

Abstract
To study the relationship between homologous recombination and DNA replication in Escherichia coli, we monitored the behavior of phage λ chromosomes, repressed or not for λ gene activities. Recombination in our system is stimulated both by DNA replication and by experimentally introduced double-strand ends, supporting the idea that DNA replication generates occasional double-strand ends. We report that the RecBC recombinational pathway of E. coli uses double-strand ends to prime DNA synthesis, implying a circular relationship between DNA replication and recombination and suggesting that the primary role of recombination is in the repair of disintegrated replication forks arising during vegetative reproduction.
Keywords: DSB repair, homologous recombination, DNA replication, recA, recD, recBC
Homologous recombination was first seen as the crossing-over of chromosomes in cells undergoing meiosis. The first scheme of crossing-over (Janssens 1909) was of the break–join type, according to which homologous chromosomes would break at the same position and swap arms (Fig. 1). The copy-choice scheme (Belling 1931a, b) was the first to link homologous recombination with chromosomal replication (Fig. 1): Parental chromosomes served as templates for the synthesis of daughter chromosomes, which could exchange templates at their growing points. A hybrid scheme, dubbed break–copy (Lederberg 1955) envisioned a broken piece of a chromosome to copy its missing part from an intact homologous chromosome (Fig. 1). Thus, in the earliest schemes, there was already a variety of proposals on the relationship between replication and recombination. The copy–choice scheme viewed DNA replication as a prerequisite for recombination, the break–copy scheme made recombination a stimulator of DNA replication, whereas the break–join scheme ignored DNA replication altogether.
Figure 1.

Ancient ideas about chromosomal recombination and crossing-over in meiosis. Pre-existing chromosomes (DNA duplexes) are shown as either solid or open single lines; newly synthesized chromosomes are shown as broken single lines. Explanations are in the text.
With the demonstration of recombination of linked markers (Hershey and Rotman 1949), bacteriophages emerged as attractive tools for addressing the relationships between replication and recombination (Delbrück and Stent 1957), but the answer was elusive. The study of T4, the early favorite for recombination experiments, was hampered by the circularity of its map and by the dependence of the phage on its recombination function for completion of its life cycle (for review, see Mosig 1998). λ soon emerged as a promising system, following the finding that it has homologous recombination (Jacob and Wollman 1954; Kaiser 1955) and that its growth is only partly dependent on recombination (Manly et al. 1969; Enquist and Skalka 1973). The experiments of Meselson and Weigle showed the presence of parental DNA in λ recombinants, ruling out copy–choice as the only recombinational mechanism (Meselson and Weigle 1961). In subsequent experiments, Meselson demonstrated recombinant chromosomes that contained little or no newly synthesized DNA, ruling out break–copy as the only mechanism of recombination (Meselson 1964). However, as genetic requirements of λ’s recombination were revealed, it became clear that the break–join events observed by Meselson might be attributable to the phage’s site-specific recombination system (Kellenberger-Gujer and Weisberg 1971), which is of the break–join type.
Using mutants lacking λ’s own recombination enzymes, Stahl and his colleagues investigated the influence of DNA synthesis on λ recombination effected by Escherichia coli’s system. They used phage chromosomes as recombinational substrates, allowed the phage life cycle to go to completion, and analyzed the packaged recombinant chromosomes genetically. The evidence obtained suggested that E. coli recombination is of the break–join type, because the E. coli pathway produced recombinants throughout λ’s chromosome with little or no DNA synthesis (Stahl et al. 1973, 1974).
When the principal exonuclease of the E. coli cells was inactivated by recD mutations, E. coli-mediated recombination in the center of λ’s chromosome became dependent on DNA synthesis (Thaler et al. 1989). However, that dependence could not be taken as evidence of copy–choice or break–copy recombination because of the possibility that the role of DNA replication was to create recombinagenic breaks in the middle of the λ chromosome (Skalka 1974; Wilkins and Mistry 1974; Stahl et al. 1985). Thus, the experiments with λ demonstrated that E. coli recombination can proceed by break–join, but they did not rule out the possibilities of break–copy or copy–choice, if DNA replication is permitted.
