

Abstract
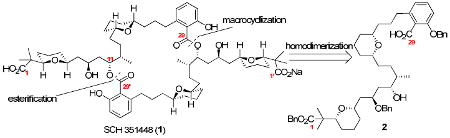
A convergent synthesis of (+)-SCH 351448 (1), a monosodium salt of a C2-symmetric macrodiolide, is described. Our approach is based on a [4+2] annulation with a chiral allyl silane (anti-5c) to assemble the pyran subunits. Homodimerization was carried out in a stepwise fashion; initial esterification at C29′ followed by macrocyclization at C29 afforded the desired macrodiolide.
In 2000, Hedge and coworkers reported the bioassay-guided isolation of a microbial metabolite, named SCH 351448 (1), from the organic extract of Micromonospora sp.1 SCH 351448 is a novel activator (ED50 = 25 μM) for low-density lipoprotein receptor promoter, which is important for the treatment of hypercholesterolemia.2
The structure of SCH 351448 (1) was determined by single-crystal X-ray analysis, and exhibited a hepta-coordinated sodium ion positioned in the interior cavity of the hydrophobic skeletal array.1 Its structure consisted of a 28-membered macrodiolide comprised of two identical hydroxy carboxylic acid monomeric subunits. The intriguing structure and unique bioactivity of 1 has led to several total synthesis programs being initiated by the synthetic community.3
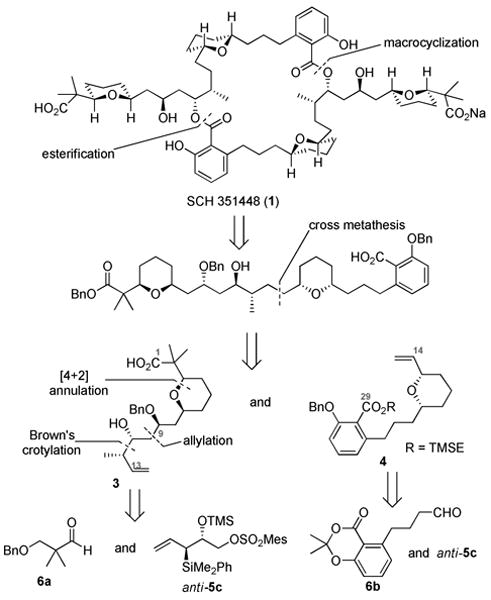
Our retrosynthetic analysis of this target (Figure 1) began with a disconnection of the C29/C29′ ester bonds to yield the monomeric subunit 2. The latter was envisioned to come from an olefin cross metathesis of fragments 3 and 4. Fragment 3 could arise from an asymmetric allylation and crotylation of the cis-2,6-dihydropyran core, which would be formed from a [4+2] annulation reaction4 of allylsilane anti-5c with aldehyde 6a. Similarly, fragment 4 would be derived from silane anti-5c and aldehyde 6b.
Figure 1. Retro synthetic analysis of SCH 351448 (1).
We have previously reported a highly diastereo- and enantio-selective [4+2] annulation between aldehydes and syn allylsilanes.4 However, early experiments at applying the annulation to form dihydropyran products 7 from silanes syn-5b/syn-5c and aldehyde 6a (Scheme 1, eq 1) gave inconsistent results and thus were not synthetically useful. Previously, Roush had reported the construction of cis-2,6-disubstituted dihydropyrans using anti-allylsilanes derived from the asymmetric γ-silyl allylboration of an aldehyde.5 In that report, the favored pathway was thought to proceed via a boat-like TS-A which fashioned the cis-isomer as the major product, while the trans-isomer was suggested to form via the unfavored chair-like TS-B (Scheme 1). In that context, we have observed that anti-silanes such as 5, participate in a [4+2]-annulation with aldehydes to produce 2,6-cis dihydropyrans 7; the results are summarized in Table 1. One proposed mechanism that accounts for the stereochemical course of the annulation involves the equilibration between a twist boat-like TS-C and a chair-like TS-D, where TS-C avoids the steric destabilizing trans-diaxial orientation versus TS-D (Scheme 1, eq 4 and eq 5).
Scheme 1. Possible transition states for the [4+2] annulation.

Table 1. Synthesis of cis-dihydropyrans via [4+2] annulation.
 | |||||
|---|---|---|---|---|---|
| entry | aldehyde | anti-silane | major isomera | yield(%)b | dr (cis:trans)c |
| 1 | R = PhCH2 | 5a | 7a | 30 | 10:1 |
| 2 | R = PhCH2 | 5b | 7b | 60 | 13:1 |
| 3 | R = PhCH2 | 5c | 7c | 83 | 17:1 |
| 4 | R = n-C4H9 | 5a | 7d | 25 | 10:1 |
| 5 | R = n-C4H9 | 5b | 7e | 58 | 12:1 |
| 6 | R = n-C4H9 | 5c | 7f | 81 | 18:1 |
Stereochemistry of the dihydropyrans was assigned by NOE experiments.
