

Abstract
The response to extracellular stimuli often alters the phosphorylation state of plasma membrane-associated proteins. In this regard, generation of a comprehensive membrane phosphoproteome can significantly enhance signal transduction and drug mechanism studies. However, analysis of this subproteome is regarded as technically challenging, given the low abundance and insolubility of integral membrane proteins, combined with difficulties in isolating, ionizing and fragmenting phosphopeptides. In this article, we highlight recent advances in membrane and phosphoprotein enrichment techniques resulting in improved identification of these elusive peptides. We also describe the use of alternative fragmentation techniques, and assess their current and future value to the field of membrane phosphoproteomics.
Keywords: CID, enrichment, ETD, HCD, neutral loss, phosphopeptide, phosphoproteomics, plasma membrane
The importance of sequence-specific protein phosphorylation events in cellular physiology cannot be overstated. These fundamentally important post-translational modifications represent ‘binary switches’ controlling functions such as enzymatic activity, rate of degradation (turnover), interactions with other proteins and subcellular localization (reviewed elsewhere [1]). Importantly, deregulation of phosphorylation-based signaling networks is the basis of many diseases, including cancer (reviewed elsewhere [2]). For example, a significant number of protein kinases (serine, threonine and tyrosine) have been implicated as driving oncogenesis, including EGFR, HER2, ABL and SRC (reviewed elsewhere [3]). With an estimated 100,000+ phosphorylation motifs in the human proteome (reviewed elsewhere [4]), global scrutiny of phospho-state changes has the potential to provide far more biologically relevant data than simple monitoring of abundance changes.
Mass spectrometry (MS)-based phosphoproteomic analysis is emerging as an essential tool in the unbiased monitoring of phospho-state changes. In recent years, phosphoproteome analysis has been conducted using nearly every available fractionation and MS technique [5-10]. As a whole, these studies have been successful in identifying new phosphorylation sites and at uncovering a multitude of downstream signal transduction events. However, relative to standard proteomic approaches, challenges in terms of phosphopeptide enrichment and MS analysis seriously limit the number of events that can be identified.
A further layer of complexity is that in many studies, it would be preferable to analyze the phospho-state changes for plasma membrane (PM) proteins. The importance of PM proteins in modern research is highlighted by the fact that they are the target of 70% of approved pharmaceutical agents [11]. As such, drug development and signal transduction studies (among others) can benefit from quantitative monitoring of membrane phospho-state changes occurring immediately after treatment. These initiating events would obviously be more difficult to detect if phosphoproteomic analysis was performed using crude cell lysate.
Proteins at the PM either span the lipid bilayer, are covalently bound to lipid molecules (integral membrane proteins) or are loosely associated on the cytoplasmic or extracellular side of the membrane (peripheral membrane proteins). Thus, PM proteins can contain both hydrophobic (transmembrane) domains, as well as hydrophilic domains. Unfortunately, the very characteristics that lead proteins to associate with the PM make them difficult to analyze with MS. Most troublesome are the integral membrane proteins containing both hydrophobic domains and, quite often, glycosylated extracellular domains (reviewed elsewhere [12,13]). Therefore, proteomic analysis of the PM is itself uniquely problematic given the challenges in protein purification and recovery. Taken together, these limitations suggest that successful analysis of the PM phosphoproteome is reliant on the efficient integration of protocols for protein purification, phosphopeptide enrichment and analysis.
In this article, we discuss some of the challenges inherent in PM phosphoproteomics. We begin by describing the difficulties in isolating peptides from the PM, along with highlighting traditional and more recent methods used to enhance coverage of these elusive proteins. This is followed by a summary of some successful approaches for phosphopeptide identification, including multimode enrichment techniques and promising contemporary MS/MS fragmentation methods that increase numbers of identified phosphorylation events. It is hoped that this survey of current approaches will assist those interested in developing PM phosphoproteomic workflows. Figure 1 is a summary of steps typically involved in PM phosphoproteomics, along with some more recent methodological advances.
Figure 1. Steps and possible approaches detailed in this review for a membrane phosphoproteomics experiment.
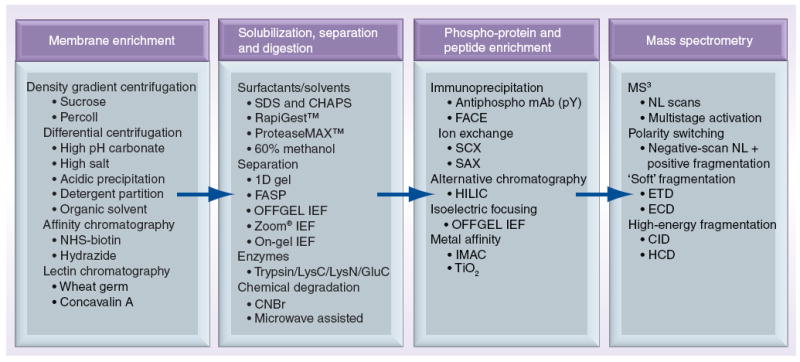
CHAPS: 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; CID: Collision-induced dissociation; CNBr: Cyanogen bromide; ECD:Electron capture dissociation; ETD: Electron transfer dissociation; FACE: Filter-based affinity capturing and elution; FASP: Filter-aided sample preparation; HCD: Higher-energy collisional dissociation; HILIC: Hydrophilic interaction chromatography; IEF: Isoelectric focusing; IMAC: Immobilized metal affinity chromatography; mAb: Monoclonal antibody; MS: Mass spectrometry; NHS: N-hydroxysuccinimide; NL: Neutral loss; SAX: Strong anion chromatography; SCX: Strong cation chromatography; SDS: Sodium dodecyl sulfate; TiO2: Titanium dioxide.
Plasma membrane purification/enrichment
Several hurdles are associated with the purification of PM proteins including: low recovery rates, protein hydrophobicity and problems with contamination from other organelles. The traditional approach to PM enrichment involves cell lysis and gradient centrifugation. In this regard, cells are harvested and disrupted by one of several methods including Dounce, glass-bead or ball- bearing homogenization, where care is taken that nuclei are not broken as the released DNA will cause a significant loss of components during the first centrifugation step. Homogenization is followed by low-speed centrifugation to pellet nuclei and intact cells [14-17]. Methodologies that combine high pH carbonate, high salt, detergents such as deoxycholate or Triton® X-114, urea and chlorform/methanol have been used in a stepwise manner to further increase the PM purity by removing nonintegral membrane proteins and to aide in the solubilization of hydrophobic proteins [14,16]. Selective acid precipitation (e.g., 1M acetic acid) has also been reported to remove contaminating organelles such as mitochondria [14]. As a last step, the partially pure PM preparation is subjected to a density gradient centrifugation that most commonly utilizes sucrose, although other gradient types such as Ficoll or Percoll are effective [15]. Fractions are then collected in small volumes and analyzed for purity either by western blotting for PM-specific markers or, by enzymatic assays for the presence of organelle-specific enzymatic activities. The fractions containing the PM preparation are then pooled, washed and pelleted.
A focus of many recent proteomic studies has been on ‘lipid rafts’, microdomains of detergent-resistant sphingolipids and cholesterol within the PM (reviewed elsewhere [18]). Multiple studies have shown that these regions are selectively enriched for signaling molecules including Src family kinases. In order to obtain the proteins located in these domains by gradient centrifugation methods, it is important to pretreat the sample with cholesterol-disrupting chemicals, such as nystatin or filipin, prior to or during the harvesting process [19].
A number of commercially available kits exist to help streamline the gradient centrifugation-based enrichment of the PM, such as the Qproteome® Plasma Membrane Protein Kit sold by Qiagen (CA, USA) and the Mem-PER® Eukaryotic Membrane Protein Extraction Kit, a product of Pierce (Thermo Fisher Scientific, IL, USA). However, even with all the differential centrifugation steps to remove the soluble proteins, nuclei and mitochondria, and even with the final sucrose gradient centrifugation step to separate membranes with different densities, the final PM preparation is still typically contaminated with significant levels of cellular proteins and yields are low. The PM is a difficult-to-isolate organelle in that the membranes of many other organelles (endoplasmic reticulum, Golgi, mitochondria and, to a lesser extent, the nuclei) have a similar composition. In the context of membrane phosphopeptides, another cause of concern in this process is that the crude nature of this approach may result in proteins that appear to be downregulated in phosphorylation when, in fact, the protein did not effectively partition into the membrane fraction. This is less of a concern when techniques such as stable isotope labeling with amino acids in cell culture (SILAC) or deuterium labeling are employed and the two samples (control and treated) are harvested in an identical manner [9,20]. For these reasons, the classical methods of isolation are far from ideal (reviewed elsewhere [21]).
