

Abstract
A series of oligomers, containing oligo(ethylene glycol) (OEG) moieties, with the same composition of amphiphilic functionalities has been designed, synthesized, and characterized for their temperature sensitive behavior. The non-covalent amphiphilic aggregates, formed from these molecules, influence the temperature sensitivity of these molecules. Moreover, the covalent tethering of the amphiphilic units also has significant influence on their temperature sensitivity. The lower critical solution temperatures (LCST) of these oligomers show increasingly sharp transitions with increasing numbers of OEG functional groups, indicating enhanced cooperativity in dehydration of the OEG moieties when covalently tethered. These molecules were also engineered to be concurrently sensitive to enzymatic reaction and pH. This possibility was investigated using porcine liver esterase as the enzyme, where we show that enzymatic action on the pentamer lowers its temperature sensitivity. The product moiety from the enzymatic reaction renders the amphiphilic oligomer exhibit a pH-dependent temperature sensitivity.
Introduction
Stimuli-sensitive systems have generated interests in a variety of areas including controlled drug delivery vehicles,1 sensing,2 tissue engineering,3 coatings,4 catalysis,5 and separations.6 The stimulus employed for these so-called smart materials can be broadly divided into two categories: physical and chemical stimuli. Among the physical stimuli, materials that respond to temperature variations have garnered particular interest due to implications in biomedical applications such as drug delivery and tissue engineering.7,8 Additionally, thermo-responsive materials have also found utility in areas such as thermal affinity separation, enzyme recycling, and protein chromatography.9
Thermal sensitivity is endowed into a material, especially polymeric ones, by engineering the molecular structures so that the polymer undergoes a coil-to-globule transition upon change in temperature.9d,10 When this transition results in solubility differences, the material is thought to exhibit lower critical solution temperature (LCST) behavior. For experimental convenience, the onset temperature at which the polymer containing solution becomes turbid is commonly probed to extract relationships between structure and LCST properties. Among the temperature-sensitive polymers, poly-N-isopropylacrylamide (p-NIPAM) has attracted a great deal of interest.11 Another class of materials that has also attracted substantial interest in temperature-sensitive materials, due to their biocompatible and anti-fouling features, involves ethyleneglycol based polymers. Both poly (ethylene glycol) (PEG) and oligo(ethyleneglycol) (OEG) have been used as components of polymeric scaffolds that exhibit temperature sensitive behavior.12 At higher temperatures, breakage of the hydrogen bonding between the ethylene glycol units and the water molecules renders them lipophilic. This is thought to be the reason for the thermal sensitivity of these functional groups.
It is interesting that incorporating a single oligoethyleneglycol unit, such as the pentaethyleneglycol moiety, by itself or as part of a small molecule does not exhibit any noticeable thermal sensitivity. However, when pentaethylene glycol is attached onto a scaffold that presents these units in a multimeric form due to self- assembly, the system exhibits significant temperature sensitivity. We have observed this phenomenon while comparing amphiphilic dendrimers with the corresponding amphiphilic small molecules.13 We were interested in identifying the underlying reasons that render a polymer more sensitive than the small molecule. We approach this by studying well-defined oligomers and compare them to the corresponding monomer. This manuscript outlines our findings, where we have designed, synthesized, and characterized the temperature sensitive behavior of a set of amphiphilic oligomers containing pentaethyleneglycol as the hydrophilic OEG moiety.
Additionally, systems that are concurrently sensitive to more than one stimulus have attracted significant interests in recent years, because these would provide a unique opportunity to fine-tune their response to each stimulus independently, as well as precisely regulate release profile during the combined effect of multiple stimuli.14 Our molecular design provides a great opportunity for testing the sensitivity of these oligomers to multiple stimuli, i.e. temperature and enzyme. We take advantage of this opportunity and test our oligomers for concurrent sensitivity to these stimuli. We particularly focus on esterase-based enzyme, because (a) they are convenient model systems to study enzyme sensitivity; (b) esterases are ubiquitous in biology and thus are relevant to temperature-sensitive systems that are of interest in several biomedical applications.