Yields were based on pure materials isolated by chromatography on SiO2.
The product ratios were determined by 1H NMR (400 MHz).
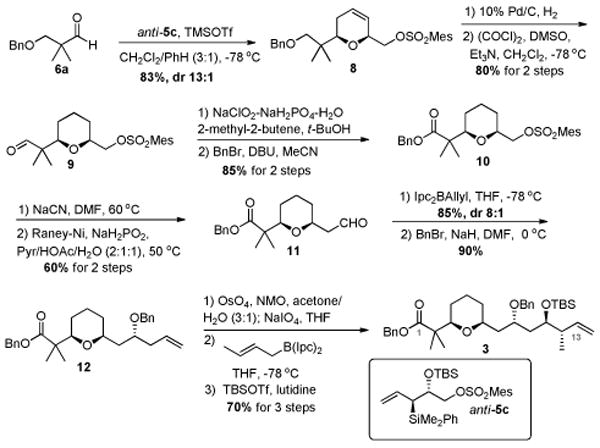
Synthesis of the C1-C13 fragment began with the known α,α′-dimethyl aldehyde 6a6 (Scheme 2). Annulation of silane anti-5c and aldehyde 6a proceeded smoothly in the presence of TMSOTf to afford the desired dihydropyran 8 in 83% yield (dr 13:1). Hydrogenation of 8 afforded a primary alcohol which was later oxidized to aldehyde 9 in 80% yield over two steps. Further oxidation under Pinnick oxidation conditions7 and protection afforded benzyl ester 10. An SN2 displacement of the mesitylate in compound 10 with NaCN followed by Raney-nickel mediated partial reduction8 of the resulting nitrile afforded aldehyde 11 in 60% yield, after hydrolysis of the intermediate imine. Asymmetric allylation of 11 using Brown's protocol 9 furnished the desired secondary homoallylic alcohol, which was subsequently protected as benzyl ether 12. Oxidative cleavage of alkene 12 followed by asymmetric crotylation of the resulting aldehyde using Brown's (E)-crotyl borane10 afforded the anti-homoallylic alcohol, which was protected as its TBS ether to provide olefin 3 as one of the coupling partners in 60% yield over three steps.
Scheme 2. Synthesis of C1-C13 fragment.
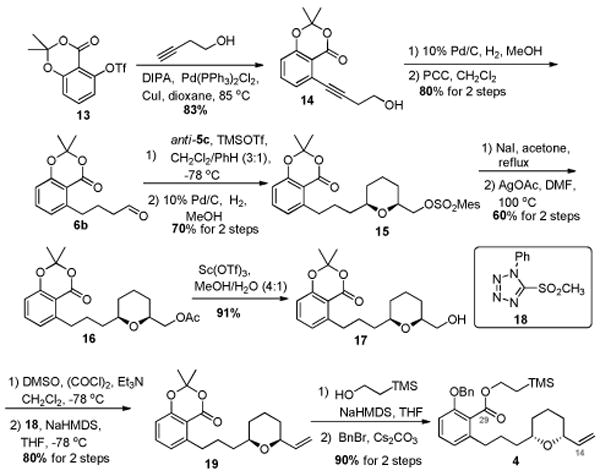
Synthesis of the C14-C29 fragment (Scheme 3) began with aryl triflate 13,11 which was subjected to a Sonogashira cross-coupling to afford propargylic alcohol 14 in 85% yield. Catalytic hydrogenation of alkyne 14 in the presence of Pd/C followed by PCC oxidation provided aldehyde 6b. Annulation between aldehyde 6b and silane anti-5c furnished the desired dihydropyran, which was hydrogenated to give 15 in 70% yield over two steps. Subsequent SN2 displacement of the mesitylate in 15 yielded an iodide, which was further converted to acetate 16 in 60% yield over two steps. A Sc(OTf)3 catalyzed hydrolysis12 of acetate 16 provided primary alcohol 17 in 91% yield, which was then subjected to a Swern oxidation, followed by a Julia-Kociénski olefination3e with sulfone 18,13 to give alkene 19 in 80% yield. Opening of the dioxinone ring in 19 afforded the intermediate phenol, which was converted to the β-silyl ester 4.
Scheme 3. Synthesis of C14-C29 fragment.
With advanced intermediates 3 and 4 available in useful amounts, we were now positioned to investigate methods for their union. Cross metathesis between 3 and 4 (Scheme 4) proceeded smoothly using the Grubbs-Hoveyda second generation catalyst,14 which delivered the (E)-olefin. This material was then subjected to diimide reduction3a to afford advanced intermediate 20. Deprotection of 20 provided seco acid 2, which was poised for the homodimerization experiments.