Smith noted that, if recombination is initiated by a double-strand end, break–join recombination becomes an endless game, with every recombination act generating a new end ready to begin another round of recombination (Smith 1991). Smith solved the paradox by allowing the recombination intermediates in E. coli to be resolved by DNA replication, in a break–copy manner, although no direct evidence for such a possibility was available at that time.
Kogoma and associates suggested that the mechanistic basis for inducible stable DNA replication, the phenomenon observed after DNA-damaging treatments, is DNA synthesis primed by recombinational repair of double-strand breaks (Magee et al. 1992; Asai et al. 1993). Using plasmids carrying λ cos in combination with a plasmid supplying terminase (the λ enzyme that makes a double-strand break at cos), they attempted a direct demonstration that recombinational repair of double-strand breaks in E. coli primes DNA synthesis (Asai et al. 1994). However, closer examination of their experimental protocol reveals that terminase was unlikely to have been produced under the following conditions: (1) Their expression plasmid proved not to be demonstrably active for terminase production (A. Kuzminov and F.W. Stahl, in prep.); (2) even if the plasmid were active, the addition of high concentrations of both transcription and translation inhibitors before the experiment (Asai et al. 1994) made any expression during the experiment unlikely. Because no evidence for terminase production was presented by the authors (Asai et al. 1994), the DNA synthesis they report cannot be attributed with confidence to double-strand break repair.
In summation, the evidence is strong that recombination in E. coli can occur in the break–join manner, and yet it is appealing to think that it proceeds by break–copy when DNA replication is allowed. Genetic experiments using the full life cycle of the phage λ were unable to clarify the issue. The major shortcoming of these experiments was that they allowed λ packaging, which begins with a double-strand break at cos. This double-strand break not only makes phage DNA ready for encapsidation, but also induces homologous recombination in its vicinity (Stahl et al. 1985; Thaler et al. 1989). When DNA synthesis is inhibited, packaging creates an even bigger complication, as chromosomes dimerized by recombination are packaged preferentially. Another confounding aspect of λ’s life cycle is that its DNA replication seems to induce double-strand breaks throughout the chromosome, stimulating recombination (see above).
And yet, λ is the ultimate delivery vehicle, offering synchronized injection of any number of phage chromosomes per cell. To circumvent the problems inherent in λ’s life cycle, one can analyze phage DNA directly, purifying it from infected cells (Wilkins and Mistry 1974; Poteete and Fenton 1993; Stahl et al. 1997) rather than allowing it to be packaged and then analyzing it genetically. We used this approach and, in addition, also repressed λ’s own expression, using λ’s chromosome as an in vivo DNA substrate for the E. coli recombination system. Here we report that, in this bacterium, recombination between an intact DNA and a double-strand end primes DNA synthesis.
Results
The substrates to monitor bacterial recombination in vivo
Kinetic studies of homologous recombination require synchronous introduction of adequate amounts of substrate into cells. We used the efficient adsorption and controlled injection of phage λ to reach this end. We simultaneously injected two kinds of λ chromosomes containing nonoverlapping deletions (Fig. 2A), purified total DNA from the infected E. coli cells, and used blot hybridization to follow the fate of the injected chromosomes as well as the formation of recombinants (Fig. 2B).
Figure 2.
