

Abstract
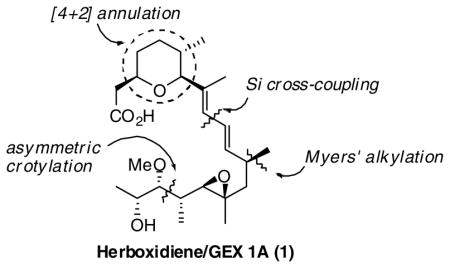
A convergent enantioselective synthesis of herboxidiene/GEX 1A (1) is described, which features a double stereodifferentiating crotylation, [4+2]-annulation and a silicon-based sp2-sp2 cross-coupling to assemble the conjugated diene.
Herboxidiene/GEX 1A (1), a secondary metabolite originally isolated from Streptomyces sp. A7847, displayed selective phytotoxicity against a range of broadleaf annual weeds while remaining harmless to coplanted wheat.1 In a search for new antitumor agents, 1 was reisolated from a fermentation culture broth with five other structurally related GEX 1 members (2–6, Figure 1).2 While several members of this family showed potent cytotoxicity (IC50 values ranging from 3.7 nM to 0.99 μM) against several human tumor cell lines in vitro, GEX 1A is the only one possessing antitumor activity in vivo. The entire family of GEX 1 compounds displayed cytotoxicity via up-regulating luciferase reporter gene expression as well as inducing both G1 and G2/M arrest in human tumor cell line WI-38.3 The stereochemical assignment of 1 was obtained through a combination of degradation studies, partial synthesis and crystallographic analysis.4 The class of natural products possesses several synthetically challenging structural features, including the trisubstituted tetrahydropyran core, the conjugated diene moiety and the poly-oxygenated side chain. Two total syntheses and several synthetic approaches toward herboxidiene/GEX 1A have recently been published.5
Figure 1.

Structures of GEX 1 family members (1–6).
Herein, we describe a convergent enantioselective total synthesis of herboxidiene/GEX 1A (1) that makes use of organosilane based bond construction methodology in three crucial ways (Scheme 1). The first disconnection at C9-C10 leads to a functionalized pyran core and an oxygenated side chain. We anticipated using a silicon-assisted sp2-sp2 cross-coupling for the union of intermediates 7 and 8. In this plan, the conjugated (E, E)-diene could be accessed with high levels of selectivity. Dihydropyran 7 could be obtained from syn-silane reagent 9 and the silyl-substituted methacrolein 10 utilizing our stereoselective [4+2]-annulation strategy.6 Further, we hoped that side chain 11 could be rapidly constructed from silane reagent (S)-12 and α-silyloxy acetal (S)-13.
Scheme 1.

Retrosynthetic analysis of herboxidiene/GEX 1A (1)
Synthesis of the C10-C19 fragment was initiated with a double stereodifferentiating crotylation7 based on the use of a newly developed crotylsilane reagent (S)-12 that bears a fully substituted stereocenter (Scheme 2).8 Since the C18 hydroxyl would be properly configured for a directed epoxidation later in the synthesis, we chose (S)-silyloxy acetal 13, as the crotylation electrophile.9 Thus, a matched crotylation between (S)-12 and (S)-13 promoted by TMSOTf, provided the desired syn-homoallylic ether 11 containing a trans trisubstituted olefin in 62% yield and high diastereoselectivity (dr > 30:1). When subjected to Arndt-Eistert conditions10 11 was converted to diazoketone 14 in 97% yield over two steps. Rearrangement of 14 followed by trapping of the ketene with (+)-pseudoephedrine gave the homologated amide 15 in 80% yield. At this stage, the C12 methyl bearing stereocenter was introduced with high selectivity using Myers pseudoephedrine derived auxiliary11 and afforded 16 in 96% yield. The magnitude of diastereoselectivity of the alkylation was determined to be >10:1 after reductive removal of the auxiliary using lithium amidotrihydroborate (LAB).12 The resulting primary alcohol 17 was then oxidized to the corresponding aldehyde under Swern conditions13, which was subjected to Takai iodoolefination to give the (E)-vinyl iodide 8 (E/Z > 20:1, 75% yield).14
Scheme 2.

