate binding by a group II intron ribozyme. J Mol Biol. 1999;291:15–27. doi: 10.1006/jmbi.1999.2922. [DOI] [PubMed] [Google Scholar]
- Saldanha R, Chen B, Wank H, Matsuura M, Edwards J, Lambowitz AM. RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry. 1999;38:9069–9083. doi: 10.1021/bi982799l. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Wank H, SanFilippo J, Singh RN, Matsuura M, Lambowitz AM. A reverse transcriptase/maturase promotes splicing by binding at its own coding segment in a group II intron RNA. Mol Cell. 1999;4:239–250. doi: 10.1016/s1097-2765(00)80371-8. [DOI] [PubMed] [Google Scholar]
- Xiang Q, Qin PZ, Michels WJ, Freeland K, Pyle AM. Sequence specificity of a group II intron ribozyme: Multiple mechanisms for promoting unusually high discrimination against mismatched targets. Biochemistry. 1998;37:3839–3849. doi: 10.1021/bi972661n. [DOI] [PubMed] [Google Scholar]
- Yang J, Zimmerly S, Perlman PS, Lambowitz AM. Efficient integration of an intron RNA into double-stranded DNA by reverse splicing. Nature. 1996;381:332–335. doi: 10.1038/381332a0. [DOI] [PubMed] [Google Scholar]
- Yang J, Mohr G, Perlman PS, Lambowitz AM. Group II intron mobility in yeast mitochondria: Target DNA-primed reverse transcription activity of aI1 and reverse splicing into DNA transposition sites in vitro. J Mol Biol. 1998;282:505–523. doi: 10.1006/jmbi.1998.2029. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, Guo H, Perlman PS, Lambowitz AM. Group II intron mobility occurs by target DNA-primed reverse transcription. Cell. 1995a;82:545–554. doi: 10.1016/0092-8674(95)90027-6. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, Guo H, Eskes R, Yang J, Perlman PS, Lambowitz AM. A group II intron RNA is a catalytic component of a DNA endonuclease involved in intron mobility. Cell. 1995b;83:529–538. doi: 10.1016/0092-8674(95)90092-6. [DOI] [PubMed] [Google Scholar]