Alternative strategies to increase the purity of the PM preparation have been developed. The most common involves affinity chromatography of selectively tagged cell-surface proteins. Reactive biotin-containing linkers targeting amino, sulfhydryl or aldehyde groups are widely used to label proteins or oligosac-charide groups on glycoproteins, and these protocols can easily be integrated into proteomic workflows. One example is the use of sulfo-NHS-SS-biotin, an agent that is reactive with primary amines and cannot cross membranes [22]. In order to enrich membrane proteins, the compound is added to extensively washed cells and allowed to bind to free amine groups present on cell-surface proteins. Once labeled, cells are lysed and biotin-tagged proteins are captured via streptavidin- (or avidin-) coated supports. The enriched membrane proteins may be eluted using solutions of high urea and dithiothreitol (DTT) [23]. An advantage to this method is that differential centrifugation can also be used to remove non-labeled contaminants. A disadvantage is the potential labeling of extracellular proteins that are intrinsic to the cell growth media or are released by lysed cells. Likewise, this technique can only label proteins with exposed extracellular domains, leading to an under-representation of intracellular PM-associated proteins (reviewed elsewhere [24]).
A similar purification technique involves lectin chromatography to target the carbohydrate groups of glycosylated proteins. Lectin affinity chromatography is effective in PM protein preparation, as many transmembrane PM proteins are glycosylated. Lectins such as concanavalin A (targets high-mannose structures) and wheat germ agglutinin (WGA; targets β-d-GlcNAc and sialylated glycans) have been used effectively as an enrichment step for purification of membrane-bound glycoproteins [25,26]. Vercoutter-Edouart et al. used lectin chromatography to study glycosylation changes during colon epithelium differentiation and proliferation [25,26]. Here, a crude membrane preparation was derived using differential centrifugation to remove nuclei and a pH 11 carbonate wash to remove cytoskeletal proteins. This was followed by Triton X-100 to extract membrane proteins. This crude membrane preparation was applied to a Concanavalin A Sepharose column, the column washed extensively before the retained glycoproteins were eluted with methyl-β-d glucose. Among the eluted proteins, 65% were identified as membrane proteins. To improve coverage, McDonald et al. coupled lectin chromatography with hydrazide chemistry [27]. They treated intact HeLa cells with periodate to oxidize vicinal hydroxyls of carbohydrate groups to aldehydes, lysed the cells and coupled the glycoproteins carrying the oxidized glycans to hydrazide resin. Following extensive washing, the covalently linked proteins were digested with trypsin and the N-linked glycopeptides released by cleavage with PNGase. The authors also performed lectin chromatography on the HeLa cell lysate. Of the 240 glycoproteins identified, only 42% were identical between the periodate/hydrazide and lectin affinity chromatography; therefore, combining the two separate methods was advantageous.
Protein solubilization, separation & digestion
Solubilization of cellular extracts with the anionic surfactant sodium dodecyl sulfate (SDS) remains the benchmark by which all other methods are judged [28,29]. Unfortunately, SDS is incompatible with many stages of downstream processing and is difficult to remove from aqueous solutions. A recent strategy described by Wisniewski et al. has shown promise in identifying and quantifying membrane proteins that are solubilized under high detergent conditions. The technique, filter-aided sample preparation (FASP), was employed in a study that successfully identified more than 1000 membrane proteins in samples taken from a murine hippocampus [30]. In the FASP method, cells are lysed and the cellular components solubilized by heating in a buffer containing 4% SDS and 0.1 M DTT. The solution is then cleared by centrifugation and mixed with a solution of 8 M urea before being placed in a filtration cartridge with a high-molecular-weight cut-off. All steps of reduction, alkylation and digestion take place within this cartridge before the digested peptides are ultimately released by centrifuging the cartridge, leaving behind the large, undigested molecules, such as DNA [31]. The FASP technique appears to circumvent the pitfalls of earlier techniques by solubilizing all cellular proteins at once. The use of one reaction cell clearly reduces the losses incurred in previous techniques (by multiple washes and centrifugation steps) and stands as a major advancement in obtaining high peptide yields at high purity with relative ease. The primary drawback with the FASP technique is that while it allows the level of membrane proteins to be more accurately represented in the mixture of tryptic peptides, it is not a method of membrane protein enrichment. Fundamentally, the issues of relatively low abundance of many membrane proteins relative to proteins found in other cellular fractions still remain, and with the FASP technique there is no way to enrich for PM proteins. The success attributed to this method has been mainly due to the use of the most sensitive MS platforms – technologies that are not yet available to all groups. In order for this technique to successfully showcase low-abundant membrane proteins found with other platforms, preceding membrane enrichment step(s) would be required before use of the FASP reaction chamber.
Alternatively, several MS-compatible surfactants have recently been introduced. For example RapiGest™ (Waters, MA, USA) is an acid labile surfactant that does not inhibit protease activity and is MS compatible [32]. Both RapiGest and similar products, such as ProteaseMAX™ (Promega, WI, USA) have been shown to result in more efficient digestion and higher coverage of membrane proteins, as well as allowing digestions to complete in a much shorter time. Unfortunately, RapiGest and similar products have been shown to produce an insoluble film on the sample surface, a result of poor solubility of a cleavage byproduct. The removal of these films increases sample preparation time and may be a cause of sample contamination (reviewed elsewhere [33]). Another approach to the problem of membrane protein solubility and digestion is the use of organic solvents to break down the phospholipid bilayer and hold the proteins in solution until they can be digested. Blonder et al. describe the successful extraction, solubilization and tryptic digestion of proteins from both Halobacterium halobium purple membranes and human epidermal membrane proteins using a buffered solution of 60% methanol [34]. Although the efficiency of the trypsin was reduced to 20% that of the aqueous buffer, SDS-PAGE analysis demonstrated that it was more than sufficient for the digestion of the well-characterized Halobacterium purple membrane proteins and for the high coverage sequencing of human membrane proteins [34].
In reality, yields from purification of PM are low due to the hydrophobic nature and low abundance of many of these proteins [35]. Therefore, maximizing the amount of protein that can be applied to phosphopeptide enrichment techniques becomes a primary concern. Although classical protein separation techniques can be employed at this point (2D-PAGE, isoelectric focusing [IEF]), problems with membrane protein incompatibility and further sample loss make it considerably more efficient to directly solubilize and digest protein [36]. Protein fractionation using 1D-PAGE, with subsequent gel cutting, remains a widely used, if imprecise, technique if a peptide fractionation strategy is unavailable (see later).