Synthesis of C10-C19 fragment 8
The short sequence required for the elaboration of the cis-2,6-trans-5,6-tetrahydropyran core is summarized in Scheme 3. A TMSOTf promoted [4+2]-annulation between syn-crotylsilane 9 and (E)-vinylsilyl aldehyde 1015 provided cis-2,6-dihydropyran 18 in 65% yield and with high selectivity (dr > 30:1).6 Initially, this annulation was plagued with significant amounts of protodesilylation giving terminal olefin 18a as the major product. After screening several bases and solvent systems, we learned that with catalytic amounts of 2,6-di-tert-butylpyridine (DTBP) and 1.0 equiv TMSOTf in a mixture of CH2Cl2:MeCN (3:1, −20 °C), the reaction proceeded smoothly to provide the desired product in a useful yield. Reduction of 18, followed by a hydroxyl-directed chemoselective hydrogenation using Wilkinson’s catalyst16, afforded the tetrahydropyran product 19 in 87% yield over both steps. Alcohol 19 was then converted to a tosylate followed by displacement with sodium cyanide to give the desired nitrile 20, thereby reconstituting the oxidation state of a carboxylate.
Scheme 3.

Synthesis of C1-C9 fragment 20
With workable amounts of advanced intermediates 8 and 20 available, we were positioned to carry out the crucial silicon-based sp2-sp2 cross-coupling.17 Preactivation of vinylsilane 20 with 2.2 equiv of TBAF18 followed by addition of [AllyPdCl]2 and vinyliodide 8, the desired product 21 was obtained in a good yield and exclusively as the (E, E)-diene isomer. Nitrile 21 was then partially reduced to the aldehyde by DIBAL-H. Pinnick oxidation19 followed by methylation with TMS stabilized diazomethane provided methyl ester 22 in three steps, 59% yield. Removal of the TBDPS group using TBAF was followed by a directed epoxidation of the bishomoallylic alcohol 23.20 As reported, C18 hydroxyl-directed epoxidation, catalyzed by VO(acac)2 gave a single diastereomer in 48% yield.5a Inversion of the C18 hydroxyl under modified Mitsunobu conditions21 and saponification of the resulting di-ester5 completed the total synthesis of herboxidiene/GEX 1A (1).
In summary, we have described a highly convergent and enantioselective synthesis of herboxidiene/GEX 1A (1) in 16 steps from the crotysilane 12. The construction of pyran fragment and oxygenated side chain, and the utility of vinylsilane in the union of 8 and 20 demonstrate the versatility of organosilanes in natural product synthesis. This work also illustrates the use of silicon-based cross-coupling as an alternative to vinylstannane and other more sensitive metal based cross-coupling reactions in complex molecule synthesis.
Supplementary Material
Scheme 4.