Results and Discussion
Molecular Design and Synthesis
The effects of compositional variations of OEG-based monomer and another monomer upon the temperature sensitive behavior of polymers have been previously investigated.15 However, since our interest lies in developing an understanding on the cooperative effect of OEG units, when covalently tethered together, our self-imposed design criterion is that the hydrophilic-lipophilic balance (HLB) in all oligomers should be the same. In our study, we have focused on variations ranging from monomeric to hexameric amphiphiles. Pentaethyleneglycol and alkyl moieties were attached to the meta- positions of a benzoyl building block as the hydrophilic and lipophilic moieties respectively. This basic building block was then converted to an oligomer using the corresponding commercially available oligoamine scaffolds (Chart 1). Note that the lipophilic alkyl moiety is also terminated with an ester functionality. This ester functionality provides the substrate handle for esterases, the reaction between which affords the corresponding carboxylic acid. The conversion of the ester moiety to the corresponding carboxylic acid functionality, in the presence of the enzyme, alters the HLB of the amphiphilic molecule and thus the LCST of the oligomers. Additionally, it is also easy to imagine the carboxylic acid moiety, thus generated, would provide an avenue for pH-sensitivity.
Chart 1.

Structures of the amphiphilic oligomers used in this study
In all these cases, the commercially available oligoamines were treated with the benzoyl chloride molecule 9 (Scheme 1). To synthesize 9, 3,5-dihydroxybenzoic acid (7) was first converted to its corresponding ethyl ester followed by the installation of a propargyl functionality on one of the phenolic groups through an alkylation reaction in the presence of potassium carbonate, as shown in Scheme 1. Treatment of the mono-substituted product with pentaethyleneglycol tosylate under similar alkylation conditions provided the precursor 8. Hydrolysis of the ethyl ester followed by treatment with oxalyl chloride, afforded the targeted aryloyl chloride molecule 9. Treatment of this molecule with oligoamines in the presence of a base afforded the targeted oligomers. This is exemplified with the synthesis of the dimer 2 in Scheme 1. Note that the molecule 10 does not contain the ester-based lipophilic functionality installed in the molecule yet. This functionality was attached to the oligomers in the last steps of the synthesis using the Huisgen 1,3-dipolar cylcoaddition reaction, the so-called click chemistry. We had to introduce the ester moiety in the last steps of the syntheses through the Huisgen reaction, because we had to generate the acid chloride species to generate that targeted benzamide molecules. The azide counterpart in the Huisgen reaction is ethyl-5-azidovalerate (11), which was synthesized from the corresponding alkyl bromide by treating it with sodium azide. Higher oligomers were synthesized through a similar route using the corresponding oligoamine, the details of which are shown in the Supporting Information. All products were characterized using 1H NMR, 13C NMR and mass spectrometry (see Supporting Information for details).
Scheme 1.

Synthetic route for the amphiphilic oligomers, exemplified with the dimer 2.
Since these are amphiphilic oligomers, we also anticipated that these molecules would aggregate in aqueous media above a certain concentration, the critical aggregation concentration (CAC). We were specifically interested in this concentration, since this feature could further influence the temperature-sensitive behavior of these molecules. It is reasonable to anticipate that the assemblies obtained in the aqueous phase from these amphiphilic molecules will contain a hydrophobic interior, which can potentially sequester lipophilic guest molecules. The lipophilic guest sequestration event is typically used as the pathway for identifying the CAC. We have used Nile red as the lipophilic guest molecule for this purpose; note that Nile red by itself is not soluble in water (Figure 1a). A plot of concentration of the surfactant molecules against the Nile red fluorescence provides a sigmoidal curve. The point at which there is a significant change in the slope of the curve is taken to be the critical aggregation concentration. This is exemplified in Figure 1b and the CACs of the molecules 1-6 are shown in Table 1. CACs are usually provided in concentrations. However, since the amphiphilic repeat unit in each of these molecules is the same, it is more relevant to assess the relative CACs in terms of mg/mL. Accordingly, we have also tabulated the CACs in mg/mL. We found that while there is a significant gain in CAC between the monomer and the dimeric amphiphile, there is no real difference in CACs among other oligomers.