Focusing on proteolysis, another unique consideration in phosphoproteomics is in obtaining adequate peptides that encompass a potential phosphorylation site. This is rarely a problem facing PM proteomics, as digestion of a protein with any proteolytic enzyme should produce at least a few peptides that fall within the appropriate size range for MS/MS analysis. However, in phosphoproteomics, one is often looking for a specific peptide that covers a potential phosphorylation site, wherein a tryptic digest may be suboptimal. For example, phosphorylation of human STAT proteins mediates regulation of several essential events and is of particular interest to our laboratory. Owing to the presence of multiple lysines and arginines in the proximity of the known phosphorylation sites of these proteins, digestion with trypsin results in peptides that are too small for most MS/MS analysis techniques. For example, the phosphorylation site of STAT1 at Y701 has the sequence ‘GTGYI’ flanked on both sides by lysine residues. Peptides of this size are unlikely to accept more than one positive charge and will be excluded from MS/MS analysis by most shotgun sequencing methods [37]. In some cases, simply changing to a new proteolytic enzyme can result in increased protein coverage. For this reason, enzymes that generate relatively larger proteolytic fragments, such as LysC, are useful [17]. There is also evidence that by using different proteases, phosphopeptide coverage will be increased. Gauci et al. evaluated the complementarity of phosphopeptides obtained in replicate experiments using Lys-N, Lys-C and trypsin to digest Hek293 cell lysates [38]. Analysis of the Lys-N generated peptides resulted in the identification of 2303 nonredundant phosphopeptides, whereas the tryptic and Lys-C digests resulted in 2719 and 861 phosphopeptides, respectively. These results suggest that Lys-C is not as efficient as Lys-N or trypsin for handling regions containing phosphorylation sites. A comparison of the data obtained from a total of 5036 non-redundant phosphopeptides revealed that there was only a 6–8% overlap between the three phosphopeptide populations. Further analysis revealed that Lys-N and trypsin enabled a 72% increase in phosphopeptides that were enriched in significantly different phosphorylation motifs, compared with a second replicate tryptic digest that only resulted in a 25% increase. In addition, the choice of enzyme may depend on the buffer. For example, Lys-C, an enzyme that cleaves on the C-terminal side of lysine residues, is highly efficient in high concentrations of urea. Alternatively, some groups have found success with chemical degradation methods such as cyanogen bromide (CNBr) [39], which hydrolyze peptide bonds at the C-terminus of methionine residues, amino acids that are particularly prevalent in membrane proteins. Washburn et al. used CNBr to cleave insoluble integral and peripheral membrane proteins isolated from Saccharomyces cerevisiae [40]. From this study they identified 131 proteins with three or more predicted transmembrane domains. In a study by van Montfort et al., a combination of tryptic and CNBr digestion of the integral membrane LacS resulted in twice the sequence coverage of either method alone [39]. Unfortunately, CNBr and its byproducts are toxic and may be absorbed through the skin, leading the technique to fall out of favor in most laboratories (reviewed elsewhere [32]). Recently, other groups have used microwave techniques to speed up the enzymatic digestion of proteins [41]. In addition, as an alternative to enzymatic digestion, microwave irradiation has also been applied to digestion of the protein by acid hydrolysis [42]. Compared with the traditional protein digestion techniques, microwave irradiation offers the following advantages: it is not time consuming (seconds instead of hours); does not require the presence of detergents; and does not require extensive sample clean-up before analysis by MS. In a recent variation of this technique, functionalized zinc oxide-coated iron oxide (Fe3O4@ ZnO) and TiO2 magnetic nanoparticles (MNPs) have been used to enrich phosphopeptides from complex samples and accelerate tryptic digestion of the trapped proteins under microwave heating. Chen and Chen used Fe3O4@ZnO MNPs to analyze complex saliva samples [43]. The Fe3O4@ZnO MNPs served as affinity probes to selectively enrich phosphoproteins and, additionally, the MNPs served as a microwave absorption medium to assist in the tryptic digestion. Using MALDI MS, they were able to shorten the entire analysis time to 20 min with a detection limit of 250 pM for a monophosphopeptide. Similar results were obtained by Hasan et al. for TiO2 MNPs for milk [44]. Table 1 contains a list of proteolytic degradation enzymes and techniques, as well as their cut sites. In order to obtain the full sequence coverage necessary for a true global map of the phosphoproteome, the same protein extract would need to be digested with multiple enzymes with different, nonoverlapping target sites.
Table 1.
Methods for protein digestion and the site of cleavage.
| Method of protein digestion | Cleavage site |
|---|---|
| Trypsin | K, R |
| Lys-C | K |
| Arg-C | R |
| Lys-N | N |
| Glu-C | C |
| Cyanogen bromide | M |
| Acid + irradiation | Varies with acid |
Enrichment of phosphopeptides
As with membrane proteomics, a major challenge facing the field of phosphoproteomics is the relative abundance of phosphopeptides and low phosphorylation stoichiometry (reviewed elsewhere [1,45]). As protein phosphorylation is a transient and reversible post-translational modification, the level of modified protein may be very low. Thus, relatively large amounts of starting material are required to enrich first for PM proteins and, subsequently, for phosphorylated PM proteins [46,47].
Several chromatographic methods using antibody-based, strong ionic, hydrophilic or metal-based resins and, to a lesser extent, IEF have been used to both enrich for phosphorylated peptides and to decrease the complexity of the sample. Phosphoproteins can be enriched using antibodies specific for phosphoserine, phosphothreonine or phosphotyrosine residues. However, the lack of broad spectrum, high-affinity antiserine and antithreonine antibodies available for immunoprecipitation studies has limited the study of serine/threonine phosphorylation events with this approach [48]. However, the existence of several high-affinity anti-pY monoclonal antibodies has allowed for extensive characterization of tyrosine phosphorylated proteins, even though the abundance of these events is substantially lower than that of serine or threonine modification (1800:200:1, pSer:pThr:pTyr) [49]. As a particular antibody may be biased towards a specific phosphotyrosine peptide, two or more antibodies can be used to improve coverage. Rush et al. used a phosphotyrosine-specific antibody to immunoprecipitate phosphotryosine peptides from digested cellular extracts of several cell lines followed by liquid chromatography (LC)–MS/MS [50]. From pervanadate-treated Jurkat cells, they identified 194 phosphotyrosine sites in 185 phosphorylated peptides in a single analysis. They then repeated this approach with several other cancer cell lines and obtained similar results. As this method does not detect phosphoserine- or phosphothreonine-containing proteins, Nagano et al. integrated this method with metal oxide affinity chromatography (discussed later) to enhance the coverage of phosphorylation signaling [51]. The overlap in phosphotyrosine sites between the two methods was low (24 tyrosine phosphorylation sites out of 325). Zheng et al. used a double enrichment method to study tyrosine phosphorylation in the IFN-α signaling pathway [52]. Tyrosine phosphorylated proteins were immunoprecipitated from control and IFN-α-treated Jurkat cell lysates using a mixture of two pTyr-specific monoclonal antibodies. Following digestion and methyl-esterification, the phosphopeptides were enriched by immobilized metal affinity chromatography (IMAC) and analyzed by reverse-phase LC–MS/MS. Using this method, Zheng et al. observed enhanced phosphorylation on characterized as well as novel tyrosine phosphorylation sites in proteins involved in IFN-α signal transduction. Without specifically enriching for proteins with phosphotyrosine residues, which have been estimated to be 0.05–0.5% of the protein phosphorylation events [53], it is likely that many of these events would have been masked by the higher abundant phosphoserine residues during MS analysis [49,54].
A recently reported variation on antibody-based enrichment is the filter-based affinity capturing and elution (FACE) system [55]. This technique uses antiphosphotyrosine antibodies to bind digested peptides, not whole proteins, and is an extension of the FASP technique discussed earlier. In this method, peptides are incubated with the antibodies in a centrifugation chamber with a 50 kDa molecular weight cutoff filter. Centrifugation elutes all unbound peptides, which can be further enriched for peptides carrying the more prevalent phosphoserine or phosphothreonine residues using IMAC, titanium dioxide (TiO2), strong anion exchange chromatography (SAX) or another method of choice as described below. Following several washes, the enriched phosphotyrosine peptides can be eluted from the filter by trifluoroacetic acid and centrifugation, leaving the antiphosphotyrosine antibody behind in the filtration device [55].