Completion of the total synthesis of 1
Acknowledgments
Financial support for this research is obtained from the NIH NIGMS GM55740. The authors are grateful to Dr. Qibin Su (AstraZeneca, Inc. Boston), Dr. Fredrik Lake (AstraZeneca, Inc. Södertälje) and Dr. Frauke Pohlki (Boston University) for useful suggestions and discussions.
Footnotes
Supporting Information Available Experimental details and new selected spectral for all new compounds. This material is available free of charge in the Internet at http://pubs.acs.org.
References
- 1.Isaac BG, Ayer SW, Elliott RC, Stonard RJ. J Org Chem. 1992;57:7220. [Google Scholar]
- 2.Sakai Y, Yoshida T, Ochiai K, Uosaki Y, Saitoh Y, Tanaka F, Akiyama T, Akinaga S, Mizukami T. J Antibiot. 2002;55:855. doi: 10.7164/antibiotics.55.855. [DOI] [PubMed] [Google Scholar]
- 3.Sakai Y, Tsujita T, Akiyama T, Yoshida T, Mizukami T, Akinaga S, Horinouchi S, Yoshida M, Yoshida M. J Antibiot. 2002;55:863. doi: 10.7164/antibiotics.55.863. [DOI] [PubMed] [Google Scholar]
- 4.Edmunds AJF, Trueb W, Oppolzer W, Cowley P. Tetrahedron. 1997;53:2785. [Google Scholar]
- 5.Total synthesis, see: Blakemore PR, Kocieński PJ, Morley A, Muir KJ. Chem Soc, Perkin Trans I. 1999:955.Banwell M, McLeod M, Premraj R, Simpson G. Pure Appl Chem. 2000;72:1631.Diastereomeric synthesis, see: Smith ND, Kocieński PJ, Street SDA. Synthesis. 1996:652.Fragment synthesis, see: Banwell MG, Bui CT, Simpson GW, Watson KG. Chem Commun. 1996:723.Banwell MG, Bui CT, Hockless DCR, Simpson GW. J Chem Soc, Perkin Trans I. 1997:1261.Banwell MG, Bui CT, Simpson GW. J Chem Soc, Perkin Trans I. 1998:791.SAR studies, see: Edmunds AJF, Arnold G, Hagmann L, Schaffner R, Furlenmeier H. Med Chem Lett. 2000:1365. doi: 10.1016/s0960-894x(00)00230-4.
- 6.Huang H, Panek JS. J Am Chem Soc. 2000;122:9836. [Google Scholar]
- 7.(a) Jain NF, Takenaka N, Panek JS. J Am Chem Soc. 1996;118:12475. [Google Scholar]; (b) Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed. 1985;24:1. [Google Scholar]
- 8.Lowe JT, Panek JS. Org Lett. 2005;7:1529. doi: 10.1021/ol0501875. [DOI] [PubMed] [Google Scholar]
- 9.(S)-silyloxy acetal 13 was prepared in two steps from the commercially available (S)-ethyl lactate. See Supporting Information for details.
- 10.Kirmse W. Eur J Org Chem. 2002:2193. [Google Scholar]
- 11.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky D, Gleason JL. J Am Chem Soc. 1997;119:6496. [Google Scholar]
- 12.For preparation of LAB reagent, see: ref. 11
- 13.Omura K, Swern D. Tetrahedron. 1978;34:1651. [Google Scholar]
- 14.Takai K, Nitta K, Utimoto K. J Am Chem Soc. 1986;108:7408. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 15.(a) Mohamed M, Brook MA. Helv Chim Acta. 2002;85:4165. [Google Scholar]; (b) Spino C, Gobdout C. J Am Chem Soc. 2003;125:12106. doi: 10.1021/ja037078a. [DOI] [PubMed] [Google Scholar]
- 16.For a review of hydroxyl-directed hydrogenation, see: Brown JM. Angew Chem Int Ed. 1987;26:190.
- 17.For recent reports of Si-based X-couplings, see: Denmark SE, Neuville L, Christy MEL, Tymonko SA. J Org Chem. 2006;71:8500. doi: 10.1021/jo061481t.Denmark SE, Sweis RF, Wehrli D. J Am Chem Soc. 2004;126:4865. doi: 10.1021/ja037234d.Rendler S, Oestreich M. Synthesis. 2005:1727.Spivey AC, Gripton CJG, Hannah J. Current Organic Synthesis. 2004;1:211.Denmark SE, Liu JHC. J Am Chem Soc. 2007;129:3737. doi: 10.1021/ja067854p.
- 18.For examples using benzyldimethylsilyl group in cross-coupling, see: Trost BM, Frederiksen MU, Papillon JPN, Harrington PE, Shin S, Shireman BT. J Am Chem Soc. 2005;127:3666. doi: 10.1021/ja042435i.Trost BM, Machacek MR, Ball ZT. Org Lett. 2003;5:1895. doi: 10.1021/ol034463w.
- 19.Bal BS, Childers WE, Jr, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
- 20.(a) Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307. [Google Scholar]; (b) Nakata T, Schmid G, Vranesic B, Okigawa M, Smith-Palmer T, Kishi Y. J Am Chem Soc. 1978;100:2933. [Google Scholar]
- 21.Martin SF, Dodge JA. Tetrahedron Lett. 1991;32:3017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.