Figure 1.

(a) Fluorescence of Nile red in water and Hexamer 6 solution. (b) CAC calculation for Hexamer.
Table 1.
CAC and DLS size of Oligomers
| Oligomers | CAC, mg/mL | CAC, mM | Size, nm (PDI) |
|---|---|---|---|
| Monomer (1) | 3.84 | 6.14 | NA |
| Dimer (2) | 1.55 | 1.27 | 18 (0.24) |
| Trimer (3) | 1.75 | 0.95 | 23 (0.26) |
| Tetramer (4) | 1.66 | 0.67 | 25 (0.24) |
| Pentamer (5) | 1.65 | 0.54 | 30 (0.24) |
| Hexamer (6) | 1.40 | 0.38 | 60 (0.19) |
The sizes of our amphiphilic aggregates, estimated using dynamic light scattering (DLS) measurements, are shown in Table 1 for comparison. We used 3 mM concentration of the molecules and the sizes seem to increase with increasing oligomeric length. It should be noted that the sizes of our assemblies are somewhat larger than those of small molecule based surfactant micelles. Thus, we believe that our assemblies are higher order aggregates with micelle-like lipophilic interior. Also, the rather shallow transition in the CAC estimation in Figure 1 further suggests that these are higher order aggregates.
The temperature sensitive behavior of the oligomers was studied using turbidity measurements, by measuring the high tension (HT) voltage response of the photomultiplier on a circular dichroism (CD) spectrometer.13,16 The CD spectrometer was used, simply because of the conveniently available temperature control in this equipment in our laboratories. Aqueous solutions of molecules 1-6 were monitored for solution turbidity with increasing temperature at 650 nm. The temperature increase was done at a rate of 1 °C/min.
In all these studies, we kept the concentration of the OEG units in solution constant, as this allows for an understanding of the possible cooperativity due to covalent tethering of the OEG units. The concentrations of the oligomer solutions were consistently kept at 3 mM with respect to the OEG unit; i.e. since the monomer 1 contains only one OEG unit, 3 mM solution of molecule 1 was used. However, we used 1.5 mM concentration of 2 when using the dimer, as this solution provided an overall OEG concentration of 3 mM. Note that, with the exception of 1, all these molecules are well above their CACs and therefore we study the behavior of the aggregates in all these cases.
Figure 2a shows the temperature sensitivity plots for the amphiphilic molecules 1-6. Two observations can be immediately made from these plots: (i) there is a systematic change in the temperature sensitivity of the oligomeric amphiphiles. Monomer 1 is less sensitive to temperature, i.e. it exhibits a much higher transition temperature compared to the higher oligomers; (ii) the transition itself seems sharper in higher oligomers. Both of these features can be more clearly visualized and better quantitative data can be acquired, if we analyze the first differential of these plots. Figures 2c and 2d show the first differential of the plots for dimer 2 and hexamer 6 respectively. It is readily seen that the transition in the hexamer is indeed sharper than the dimer. A Gaussian function was applied to fit the non-linear graph, leading to the temperature at the sharpest transition point that is taken to be the transition temperature (Tt) (Table 2). A full width-half maximum (FWHM) can also be extracted from these curves, which provides an important insight into the rate of the transition, as this is a measure of the steepness of the transition (Table 2). This rate can be correlated with the cooperativity among the OEG units, because the concentrations of OEG units in all these solutions are the same.
Figure 2.

(a) Plot of HT voltage vs. temperature for molecules 1–6 in water. (b) LCST of Momomer 1 at 6.4 mM and 3.0 mM. (c) First differential plot for dimer 2. (d) First differential plot for hexamer 6.
Table 2.