Strong cation chromatography (SCX) is a technique that has been successfully utilized to separate phosphorylated from unphosphorylated peptides. SCX enrichment works because the vast majority of phosphopeptides elute early in the gradient due to the charge differential imparted by the phosphate group [5,56]. Beausoleil et al. determined that at a pH of 2.7, nearly 70% of all tryptic peptides from an in silico tryptic digest of human proteins in the National Center for Biotechnology Information protein database have a predicted charge of +2 [5], while only 3% have a charge of +1 and the remainder carry a charge of more than +2. Mono-phosphopeptides have a charge of +1 due to the negative charge a phosphate group carries at low pH. Thus, with increasing salt concentration, phosphopeptides elute earlier than unphosphorylated peptides and multiply phosphorylated peptides are not retained on the column. Although there is little decrease in the complexity of the phosphopeptide fraction, SCX chromatography will result in highly enriched phosphopeptide fractions with removal of the majority of the unphosphorylated peptides. Using this method followed by LC–MS/MS, Ballif et al., identified over 500 protein phosphorylation sites in the developing mouse brain [56], while Beausoleil et al. identified 2002 phosphorylation sites from the nuclear fraction of a HeLa cell lysate [5]. The disadvantage of this approach is the amount of starting material that is required for effective separation on an SCX column. Groups using this approach generally start with 5–20 mg of whole peptide digestion [7,10,57,58]. Starting with less than this amount of protein has been shown to result in poor yields of identified phosphopeptides [47], an observation we have verified in our laboratory (data not shown). A peptide and its phosphorylated counterpart may have more differences than just the added mass of the phosphate group. The downward shift in isoelectric point (pI) imposed by the phosphate can range from minor to significant, depending on the structure of the individual peptide. The phosphate can also increase the hydrophilicity of the peptide. Together, these three changes can make standard fractionation procedures unreliable for predictable fractionation of phosphopeptides. Unfortunately, most SCX buffers are not directly compatible with in-line LC–MS systems, although optimized 2D separations such as MUDPIT have been successfully utilized in a number of studies (reviewed elsewhere [59]). It is worth noting that several groups have recently begun using SAX in lieu of SCX, with similar results [60-62]. However, it has been reported that acidic peptides that bind strongly to SAX and phosphopeptides that bind with an even greater affinity can be difficult to elute [4].
One alternative to SCX peptide fractionation of peptides is liquid-based isoelectric focusing, such as the Agilent OFFGEL system. The OFFGEL uses IEF strips of varying pH gradients beneath fluid cells containing buffers and the peptide mixtures. Isoelectric focusing at high current causes peptides to migrate through the gel and into the wells most closely representing the pI of the individual peptides [63,64]. This platform has been used by a number of different studies for both the separation and enrichment of phosphopeptides. The lowered pI of phosphopeptides causes them to aggregate near the anode and become enriched in those wells [65]. Since phosphopeptides have been shown to focus in the pH range from 3–6 [66], the use of the manufacturer’s low pH range strips can be used to efficiently separate these peptides into as many as 20 fractions in a single run. In evaluations of this platform in our laboratory, we found that the number of phosphopeptides in each well was the highest closest to the anode, with decreasing number of phosphopeptides in each subsequent well approaching the cathode (data not shown). The low pI of phosphopeptides can also be exploited as a crude enrichment technique. Chemical modification of the peptides, such as methyl esterification, has been shown to further enhance and refine this effect. In two separate studies, tryptic digests were subjected to methyl esterification and separated by IEF. Both groups showed that phosphopeptides were enriched at neutral to acidic pH, and that unphosphorylated peptides were removed from the gel because the peptide pIs exceeded the pH gradient of the gels. In this way, they were able to both enrich and separate phosphopeptides within a single procedure, using in-gel IEF [67,68]. The technique was further extended and shown to work off gel, using a modified ZOOM® IEF apparatus (Invitrogen, CA, USA) [69,70].
Another separation technique that has recently been explored for enriching phosphopeptides is hydrophilic interaction chromatography (HILIC) [71]. Peptide retention is based on the hydrophilicity of the peptide. Hydrogen bonding occurs between the peptide and the neutral hydrophilic stationary phase, with retention increasing with increasing hydrophilicity or polarity of the peptide. Samples are bound to the resin at high organic solvent concentration and eluted with an inverse gradient of acetonitrile in water. McNulty et al. showed that phosphopeptides had an increased retention compared with unphosphorylated peptides, and that peptides were uniformly distributed throughout the gradient with short hydrophobic peptides in the earlier fractions and the larger hydrophilic and highly acidic peptides in the later fractions [72]. Approximately 50% of unphosphorylated peptides were found in the solvent front. This is an advantage over SCX, where all the phosphorylated peptides are either not retained by the column or elute at the beginning of the gradient. In addition, the buffer, a salt-free volatile trifluoroacetic acid (TFA)/acetonitrile (ACN), is compatible with any next step in the proteomic study without an intermediate desalting step. Using HILIC in combination with IMAC, McNulty et al. achieved a phosphopeptide selectivity of more than 99% and identified over 1000 unique phosphorylation sites with just 300 μg of a HeLa cell lysate [72]. Using IMAC before HILIC, Albuquerque et al. identified 8764 unique phosphopeptides from 2278 phosphoproteins in their study of yeast S. cerevisiae following DNA damage [73]. In addition, they were able to identify approximately 50% of the phosphorylation sites of two low-abundant proteins, Rad9 and Mrc1.
Another widely used method is IMAC [6,74-76], which is generally used to enrich phosphopeptides just prior to MS analysis. IMAC makes use of the high affinity of the negatively charged phosphate groups for metals, typically Fe3+ or Ga3+, to enrich for phosphopeptides [77]. However, a problem associated with the IMAC technique is the nonspecific binding of nonphosphorylated peptides that contain multiple acidic amino acid residues that co-purify with the phosphopeptides. To overcome this, Ficarro et al. derivatized the carboxylic acids on the acidic amino acids by O-methyl esterification [74]. This led to multiple problems such as sample loss, increased complexity of the sample due to incomplete derivatization and sample loss due to extensive lyophilization [6]. Another method to decrease the contaminating nonphosphorylated peptides is to load the sample at low pH (below 1.9 – for example, 0.3% TFA in buffer). Under these conditions, the acidic peptides will become neutralized without affecting the binding of the phosphopeptide [78]. Furthermore, using a high concentration of ACN (50%) in the sample-loading buffer decreases the hydrophobic interaction of the nonphosphopeptides with IMAC. Kokubu et al. found that once nonphosphopeptides bound to IMAC, it was very difficult to separate them from phosphopeptides, thus making it important to load the sample under optimal conditions – that is, 0.3% TFA/50% ACN in water [78].
As an alternative to IMAC, resins containing metal oxides such as TiO2 were introduced [79-81]. To prevent the binding of acidic peptides to TiO2, samples are loaded under highly acidic conditions in the presence of 2,5-dihydroxybenzoic acid (DHB), phthalic acid or glycolic acid [80,81]. Phosphopeptides are then efficiently eluted at high pH (9.0 to 11.3) [80]. An advantage to TiO2 is compatibility with low pH solutions, detergents and salts in contrast to IMAC, which is adversely affected by various buffers, buffer components and detergents [82]. In a comparison study, Bodenmiller et al. compared phosphoramidate chemistry, IMAC and TiO2 to reproducibly and comprehensively isolate phosphopeptides from complex mixtures, and found the three methods to differ in their specificity of phosphopeptides isolated [83]. They conclude that while there was a partial overlap in phosphopeptides isolated, no one single method is sufficient in providing a comprehensive phosphoproteome. To increase the size of the phosphoproteome, Thingholm et al. have recently introduced sequential elution from IMAC (SIMAC) to sequentially separate monophosphopeptides from multiply phosphorylated peptides using small amounts of complex samples [84]. They used acidic conditions to elute primarily monophosphopeptides from IMAC and subsequent basic conditions to elute multiply phosphorylated peptides. Following IMAC, TiO2 removes the unphosphorylated peptides contaminating the monophosphorylated peptide fraction. Both mono- and multi-phosphorylated fractions are then analyzed separately using MS conditions that are optimal for each sample. In this study, they report greater than twice the number of phosphopeptides identified from 120 μg human mesenchymal stem cells using SIMIC versus an optimized TiO2 chromatographic method. Thus, in all of the studies described above, the addition of a second chromatography step substantially increases the total number of phosphopeptides discovered, leading several companies to recently develop IMAC columns that synchronously employ more than one type of material, such as PhosphoCatch™ sold by Promega (WI, USA), which comprises two metal resins in one spin column (titanium and zirconium).
MS analysis of phosphopeptides
Three final challenges exist that make sequencing of phosphopeptides by MS a nontrivial issue: the low ionization efficiency of phosphorylated peptides; the loss of the labile phosphate group; and the inadequate fragmentation pattern (reviewed elsewhere [85]). First, most mass spectrometers are set to detect positively charged ion species that occur at low pH (e.g., the N-terminus of peptides or the amino groups of histidine, lysine and arginine); however, in the positive ion mode, phosphorylated peptides have a suppressed response. Thus, in the analysis of a mixture of peptides, there would appear to be an enhancement in the signal of the unphosphorylated peptide compared with it is phosphorylated counterpart. Second, in collision-induced dissociations (CID) there is a neutral loss of phosphoric acid from threonine and serine residues due to the β-elmination of the phosphodiester bond [86]. The neutral loss of phosphoric acid occurs before fragmentation of the peptide backbone, thus resulting in the incomplete identification of the peptide. In the last decade, many techniques have been developed to address these challenges.