Scheme 4. Attempted template-directed macrodimerization.
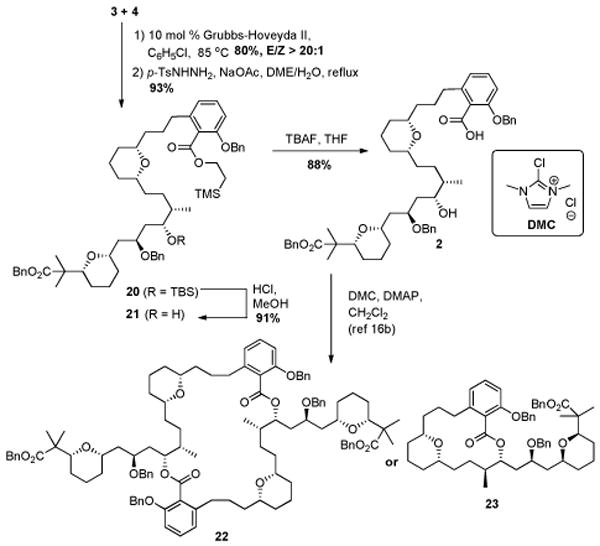
A synthetic strategy to construct the C2-symmetrical macrodiolide core of cycloviracin B1 has been described by Furstner. 15 It involved a template-directed macrodilactonization reaction promoted by 2-chloro-1,3-dimethylimidazolinium chloride (DMC). 16 Inspired by this work, we investigated a similar strategy for macrodiolide formation. Unfortunately, treatment of seco acid 2 with DMC/DMAP and suitable additives17 only led to the undesired 14-membered lactone 23 18 without formation of dimeric product 22. After these disappointments, we evaluated a stepwise pathway to complete the synthesis, as illustrated in Scheme 5.
Scheme 5. Assemly of 1 by dioxinone ring-opening and macrocyclization.
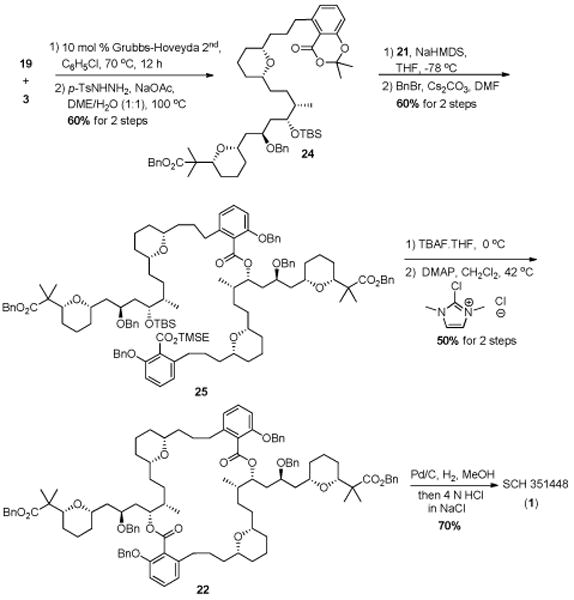
The reaction sequence that ultimately proved successful utilized Lee's method of esterification, 3a which was facilitated by dioxinone ring-opening. Cross metathesis between 19 and 3 afforded the intermediate alkene, which was reduced with diimide to deliver dioxinone 24. TBS deprotection of 20 gave alcohol 21 in 91% yield (Scheme 4). Deprotonation of alcohol 21 with NaHMDS and addition of dioxinone 24 led to the desired mono-ester product, which was protected to afford 25 in 60% yield over two steps. Deprotection of the monoester provided the seco acid, which was subjected to a DMC/DMAP-promoted esterification16 reaction to achieve mycrocycle 22 in 50% yield over two steps. Lastly, deprotection followed by work up with 4 M HCl saturated with NaCl, 3a delivered SCH 351448 (1) as its monosodium salt in 70% yield. The spectral data for our synthetic material matched those reported for the natural product.3
In summary, we have described a convergent, enantioselective total synthesis of (+)-SCH 351448 with a 2.3% overall yield from readily available allylsilane anti-5c. Synthetic highlights of our route include a [4+2] annulation strategy using silane anti-5c to ultimately construct the tetrahydropyran ring systems in fragments 3 and 4. Olefin cross metathesis was utilized in the union of two advanced fragments to generate the monomeric subunit. A metal-template directed macrodilactonization strategy proved unsuccessful. Thus, the macrodiolide was assembled through a two-step sequence involving dioxinone ring-opening with concomitant esterification followed by DMC/DMAP-mediated macrocyclization.
Supplementary Material
Acknowledgments
Financial support was obtained from NIH CA 53604. We are grateful to Prof. John Snyder, Dr. Paul Ralifo and Dr. Norman Lee (Chemical Instrumentation Center at Boston University) for helpful discussions and assistance with NMR and HRMS experiments.