Tt and FWHM of oligomers
| Oligomers | Tt (°C) | FWHM |
|---|---|---|
| Monomer (1) | 51.3 | 17.2 |
| Dimer (2) | 43.4 | 11.2 |
| Trimer (3) | 45.3 | 9.9 |
| Tetramer (4) | 40.1 | 6.9 |
| Pentamer (5) | 36.4 | 4.1 |
| Hexamer (6) | 35.8 | 3.9 |
Note that the onset temperature in all oligomeric molecules 2-6 is very similar, while that of 1 is significantly different. To explain this observation, we need to first examine the underlying reasons behind the OEG units exhibiting temperature-sensitive coil-to-globule transition. It is generally accepted that the driving force involves the breaking of hydrogen bonds between OEG units and water molecules with increasing temperature.12 This is thought to occur, because of the entropy gained due to the shedding of the ordered water molecules around OEG units. Note that in our experiments, the concentrations of OEG units are the same in all cases. However, the onset temperature is different for the monomer 1 compared to other oligomers. We attribute this to the difference in LCST of OEG-based aggregates in 2-6 vs. monomeric OEG-based molecule 1. Note that the oligomers 2-6 are present in solution above its CAC, while the monomer 1 is below its CAC. Thus, a high percentage of the oligomer is present as non-covalent nanoscale assemblies, where a number of OEG units are presented on their surfaces, a feature that is not present in 1. To test this further, we measured LCST behavior of 1 at a concentration above its CAC (6.4 mM). Indeed, the Tt decreased to 21.6 °C with a corresponding sharpening of the transition (see Figure 2b). These could also be attributed to the inherent increase in the concentration of OEG units in the solution. We could not distinguish these possibilities. However, when put together with other results, this result is consistent with our hypothesis that the non-covalent nanoscale assembly formation influences the onset temperature.
Although there are very small differences, if any, in the onset temperatures among the oligomers the FWHM of the oligomers exhibit a systematic difference in these molecules. As mentioned above, FWHM is related to the rate of the coil-to-globule transition. Since the OEG concentration in all oligomers is identical, the difference in rate of the transition can be attributed to the difference in cooperativity in the OEG units shedding the water in response to the temperature variation. This, in turn, suggests that a greater cooperativity is observed in higher oligomers compared to the lower oligomers (for example, the FWHM of dimer is 11.2, while that of the hexamer is only 3.9). Overall, these results suggest that formation of non-covalent assembly provides a pathway for providing a critical change in the onset temperature in the coil-to-globule transition of the OEG units. In addition, the rate of transition in these non-covalent assemblies can be tuned by covalently tethering amphiphilic units, as this provides a useful vehicle for cooperative dehydration of the OEG units.
Multi-Stimuli Sensitivities
Next, we were interested in testing whether esterase activity will modulate the LCST behavior of these amphiphilic molecules. The appeal of this is in the fact that esterase based degradation is often the basis for degradation of biomaterials. Therefore, it is interesting to examine as to how the temperature-sensitivity of a molecule can be modulated after being subjected to an enzymatic reaction. Note that we have engineered the molecule in a fashion that an esterase reaction would alter HLB in the amphiphilic oligomers, which is expected to alter LCST behavior. This is anticipated, because esterases cleave esters to produce carboxylic acid; since the aliphatic esters are more lipophilic than the corresponding carboxylic acids, the products of the enzymatic reaction would possess markedly different HLB values as compared to the reactants (Scheme 2). In our case, the molecule is expected to become more hydrophilic resulting in a significant change in its LCST behavior.
Scheme 2.

Change in HLB in Pentamer 5 due to the enzymatic reaction.
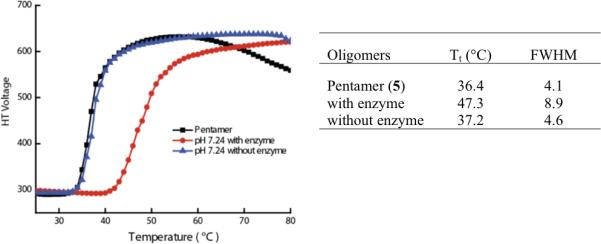
We examined this possibility with the pentamer 5 using porcine liver esterase (PLE). Accordingly, we subjected a 3 mM solution of 5 in HEPES buffer at pH 7.24 to reaction with PLE for 20 hours, after which we examined the temperature-dependent turbidity generation in the solution. Indeed, we noted that the onset temperature increased by ~10 °C in the enzyme-treated solution. As a control experiment, the LCST behavior of the same pentamer solution in HEPES buffer at pH 7.24 without enzyme was examined after 20 hours. No change in the temperature-sensitivity was observed, suggesting that the observed change in LCST is indeed due to an enzymatic reaction rather than any adventitious hydrolysis over time in aqueous phase. Also, the partial hydrolysis of the ester functionalities in the mixture due to the esterase reaction was confirmed by 1H NMR (see Supporting Information).