The classic approach to phosphopeptide analysis is the use of neutral loss scans [87]. In this MS technique, the ions of highest intensity in the MS1 scan are selected for fragmentation, just as in MS/MS sequencing experiments. Within these spectra, the software searches for neutral losses of 98.0, 49.0 or 32.7, the single-, double- and triple-charged states of phosphoric acid, respectively. If an ion selected for MS2 demonstrates this neutral loss, the resulting peptide is selected for MS3 before the device returns to perform MS2 on the next most intense ion. Recently, this approach has had a makeover with the introduction of multistage activation on some MS platforms. This technique combines the MS2 and MS3 spectra from the ion that experienced the neutral mass loss. The new combined spectrum contains more fragment information than the two individual spectra, and the peptide has a greater chance of being successfully sequenced by automated analysis software [88].
A novel approach to avoiding signal suppression was recently utilized by Old et al. in a study to identify phosphorylation events linked to B-Raf signaling [8]. This study used a combination of negative and positive scan modes to identify and sequence phosphopeptides. The ABI 4000 Q-trap used in this study was set to rapidly switch from negative scan neutral loss to positive scan fragmentation mode in order to identify phosphopeptides and to obtain sequence coverage compatible with MS/MS peptide interpretation software. Unfortunately, not all instruments are capable of rapidly reversing polarity due to differences in system architecture, and even this system required substantial optimization to negate artifacts caused by this switch.
New fragmentation techniques have been used to circumvent some of the inherent difficulties in obtaining fragmentation spectra of phosphopeptides. Electron capture dissociation (ECD) is one such technique. In this fragmentation, low-energy electrons are added to ions contained within a trap. This method results in less biased fragmentation than CID techniques and produces predominantly c and z ions [89]. While this technique has been shown to work well with fourier transformation-based devices, it has yet to be implemented with other devices. A similar technique, electron transfer dissociation (ETD), has been successfully implemented on a number of different platforms. ETD is a so-called ‘soft’ fragmentation technique that uses a chemical ionization source to fragment the backbone of multiply charged peptides. In 2007, Molina et al. used a combination of ETD and CID fragmentation techniques to identify over 1400 unique phosphorylation sites from human embryonic kidney 293T cells [86]. Interpretation of ETD fragmentation spectra was responsible for more than 60% of the total phosphopeptides identified. ETD is considerably more efficient at fragmentation of multiply charged ion species. Peptides produced by cleavage with trypsin typically are not large enough to hold the minimum number of charges essential for efficient ETD fragmentation. Groups using ETD as a primary fragmentation source typically use proteolytic enzymes, such as LysC and Glu-C, that result in relatively larger peptides [86].
Another fragmentation method showing promise in phosphoproteomics is higher-energy collisional dissociation (HCD) [90,91]. A study by Nagaraj et al. analyzed the feasibility of using this technique for global phosphoproteomics [92]. In this study, they used a Orbitrap Velos (Thermo Scientific) system to analyze a HeLa cell lysate processed by the FASP method described above, fractionated by SCX and enriched for phosphopeptides by TiO2 chromatography. The MS1 scans were performed in the Orbitrap at 30,000 MW resolution. The ten ions in each scan with the highest intensities were fragmented by HCD in the collision cell and the fragments were scanned by the Orbitrap at the much faster resolution setting of 7500 MW. Samples analyzed by this technique revealed a greater number of positive phosphopeptide identifications than the same samples analyzed by CID-based neutral loss scans [92]. However, it is worth noting that the sensitivity required to perform this analysis is unique to the Velos instrument, thanks to improvements in lens technology [93]. Although other groups have reported success with this technique on an Orbitrap XL [90,91], we were unable to replicate these results and have found the relatively reduced sensitivity of our Orbitrap detrimental to peptide analysis as compared with the same samples analyzed by CID (data not shown). Figure 2 is a comparison of fragmentation spectra of a phosphopeptide standard at the same concentration using the different fragmentation types available on an Orbitrap XL with ETD. A relatively new advance in some MS platforms involves addition of ‘decision tree’ criteria for deciding how a specific ion is to be fragmented. For example, the Orbitrap XL in our laboratory has a preprogrammed method that employs the ETD for fragmentation of peptides that are carrying more than three positive charges, while smaller peptides are fragmented by traditional CID. Decision tree-based techniques have been shown to produce higher quality data in less time than traditional MS/MS methods [94]. The development of more advanced decision algorithms on platforms capable of more than one complementary fragmentation method can only result in a higher percentage of peptide and phosphopeptide coverage.
Figure 2. Overlapping fragmentation information obtained by using the different fragmentation methods available on our Orbitrap XL plus electron transfer dissociation to fragment a synthetic phosphopeptide (Waters, MA, USA), with a m+H+ of 1368.6776.
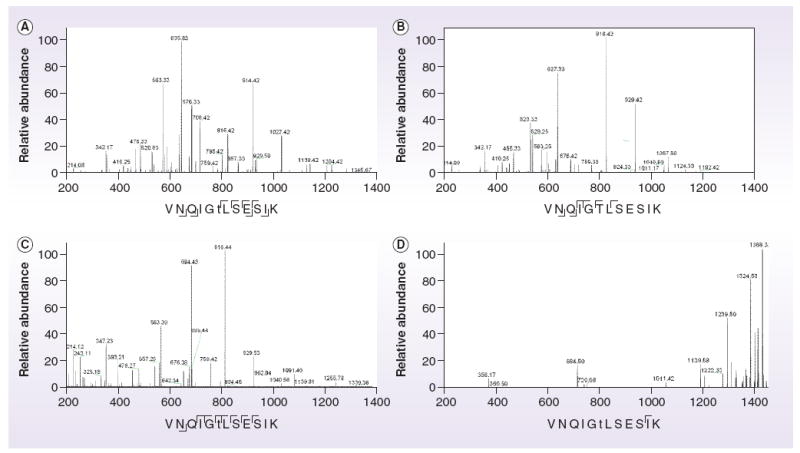
The phosphorylated threonine residue is represented by ‘t’. (A) Collision-induced dissociation of the intact phosphopeptide. (B) ‘Neutral loss’ MS3 performed on the doubly charged fragment at 635.83, representing the loss of phosphoric acid from the threonine residue, represented by ‘T’. (C & D) Higher-energy collisional dissociation and electron transfer dissociation fragmentation of the intact phosphopeptide, respectively.
Expert commentary
In this article, we describe the main difficulties confronting the field of PM proteomics, including protein abundance, insolubility and purification. In addition, the analysis of phosphopeptides is impeded by physical issues such as signal suppression and inadequate fragmentation spectra. Methodological advances continue to chip away at these obstacles and considerable progress has been made towards comprehensive monitoring of PM phosphorylation events. Table 2 highlights several noteworthy studies illustrating ‘state of the art’ technologies that have successfully uncovered large numbers of phosphorylation events. While the benefits of advances in MS fragmentation strategies cannot be denied, it is worth noting that even the most thorough study released to date relied exclusively on CID for peptide fragmentation [55]. As new fragmentation techniques, such as ETD and HCD, provide additional information in phosphopeptide structure, at the time of writing, no comprehensive phosphoproteomics study has shown these methods to be clearly superior to CID-based MS2 or neutral loss MS3. It is more likely that they will function as complementary methods. As demonstrated in Table 2, the key to obtaining a comprehensive picture of the membrane phosphoproteome appears to be the integration of multiple enrichment techniques that provide the greatest diversity of phosphopeptides.
Table 2.