Footnotes
Supporting Information Available: Experimental details and selected spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hegde VR, Puar MS, Dai P, Patel M, Gullo VP, Das PR, Bond RW, McPhail AT. Tetrahedron Lett. 2000;41:1351–1354. [Google Scholar]
- 2.(a) Brown MS, Goldstein JL. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]; (b) Brown MS, Goldstein JL. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 3.For previous total syntheses see: Kang EJ, Cho EJ, Lee YE, Ji MK, Shin DM, Chung YK, Lee E. J Am Chem Soc. 2004;126:2680–2681. doi: 10.1021/ja0315309.Kang EJ, Cho EJ, Ji MK, Lee YE, Shin DM, Choi SY, Chung YK, Kim JS, Kim HJ, Lee SG, Lah M, Lee E. J Org Chem. 2005;70:6321–6329. doi: 10.1021/jo0507993.Soltani O, De Brabander JK. Org Lett. 2005;7:2791–2793. doi: 10.1021/ol0510769.Bolshakov S, Leighton JL. Org Lett. 2005;7:3809–3812. doi: 10.1021/ol0515006.Crimmins MT, Vanier GS. Org Lett. 2006;8:2887–2890. doi: 10.1021/ol061073b.Cheung LL, Rychnovsky SD. Org Lett. 2008;10:3101–3104. doi: 10.1021/ol8011474.Backes JR, Koert U. Eur J Org Chem. 2006;12:2777–2785.Chan KP, Ling YH, Loh TP. Chem Commun. 2007:939–941. doi: 10.1039/b616558c.Park H, Kim H, Hong J. Org Lett. 2011;13:3742–3745. doi: 10.1021/ol201426c.
- 4.Su Q, Panek JS. J Am Chem Soc. 2004;126:2425–2430. doi: 10.1021/ja037957x. [DOI] [PubMed] [Google Scholar]
- 5.Roush WR, Dilley GJ. Synlett. 2001;SI:955–959. [Google Scholar]
- 6.Yang Y, Wang J, Kayser M. Tetrahedron Asymmetric. 2007;18:2021–2025. [Google Scholar]
- 7.Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 8.Ghosh AK, Moon DK. Org Lett. 2007;9:2425–2427. doi: 10.1021/ol070855h. [DOI] [PubMed] [Google Scholar]
- 9.Brown HC, Jadhav PK. J Am Chem Soc. 1983;105:2092–2093. [Google Scholar]
- 10.Brown HC, Bhat KS. J Am Chem Soc. 1986;108:5919–5923. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 11.Uchiyama M, Ozawa H, Takuma K, Matsumoto Y, Yonehara M, Hiroya K, Sakamoto T. Org Lett. 2006;8:5517–5520. doi: 10.1021/ol062190+. [DOI] [PubMed] [Google Scholar]
- 12.Kajiro H, Mitamura S, Mori A, Hiyama T. Bull Chem Soc Jpn. 1999;72:1553–1560. [Google Scholar]
- 13.Leburn ME, Le Marquand P, Berthelette C. J Org Chem. 2006;71:2009–2013. doi: 10.1021/jo052370h. [DOI] [PubMed] [Google Scholar]
- 14.(a) Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]; (b) Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 15.Fürstner A, Albert M, Mlynarski J, Matheu M, DeClercq E. J Am Chem Soc. 2003;125:13132–13142. doi: 10.1021/ja036521e. [DOI] [PubMed] [Google Scholar]
- 16.(a) Isobe T, Ishikawa T. J Org Chem. 1999;64:5832–5835. [Google Scholar]; (b) Isobe T, Ishikawa T. J Org Chem. 1999;64:6984–6887. [Google Scholar]; (c) Isobe T, Ishikawa T. J Org Chem. 1999;64:6989–6992. [Google Scholar]; (d) Fujisawa T, Mori T, Fukumoto K, Sato T. Chem Lett. 1982;11:1891–1895. [Google Scholar]
- 17.Fürstner A. In: Templated Organic Synthesis. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 2000. pp. 249–273.Typical procedure: the additive (2.0 equiv) is added at 0 °C to a solution of seco acid 2 (20 mg, 0.023 mmol) and 2-chloro-1,3-dimethylimidazolinium chloride (10 mg, 0.059 mmol) in CH2Cl2 (1.1 mL) and the resulting mixture is stirred for 1 h at that temperature. DMAP (7.2 mg, 0.059 mmol) is then introduced and stirring is continued for 16 h at ambient temperature; additives: NaH, KH, CaH2, Na2CO3, Cs2CO3.
- 18.Similar 14-membered lactones were also reported in the previous syntheses by Lee and Rychnovsky.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.