Note that the molecular design is also engineered to exhibit a pH-sensitive behavior, since the carboxylic acid formed after the enzymatic reaction can present different hydrophilicities, depending on whether the carboxylic acid is protonated or deprotonated. Thus, the pH of the solution can significantly alter the LCST behavior of the molecule. We tested this possibility by monitoring the LCST of the enzymatic product at different pHs. After the enzymatic reaction, the pH of the solution was adjusted to 5.0, 6.5, 8.5, and 10.8 by addition acid or base to the solution. The LCST behaviors were examined at these different pH values (Figure 4). Deprotonation of the carboxylic acid at basic pH increases hydrophilicity due to increase in the number of negatively charged carboxylate anions. Similarly, protonation of the carboxylates at lower pH should decrease hydrophilicity. Indeed, at pH 5.0 and 6.5, we found that Tt decreases to 39.9 and 44.4 and the FWHM decreases to 7.0 and 5.3 respectively, compared a Tt of 47.3 and FWHM of 8.9 at the neutral pH of 7.2. Similarly, at pH 8.5 and 10.8, Tt increased compared to the neutral pH, while the FWHM did not significantly change at pH 8.5 (in fact, decreased slightly) and increased greatly at pH 10.8. These observations are indeed consistent with our hypothesis.
Figure 4.

LCST profile of Pentamer 5 after enzymatic reaction with different pH.
Although consistent, there is a possibility that the esters might also be hydrolyzed at different pH and this could cause further change in HLB. We were concerned about this, especially because of the rather significant change noted at pH 10.8. Accordingly, we incubated the pentamer 5 at different pH (Figure 5). When analyzed from pH 5 to 8.5, the free pentamer 5 did not exhibit any difference. However at pH 10.8, the pentamer 5 did exhibit a significant change that indicates hydrolysis of the ester. To further test this, we analyzed the pentamer 5 solution at different time intervals and indeed the LCST systematically evolved with time, further supporting the observation that hydrolysis of the ester over time affects the observed LCST behavior (Figure 5). Under these conditions at lower pH however, there is no observable hydrolysis of the ester. Therefore, it is clear that there is indeed a pH-dependent temperature sensitivity of the enzymatic product. However, these observations are not reliable at high pH (>8.5), because there is an independent base-catalyzed hydrolysis of the ester.
Figure 5.

LCST profile of pentamer 5 control at different pH and different time for pH 10.8
Summary
We have designed, synthesized and characterized a series of amphiphilic oligomers containing pentaethylene glycol functionalities as the hydrophilic segment and esters as the hydrophobic moiety. By systematically comparing the oligomers, we note that: (i) non-covalent organization of the OEG units through aggregation causes a significant increase in temperature sensitivity; (ii) cooperativity is further enhanced, when these OEG units are covalently tethered in the oligomers, as noted by the increase in transition kinetics with increasing oligomerization; (iii) when an enzyme-sensitive functionality is incorporated onto the lipophilic segment of the amphiphile, these molecules can be rendered sensitive to both enzyme and temperature; and (iv) since the product of the enzymatic reaction provides a pH-sensitive functionality, the amphiphilic assembly is rendered responsive to three different stimuli. Overall, our studies here provide insights into the need for multimeric presentation of oligoethylene glycol units based on either non-covalent assemblies and/or covalent tethering. Our report also outlines a strategy to design a molecule that can be sequentially sensitive to three different stimuli. We believe that this work will have implications in designing molecular scaffolds for applications such as drug delivery and tissue engineering, where stimuli-sensitive materials are used.
Supplementary Material
Figure 3.
LCST profile of Pentamer 5 after enzymatic reaction.