A selection of recent phosphoproteomic studies utilizing novel technologies.
| Technology | Phosphorylation sites (n) | Ref. |
|---|---|---|
| Hydrophilic interaction chromatography | ~1000 | [72] |
| Multiprotease approach, combining electron transfer dissociation and collision-induced dissociation | 1141 | [86] |
| Multiprotease approach | >5000 | [38] |
| Sequential elution from immobilized metal ion chromatography | ~700 | [84] |
| Filter-assisted sample preparation, filter-based affinity capturing and elution, multiple enrichment techniques | ~12,000 | [55] |
Five-year view
Clearly, this is an exciting time for those wishing to undertake analysis of the PM phosphoproteome. The introduction of techniques described herein has logarithmically increased the number of potential phosphopeptides IDs derived from a single experiment. It is hoped that this ‘Moore’s Law’-like trend will continue to a point where tens of thousands of biologically relevant events can be detected from smaller quantities of starting material. Specifically, it is hoped that the standardization of phosphopeptide enrichment kits utilizing multiple IMAC resin materials synchronously will further improve phosphopeptide recovery and identification. In conjunction with this, we anticipate that digestion of protein samples with multiple proteases or chemical digestion techniques will become a standard approach for enhancing productivity. Likewise, integration and standardization of multiple fragmentation techniques such as CID and ETD within a single experiment is an emerging approach to maximize data generation. The greatest advances, however, will probably come from improvements in MS hardware, where the recent introduction of robust platforms such as the Orbitrap (Thermo Scientific) has demonstrated that phosphoproteomic analysis is an achievable goal for most laboratories.
Key issues.
Phosphoproteomic analysis of the plasma membrane requires considerable quantities of input material.
Aggressive methods are required to effectively solubilize highly hydrophobic integral membrane proteins.
Recent techniques using filter-aided digestions have resulted in higher yields in both membrane protein, as well as in membrane phosphopeptide identifications.
Proteolysis with trypsin can be complemented with digestions that result in larger peptides, and the use of multiple digestion techniques has been shown to result in increased phosphopeptide coverage.
The most successful approaches to membrane phosphoproteomics involve combinations of multiple enrichment techniques.
New fragmentation techniques, such as higher-energy collision dissociation and electron transfer dissociation, have shown some promise in aiding phosphopeptide analysis, but have yet to show significant advantages over collision-induced dissociation in a global analysis.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This research was supported (in part) by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute.
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
of interest
of considerable interest
- 1.Macek B, Mann M, Olsen JV. Global and site-specific quantitative phosphoproteomics: principles and applications. Annu Rev Pharmacol Toxicol. 2009;49:199–221. doi: 10.1146/annurev.pharmtox.011008.145606. [DOI] [PubMed] [Google Scholar]
- 2.Ding SJ, Qian WJ, Smith RD. Quantitative proteomic approaches for studying phosphotyrosine signaling. Expert Rev Proteomics. 2007;4(1):13–23. doi: 10.1586/14789450.4.1.13. [DOI] [PubMed] [Google Scholar]
- 3•.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. doi: 10.1038/35077225. Excellent review on phospho-state regulation and cancer. [DOI] [PubMed] [Google Scholar]
- 4.Thingholm TE, Jensen ON, Larsen MR. Analytical strategies for phosphoproteomics. Proteomics. 2009;9(6):1451–1468. doi: 10.1002/pmic.200800454. [DOI] [PubMed] [Google Scholar]
- 5.Beausoleil SA, Jedrychowski M, Schwartz D, et al. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA. 2004;101(33):12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brill LM, Salomon AR, Ficarro SB, Mukherji M, Stettler-Gill M, Peters EC. Robust phosphoproteomic profiling of tyrosine phosphorylation sites from human T cells using immobilized metal affinity chromatography and tandem mass spectrometry. Anal Chem. 2004;76(10):2763–2772. doi: 10.1021/ac035352d. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Gerber SA, Rudner AD, et al. Large-scale phosphorylation analysis of α-factor-arrested Saccharomyces cerevisiae. J Proteome Res. 2007;6(3):1190–1197. doi: 10.1021/pr060559j. [DOI] [PubMed] [Google Scholar]
- 8.Old WM, Shabb JB, Houel S, et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell. 2009;34(1):115–131. doi: 10.1016/j.molcel.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 10.Olsen JV, Vermeulen M, Santamaria A, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3(104):ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 11.Wu CC, Yates JR., 3rd The application of mass spectrometry to membrane proteomics. Nat Biotechnol. 2003;21(3):262–267. doi: 10.1038/nbt0303-262. [DOI] [PubMed] [Google Scholar]
- 12.Yeagle P. The Membranes of Cells. Academic Press; Buffalo, NY, USA: 1987. [Google Scholar]
- 13.Jain M. Introduction to Biological Membranes. John Wiley and Sons; NY, USA: 1988. [Google Scholar]
- 14.Navarre C, Degand H, Bennett KL, Crawford JS, Mortz E, Boutry M. Subproteomics: identification of plasma membrane proteins from the yeast Saccharomyces cerevisiae. Proteomics. 2002;2(12):1706–1714. doi: 10.1002/1615-9861(200212)2:12<1706::AID-PROT1706>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 15.Pasquali C, Fialka I, Huber LA. Subcellular fractionation, electromigration analysis and mapping of organelles. J Chromatogr B Biomed Sci Appl. 1999;722(1–2):89–102. doi: 10.1016/s0378-4347(98)00314-4. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Xie J, Wang X, et al. Proteomic analysis of mouse liver plasma membrane: use of differential extraction to enrich hydrophobic membrane proteins. Proteomics. 2005;5(17):4510–4524. doi: 10.1002/pmic.200401318. [DOI] [PubMed] [Google Scholar]
- 17.Wisniewski JR. Tools for phospho- and glycoproteomics of plasma membranes. Amino Acids. 2010 doi: 10.1007/s00726-010-0796-0798. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 18.Staubach S, Hanisch FG. Lipid rafts: signaling and sorting platforms of cells and their roles in cancer. Expert Rev Proteomics. 2011;8(2):263–277. doi: 10.1586/epr.11.2. [DOI] [PubMed] [Google Scholar]
- 19.Foster LJ, De Hoog CL, Mann M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc Natl Acad Sci USA. 2003;100(10):5813–5818. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olsen JV, Nielsen PA, Andersen JR, Mann M, Wisniewski JR. Quantitative proteomic profiling of membrane proteins from the mouse brain cortex, hippocampus, and cerebellum using the HysTag reagent: mapping of neurotransmitter receptors and ion channels. Brain Res. 2007;1134(1):95–106. doi: 10.1016/j.brainres.2006.11.082. [DOI] [PubMed] [Google Scholar]
- 21.Groen AJ, Lilley KS. Proteomics of total membranes and subcellular membranes. Expert Rev Proteomics. 2010;7(6):867–878. doi: 10.1586/epr.10.85. [DOI] [PubMed] [Google Scholar]
- 22.Hoang VM, Conrads TP, Veenstra TD, et al. Quantitative proteomics employing primary amine affinity tags. J Biomol Tech. 2003;14(3):216–223. [PMC free article] [PubMed] [Google Scholar]
- 23.Peirce MJ, Cope AP, Wait R. Proteomic analysis of the lymphocyte plasma membrane using cell surface biotinylation and solution-phase isoelectric focusing. Methods Mol Biol. 2009;528:135–140. doi: 10.1007/978-1-60327-310-7_10. [DOI] [PubMed] [Google Scholar]
- 24.Elschenbroich S, Kim Y, Medin JA, Kislinger T. Isolation of cell surface proteins for mass spectrometry-based proteomics. Expert Rev Proteomics. 2010;7(1):141–154. doi: 10.1586/epr.09.97. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh D, Krokhin O, Antonovici M, et al. Lectin affinity as an approach to the proteomic analysis of membrane glycoproteins. J Proteome Res. 2004;3(4):841–850. doi: 10.1021/pr049937f. [DOI] [PubMed] [Google Scholar]