Acknowledgment
We thank the NIGMS of the NIH (GM 065255) and the U.S. Army Research Office for partial support of this work. We also thank the Materials Research Science and Engineering Center at UMass for infrastructural support.
Footnotes
Supporting Information Available: Synthesis and other experimental details. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.a Langer R. Science. 1990;249:1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]; b Kwon IC, Bae YH, Kim SW. Nature. 1991;354:291–293. doi: 10.1038/354291a0. [DOI] [PubMed] [Google Scholar]; c Alarcon CH, Pennadam S, Alexander C. Chem. Soc. Rev. 2005;34:276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]; d Meng FH, Zhong ZY, Feijen J. Biomacromolecules. 2009;10:197–209. doi: 10.1021/bm801127d. [DOI] [PubMed] [Google Scholar]; e Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik F, Zauscher S, Luzinov I, Minko S. Nat. Mater. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 2.a Juodkazis S, Mukai N, Wakaki R, Yamaguchi A, Matsuo S, Misawa H. Nature. 2000;408:178–181. doi: 10.1038/35041522. [DOI] [PubMed] [Google Scholar]; b Liu JW, Lu Y. J. Am. Chem. Soc. 2008;127:12677–12683. doi: 10.1021/ja053567u. [DOI] [PubMed] [Google Scholar]; c Masato I, Rika O, Atsuhiko W, Itaru H. Chem. Sci. 2010;1:491–498. [Google Scholar]; d Savariar EN, Ghosh S, Gonzalez D, Thayumanavan S. J. Am. Chem. Soc. 2008;130:5416–5417. doi: 10.1021/ja800164z. [DOI] [PubMed] [Google Scholar]; e Yashima E, Maeda K. Macromolecules. 2008;41:3–12. [Google Scholar]; f Kang YJ, Walish JJ, Gorishnyy T, Thomas EL. Nat. Mater. 2007;6:957–960. doi: 10.1038/nmat2032. [DOI] [PubMed] [Google Scholar]
- 3.a Yamada N, Okano T, Sakai H, Karikusa F, Sawasaki Y, Sakurai Y. Makromol. Chem. Rapid Commun. 1990;11:571–576. [Google Scholar]; b Lee KY, Peters MC, Anderson KW, Mooney DJ. Nature. 2000;408:998–1000. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]; c Tandon N, Cannizzaro C, Chao PHG, Maidhof R, Marsano A, Au HTH, Radisic M, Gordana VN. Nat. Protoc. 2009;4:155–173. doi: 10.1038/nprot.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chung HJ, Park TG. Nano Today. 2009;4:429–437. [Google Scholar]
- 4.a Uhlmann P, Ionov L, Houbenov N, Nitschke M, Grundke K, Motornov M, Minko S, Stamm M. Prog. Org. Coat. 2006;55:168–174. [Google Scholar]; b Kim JW, Yoon J, Hayward RC. Nat. Mater. 2010;9:159–164. doi: 10.1038/nmat2606. [DOI] [PubMed] [Google Scholar]; c Shchukin DG, Grigoriev DO, Mohwald H. Soft Matter. 2010;6:720–725. [Google Scholar]; d Cole MA, Jasieniak M, Thissen H, Voelcker NH, Griesser HJ. Anal. Chem. 2009;81:6905–6912. doi: 10.1021/ac9009337. [DOI] [PubMed] [Google Scholar]; e Chen XX, Gao J, Song B, Smet M, Zhang X. Langmuir. 2010;26:104–108. doi: 10.1021/la902137b. [DOI] [PubMed] [Google Scholar]
- 5.a Diaz DD, Kuhbeck D, Koopmans RJ. Chem. Soc. Rev. 2011;40:427–448. doi: 10.1039/c005401c. [DOI] [PubMed] [Google Scholar]; b Zhang JT, Wei G, Keller TF, Gallagher H, Stotzel C, Muller FA, Gottschaldt M, Schubert US, Jandt KD. Macromol. Mater. Eng. 2010;295:1049–1057. [Google Scholar]; c He YB, Bian Z, Kang CQ, Cheng YQ, Gao LX. Chem. Commun. 2010;46:3523–3534. [Google Scholar]; d Ge ZS, Xie D, Chen DY, Jiang XZ, Zhang YF, Liu HW, Liu SY. Macromolecules. 2007;40:3538–3546. [Google Scholar]
- 6.a Kikuchi A, Okano T. Prog. Polym. Sci. 2002;27:1165–1193. [Google Scholar]; b Wahlund PO, Galaev Y, Kazakov SA, Lozinsky VI, Mattiasson B. Macromol. Biosci. 2002;2:33–42. [Google Scholar]; c Nash MA, Yager P, Hoffman AS, Stayton PS. Bioconjugate Chem. 2010;21:2197–2204. doi: 10.1021/bc100180q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Choi SW, Zhang Y, Xia YN. Angew. Chem. Int. Ed. 2010;49:7904–7908. doi: 10.1002/anie.201004057. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dickson JA, Calderwood SK. Nature. 1976;263:772–774. doi: 10.1038/263772a0. [DOI] [PubMed] [Google Scholar]; c Galiana G, Branca RT, Jenista ER, Warren WS. Science. 2008;332:421–424. doi: 10.1126/science.1163242. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Weinstein JN, Magin RL, Yatvin MB, Zaharko DS. Science. 1979;204:188–191. doi: 10.1126/science.432641. [DOI] [PubMed] [Google Scholar]; e Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Science. 1978;202:1290–1293. doi: 10.1126/science.364652. [DOI] [PubMed] [Google Scholar]
- 8.a Liu RX, Fraylich M, Saunders BR. Colloid Polym. Sci. 2009;287:627–643. [Google Scholar]; b Wei. H, Zhang XZ, Zhou Y, Cheng SX, Zhuo RX. Biomaterials. 2006;27:2028–2034. doi: 10.1016/j.biomaterials.2005.09.028. [DOI] [PubMed] [Google Scholar]; c Cammas S, Suzuki K, Sone C, Sakurai Y, Kataoka K, Okano T. J. Controlled Release. 1997;48:157–164. [Google Scholar]; d Ebara M, Yamato M, Aoyagi T, Kikuchi A, Sakai K, Okano T. Biomacromolecules. 2004;5:505–510. doi: 10.1021/bm0343601. [DOI] [PubMed] [Google Scholar]
- 9.a Furukawa H, Shimojyo R, Ohnishi N, Fukuda H, Kondo A. Appl. Microbiol. Biotechnol. 2003;62:478–483. doi: 10.1007/s00253-003-1330-7. [DOI] [PubMed] [Google Scholar]; b Kanazawa H, Kashiwase Y, Yamoamoto K, Matsushima Y. Anal. Chem. 1997;69:823–830. doi: 10.1021/ac961024k. [DOI] [PubMed] [Google Scholar]; c Kanazawa H, Sunamoto T, Matsushima Y. Anal. Chem. 2000;72:5961–5966. doi: 10.1021/ac0004658. [DOI] [PubMed] [Google Scholar]; d Lutz JF, Weichenhan K, Akdemir O, Hoth A. Macromolecules. 2007;40:2503–2508. [Google Scholar]; e Okano T, Yamada N, Okuhara M, Sakai H, Sakurai Y. Biomaterials. 1995;16:297–303. doi: 10.1016/0142-9612(95)93257-e. [DOI] [PubMed] [Google Scholar]; f Cunliffe D, de las Heras Alarcon C, Peters VSJR, Alexander C. Langmuir. 2003;19:2888–2899. [Google Scholar]