- 26.Vercoutter-Edouart AS, Slomianny MC, Dekeyzer-Beseme O, Haeuw JF, Michalski JC. Glycoproteomics and glycomics investigation of membrane N-glycosylproteins from human colon carcinoma cells. Proteomics. 2008;8(16):3236–3256. doi: 10.1002/pmic.200800151. [DOI] [PubMed] [Google Scholar]
- 27.McDonald CA, Yang JY, Marathe V, Yen TY, Macher BA. Combining results from lectin affinity chromatography and glycocapture approaches substantially improves the coverage of the glycoproteome. Mol Cell Proteomics. 2009;8(2):287–301. doi: 10.1074/mcp.M800272-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everberg H, Leiding T, Schioth A, Tjerneld F, Gustavsson N. Efficient and non-denaturing membrane solubilization combined with enrichment of membrane protein complexes by detergent/polymer aqueous two-phase partitioning for proteome analysis. J Chromatogr. 2006;1122(1–2):35–46. doi: 10.1016/j.chroma.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 29.Jones MN. Surfactants in membrane solubilisation. Int J Pharmaceut. 1999;177(2):137–159. doi: 10.1016/s0378-5173(98)00345-7. [DOI] [PubMed] [Google Scholar]
- 30.Wisniewski JR, Zougman A, Mann M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J Proteome Res. 2009;8(12):5674–5678. doi: 10.1021/pr900748n. [DOI] [PubMed] [Google Scholar]
- 31••.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. Introduction and instruction for the filter-aided sample preparation methodology. [DOI] [PubMed] [Google Scholar]
- 32.Yu YQ, Gilar M, Gebler JC. A complete peptide mapping of membrane proteins: a novel surfactant aiding the enzymatic digestion of bacteriorhodopsin. Rapid Commun Mass Spectrom. 2004;18(6):711–715. doi: 10.1002/rcm.1374. [DOI] [PubMed] [Google Scholar]
- 33•.Li M, Powell MJ, Razunguzwa TT, O’Doherty GA. A general approach to anionic acid-labile surfactants with tunable properties. J Org Chem. 2010;75(18):6149–6153. doi: 10.1021/jo100954q. Valuable resource detailing several mass spectrometry-compatible detergents. [DOI] [PubMed] [Google Scholar]
- 34.Blonder J, Conrads TP, Yu LR, et al. A detergent- and cyanogen bromide-free method for integral membrane proteomics: application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics. 2004;4(1):31–45. doi: 10.1002/pmic.200300543. [DOI] [PubMed] [Google Scholar]
- 35.Macher BA, Yen TY. Proteins at membrane surfaces-a review of approaches. Mol Biosyst. 2007;3(10):705–713. doi: 10.1039/b708581h. [DOI] [PubMed] [Google Scholar]
- 36.D’Amici GM, Huber CG, Zolla L. Separation of thylakoid membrane proteins by sucrose gradient ultracentrifugation or blue native-SDS-PAGE two-dimensional electrophoresis. Methods Mol Biol. 2009;528:61–70. doi: 10.1007/978-1-60327-310-7_4. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen DN, Becker GW, Riggin RM. Protein mass spectrometry: applications to analytical biotechnology. J Chromatogr. 1995;705(1):21–45. doi: 10.1016/0021-9673(94)01256-e. [DOI] [PubMed] [Google Scholar]
- 38.Gauci S, Helbig AO, Slijper M, Krijgsveld J, Heck AJ, Mohammed S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal Chem. 2009;81(11):4493–4501. doi: 10.1021/ac9004309. [DOI] [PubMed] [Google Scholar]
- 39.van Montfort BA, Doeven MK, Canas B, Veenhoff LM, Poolman B, Robillard GT. Combined in-gel tryptic digestion and CNBr cleavage for the generation of peptide maps of an integral membrane protein with MALDI-TOF mass spectrometry. Biochim Biophys Acta. 2002;1555(1–3):111–115. doi: 10.1016/s0005-2728(02)00264-5. [DOI] [PubMed] [Google Scholar]
- 40.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 41.Pramanik BN, Mirza UA, Ing YH, et al. Microwave-enhanced enzyme reaction for protein mapping by mass spectrometry: a new approach to protein digestion in minutes. Protein Sci. 2002;11(11):2676–2687. doi: 10.1110/ps.0213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong H, Marcus SL, Li L. Microwave-assisted acid hydrolysis of proteins combined with liquid chromatography MALDI MS/MS for protein identification. J Am Soc Mass Spec. 2005;16(4):471–481. doi: 10.1016/j.jasms.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 43.Chen WY, Chen YC. Functional Fe3O4@ ZnO magnetic nanoparticle-assisted enrichment and enzymatic digestion of phosphoproteins from saliva. Anal Bioanal Chem. 2010;398(5):2049–2057. doi: 10.1007/s00216-010-4174-x. [DOI] [PubMed] [Google Scholar]
- 44.Hasan N, Wu HF, Li YH, Nawaz M. Two-step on-particle ionization/enrichment via a washing- and separation-free approach: multifunctional TiO2 nanoparticles as desalting, accelerating, and affinity probes for microwave-assisted tryptic digestion of phosphoproteins in ESI-MS and MALDI-MS: comparison with microscale TiO2. Anal Bioanal Chem. 2010;396(8):2909–2919. doi: 10.1007/s00216-010-3573-3. [DOI] [PubMed] [Google Scholar]
- 45.Rogers LD, Foster LJ. Phosphoproteomics – finally fulfilling the promise? Mol Biosyst. 2009;5(10):1122–1129. doi: 10.1039/b905580k. [DOI] [PubMed] [Google Scholar]
- 46.Demus H. Subcellular fractionation of human lymphocytes. Isolation of two plasma membrane fractions and comparison of the protein components of the various lymphocytic organelles. Biochim Biophys Acta. 1973;291(1):93–106. doi: 10.1016/0005-2736(73)90064-3. [DOI] [PubMed] [Google Scholar]
- 47.Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protocols. 2008;3(10):1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puente LG, Megeney LA. Isolation of phosphoproteins. Methods Mol Biol. 2008;424:365–372. doi: 10.1007/978-1-60327-064-9_28. [DOI] [PubMed] [Google Scholar]
- 49.Schumacher JA, Crockett DK, Elenitoba-Johnson KS, Lim MS. Evaluation of enrichment techniques for mass spectrometry: identification of tyrosine phosphoproteins in cancer cells. J Mol Diagn. 2007;9(2):169–177. doi: 10.2353/jmoldx.2007.060031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rush J, Moritz A, Lee KA, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23(1):94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- 51.Nagano K, Shinkawa T, Yabuki N, et al. Integration of proteomic analyses to monitor the activity status of phosphorylation signaling. J Proteomics. 2011;74(3):319–326. doi: 10.1016/j.jprot.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 52.Zheng H, Hu P, Quinn DF, Wang YK. Phosphotyrosine proteomic study of interferon alpha signaling pathway using a combination of immunoprecipitation and immobilized metal affinity chromatography. Mol Cell Proteomics. 2005;4(6):721–730. doi: 10.1074/mcp.M400077-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Cooper JA, Sefton BM, Hunter T. Detection and quantification of phosphotyrosine in proteins. Methods Enzymol. 1983;99:387–402. doi: 10.1016/0076-6879(83)99075-4. [DOI] [PubMed] [Google Scholar]
- 54.Gronborg M, Kristiansen TZ, Stensballe A, et al. A mass spectrometry-based proteomic approach for identification of serine/threonine-phosphorylated proteins by enrichment with phospho-specific antibodies: identification of a novel protein, Frigg, as a protein kinase A substrate. Mol Cell Proteomics. 2002;1(7):517–527. doi: 10.1074/mcp.m200010-mcp200. [DOI] [PubMed] [Google Scholar]
- 55••.Wisniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010;9(6):3280–3289. doi: 10.1021/pr1002214. Introduction of the filter-aided antibody capturing and elution method for phosphoproteomics. [DOI] [PubMed] [Google Scholar]
- 56.Ballif BA, Villen J, Beausoleil SA, Schwartz D, Gygi SP. Phosphoproteomic analysis of the developing mouse brain. Mol Cell Proteomics. 2004;3(11):1093–1101. doi: 10.1074/mcp.M400085-MCP200. [DOI] [PubMed] [Google Scholar]
- 57.Dephoure N, Zhou C, Villen J, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA. 2008;105(31):10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci USA. 2007;104(5):1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morandell S, Stasyk T, Grosstessner-Hain K, et al. Phosphoproteomics strategies for the functional analysis of signal transduction. Proteomics. 2006;6(14):4047–4056. doi: 10.1002/pmic.200600058. [DOI] [PubMed] [Google Scholar]