- 10.a Meewes M, Ricka J, De Silva M, Nyffenegger R, Binkert T. Macromolecules. 1991;24:5811–5816. [Google Scholar]; b Wu C, Zhou SQ. Macromolecules. 1995;28:8381–8387. [Google Scholar]; c Schmaljohann D. Adv. Drug Delivery Rev. 2006;58:1655–1670. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 11.a Pelton R. Adv. Colloid Interface Sci. 2000;85:1–33. doi: 10.1016/s0001-8686(99)00023-8. [DOI] [PubMed] [Google Scholar]; b Zhang J, Peppas NA. Macromolecules. 2000;33:102–107. [Google Scholar]; c Beltran S, Baker JP, Hooper HH, Blanch HW, Prausnitz JM. Macromolecules. 1991;24:102–107. [Google Scholar]; d Stayton PS, Shimoboji T, Long C, Chilkoti A, Chen GH, Harris JM, Hoffman AS. Nature. 1995;378:472–474. doi: 10.1038/378472a0. [DOI] [PubMed] [Google Scholar]; e Ballauff M, Lu Y. Polymer. 2007;48:1815–1823. [Google Scholar]; f Haba Y, Harada A, Takagishi T, Kono K. J. Am. Chem. Soc. 2004;126:12760–12761. doi: 10.1021/ja047755g. [DOI] [PubMed] [Google Scholar]
- 12.a Badi N, Lutz JF. J. Controlled Release. 2009;140:224–229. doi: 10.1016/j.jconrel.2009.04.012. [DOI] [PubMed] [Google Scholar]; b Lian XM, Wu DX, Song XH, Zhao HY. Macromolecules. 2010;43:7434–7445. [Google Scholar]; c Bae YC, Lambert SM, Soane DS, Prausnitz JM. Macromolecules. 1991;24:4403–4407. [Google Scholar]; d Karlstrom G. J. Phys. Chem. 1985;89:4962–4964. [Google Scholar]; e Nwankwo I, Xia DW, Smid J. J. Polym. Sci., Part B: Polym. Phys. 1988;26:581–594. [Google Scholar]; f Lutz JF, Hoth A, Schade K. Des. Monomers Polym. 2009;12:343–353. [Google Scholar]; g Kojima C, Yoshimura K, Harada A, Sakanishi Y, Kono K. J. Polym. Sci., Part A: Polym. Chem. 2010;48:4047–4054. [Google Scholar]
- 13.Aathimanikandan SV, Savariar EN, Thayumanavan S. J. Am. Chem. Soc. 2005;127:14922–14929. doi: 10.1021/ja054542y. [DOI] [PubMed] [Google Scholar]
- 14.a Chen G, Hoffman AS. Nature. 1995;373:49–52. doi: 10.1038/373049a0. [DOI] [PubMed] [Google Scholar]; b Peng T, Cheng YL. Polymer. 2001;42:2091–2100. [Google Scholar]; c Kurisawa M, Terano M, Yui. N. Macromol Rapid Commun. 1995;16:663–666. [Google Scholar]; d Klaikherd A, Chikkanagari N, Thayumanavan S. J. Am. Chem. Soc. 2009;131:4830–4838. doi: 10.1021/ja809475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a Qiao ZY, Du FS, Zhang R, Liang DH, Li ZC. Macromolecules. 2010;43:6485–6494. [Google Scholar]; b Magnusson JP, Khan A, Pasparakis G, Saeed AO, Wang WX, Alexander C. J. Am. Chem. Soc. 2008;130:10852–10853. doi: 10.1021/ja802609r. [DOI] [PubMed] [Google Scholar]; c Roth PJ, Jochum FD, Forst FR, Zentel R, Theato P. Macromolecules. 2010;43:4638–4645. [Google Scholar]; d Sun GB, Guan AB. Macromolecules. 2010;43:9668–9673. [Google Scholar]; e Muh KM, Bae YH. Polymer. 1999;40:6147–6155. [Google Scholar]; f Dong Y, Gunning P, Cao HL, Mathew A, Newland B, Saeed AO, Magnusson JP, Alexander C, Tai HY, Pandit A, Wang WX. Polym. Chem. 2010;1:827–830. [Google Scholar]; g Yamamoto SI, Pietrasik J, Matyjaszewski K. J. Polym. Sci., Part A: Polym. Chem. 2008;46:194–202. [Google Scholar]
- 16.Arvinte T, Bui TTT, Dahab AA, Demeule B, Drake AF, Ethag D, King P. Anal. Biochem. 2004;332:46–57. doi: 10.1016/j.ab.2004.05.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.