- 60.Nuhse TS, Peck SC. Peptide-based phosphoproteomics with immobilized metal ion chromatography. Methods Mol Biol. 2006;323:431–436. doi: 10.1385/1-59745-003-0:431. [DOI] [PubMed] [Google Scholar]
- 61.Nuhse TS, Bottrill AR, Jones AM, Peck SC. Quantitative phosphoproteomic analysis of plasma membrane proteins reveals regulatory mechanisms of plant innate immune responses. Plant J. 2007;51(5):931–940. doi: 10.1111/j.1365-313X.2007.03192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nuhse TS, Stensballe A, Jensen ON, Peck SC. Phosphoproteomics of the Arabidopsis plasma membrane and a new phosphorylation site database. Plant Cell. 2004;16(9):2394–2405. doi: 10.1105/tpc.104.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chenau J, Michelland S, Sidibe J, Seve M. Peptides OFFGEL electrophoresis: a suitable pre-analytical step for complex eukaryotic samples fractionation compatible with quantitative iTRAQ labeling. Proteome Sci. 2008;6:9. doi: 10.1186/1477-5956-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horth P, Miller CA, Preckel T, Wenz C. Efficient fractionation and improved protein identification by peptide OFFGEL electrophoresis. Mol Cell Proteomics. 2006;5(10):1968–1974. doi: 10.1074/mcp.T600037-MCP200. [DOI] [PubMed] [Google Scholar]
- 65.Gauci S, van Breukelen B, Lemeer SM, Krijgsveld J, Heck AJ. A versatile peptide pI calculator for phosphorylated and N-terminal acetylated peptides experimentally tested using peptide isoelectric focusing. Proteomics. 2008;8(23–24):4898–4906. doi: 10.1002/pmic.200800295. [DOI] [PubMed] [Google Scholar]
- 66•.Maccarrone G, Kolb N, Teplytska L, et al. Phosphopeptide enrichment by IEF. Electrophoresis. 2006;27(22):4585–4595. doi: 10.1002/elps.200600145. First of several studies employing isoelectric focusing for phosphopeptide separation and enrichment. [DOI] [PubMed] [Google Scholar]
- 67.Xu CF, Wang H, Li D, Kong XP, Neubert TA. Selective enrichment and fractionation of phosphopeptides from peptide mixtures by isoelectric focusing after methyl esterification. Anal Chem. 2007;79(5):2007–2014. doi: 10.1021/ac061606u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hung CW, Kubler D, Lehmann WD. pI-based phosphopeptide enrichment combined with nanoESI-MS. Electrophoresis. 2007;28(12):2044–2052. doi: 10.1002/elps.200600678. [DOI] [PubMed] [Google Scholar]
- 69.Xu Y, Sprung R, Kwon SW, Kim SC, Zhao Y. Isolation of phosphopeptides by pI-difference-based electrophoresis. J Proteome Res. 2007;6(3):1153–1157. doi: 10.1021/pr060498p. [DOI] [PubMed] [Google Scholar]
- 70.Zuo X, Lee K-B, Speicher DW. Fractionation of Complex Proteomes by Microscale Solution Isoelectrofocusing Using ZOOM– IEF Fractionators to Improve Protein Profiling. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; NJ, USA: 2005. pp. 97–117. [Google Scholar]
- 71.McNulty DE, Annan RS. Hydrophilic interaction chromatography for fractionation and enrichment of the phosphoproteome. Methods Mol Biol. 2009;527:93–105. doi: 10.1007/978-1-60327-834-8_8. [DOI] [PubMed] [Google Scholar]
- 72.McNulty DE, Annan RS. Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol Cell Proteomics. 2008;7(5):971–980. doi: 10.1074/mcp.M700543-MCP200. [DOI] [PubMed] [Google Scholar]
- 73.Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics. 2008;7(7):1389–1396. doi: 10.1074/mcp.M700468-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ficarro SB, McCleland ML, Stukenberg PT, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20(3):301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 75.Ficarro SB, Salomon AR, Brill LM, et al. Automated immobilized metal affinity chromatography/nano-liquid chromatography/electrospray ionization mass spectrometry platform for profiling protein phosphorylation sites. Rapid Commun Mass Spectrom. 2005;19(1):57–71. doi: 10.1002/rcm.1746. [DOI] [PubMed] [Google Scholar]
- 76.Collins MO, Yu L, Coba MP, et al. Proteomic analysis of in vivo phosphorylated synaptic proteins. J Biol Chem. 2005;280(7):5972–5982. doi: 10.1074/jbc.M411220200. [DOI] [PubMed] [Google Scholar]
- 77.Porath J. Immobilized metal ion affinity chromatography. Protein Exp Purification. 1992;3(4):263–281. doi: 10.1016/1046-5928(92)90001-d. [DOI] [PubMed] [Google Scholar]
- 78.Kokubu M, Ishihama Y, Sato T, Nagasu T, Oda Y. Specificity of immobilized metal affinity-based IMAC/C18 tip enrichment of phosphopeptides for protein phosphorylation analysis. Anal Chem. 2005;77(16):5144–5154. doi: 10.1021/ac050404f. [DOI] [PubMed] [Google Scholar]
- 79.Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC–ESI-MS/MS and titanium oxide precolumns. Anal Chem. 2004;76(14):3935–3943. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]
- 80.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics. 2005;4(7):873–886. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
- 81.Sano A, Nakamura H. Evaluation of titanium and titanium oxides as chemo-affinity sorbents for the selective enrichment of organic phosphates. Anal Sci. 2007;23(11):1285–1289. doi: 10.2116/analsci.23.1285. [DOI] [PubMed] [Google Scholar]
- 82.Jensen SS, Larsen MR. Evaluation of the impact of some experimental procedures on different phosphopeptide enrichment techniques. Rapid Commun Mass Spectrom. 2007;21(22):3635–3645. doi: 10.1002/rcm.3254. [DOI] [PubMed] [Google Scholar]
- 83.Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods. 2007;4(3):231–237. doi: 10.1038/nmeth1005. [DOI] [PubMed] [Google Scholar]
- 84.Thingholm TE, Jensen ON, Robinson PJ, Larsen MR. SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol Cell Proteomics. 2008;7(4):661–671. doi: 10.1074/mcp.M700362-MCP200. [DOI] [PubMed] [Google Scholar]
- 85.Gafken PR, Lampe PD. Methodologies for characterizing phosphoproteins by mass spectrometry. Cell Comm Adhesion. 2006;13(5–6):249–262. doi: 10.1080/15419060601077917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86•.Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci USA. 2007;104(7):2199–2204. doi: 10.1073/pnas.0611217104. Details the use of electron transfer dissociation fragmentation of phosphopeptides in a global context. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal Chem. 2004;76(13):3590–3598. doi: 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- 88.Andreas F, Hühmer ZH, Sadygov R, Biringer RG, Schwartz JC. Analysis of phosphopeptides by linear ion trap mass spectrometry using multistage activation. Thermo Electron Corporation Application Note 30093 [Google Scholar]
- 89.Zubarev RA, Horn DM, Fridriksson EK, et al. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 2000;72(3):563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 90.Wu J, Warren P, Shakey Q, et al. Integrating titania enrichment, iTRAQ labeling, and Orbitrap CID-HCD for global identification and quantitative analysis of phosphopeptides. Proteomics. 2010;10(11):2224–2234. doi: 10.1002/pmic.200900788. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Y, Ficarro SB, Li S, Marto JA. Optimized Orbitrap HCD for quantitative analysis of phosphopeptides. J Am Soc Mass Spec. 2009;20(8):1425–1434. doi: 10.1016/j.jasms.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 92.Nagaraj N, D’Souza RC, Cox J, Olsen JV, Mann M. Feasibility of large scale phosphoproteomics with HCD fragmentation. J Proteome Res. 2010;9(12):6786–6794. doi: 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- 93.Olsen JV, Schwartz JC, Griep-Raming J, et al. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteomics. 2009;8(12):2759–2769. doi: 10.1074/mcp.M900375-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94•.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods. 2008;5(11):959–964. doi: 10.1038/nmeth.1260. Introduction of intelligent, decision-tree-based proteomics on an Orbitrap system. [DOI] [PMC free article] [PubMed] [Google Scholar]