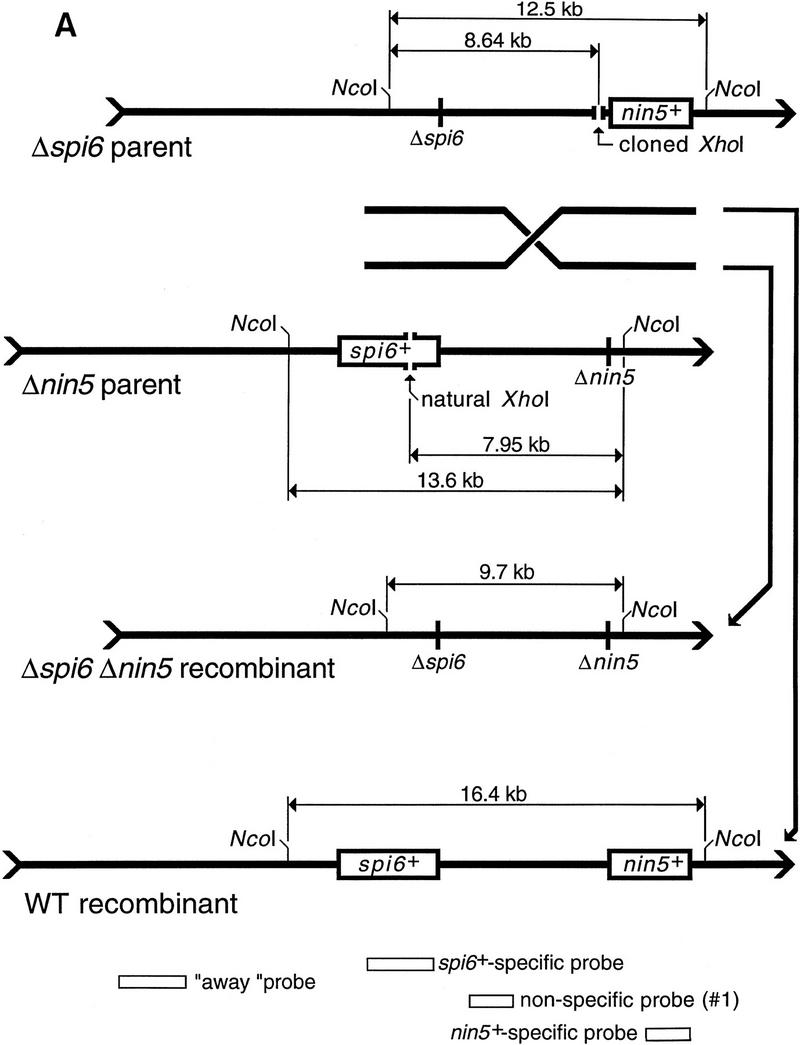

The two principal deletion substrates used in this study and the two complementary recombinant products. (A) A schematic diagram (not to scale) of the two principal substrate DNAs, the two complementary recombinants, and the probes used in this study (for probe specifications, see Materials and Methods). The spi6 and nin5 regions are boxed in the DNAs that retain them; boxes are absent in the DNAs having the corresponding deletions. Also shown are the unique XhoI restriction sites for in vivo cutting and the NcoI restriction sites used in the subsequent in vitro analysis. The sizes of the fragments generated as a result of in vivo and subsequent in vitro cutting are indicated. (B) A blot with two freely replicating parental λs, cut with NcoI and rehybridized to three region-specific probes. Nonspecific probe (1) hybridizes to the region between deletions detecting both Δspi6 and Δnin5 phages. nin5+-specific probe identifies only Δspi6 phages and the wild-type recombinant; spi6+-specific probe identifies only Δnin5 phages and the wild-type recombinant. A weak cross-hybridization in lanes j, n, and o is due to overloading, which causes a small fraction of Δspi6 DNA to migrate with the Δnin5 band and vice versa. Explanations are shown only for the window at left. (M1) MW markers for the parental phages; (M2) MW markers for the recombinant phages.
Soon after injection, λ’s chromosomes circularize. When λ’s expression and host recombination were both blocked, the injected chromosomes gradually disappeared (see below). When not repressed, the phage replicated freely, resulting in an increase in the two parental DNAs accompanied by downward smearing (Fig. 2B, lanes c–e). The two faint bands with mobilities higher and lower than that of the two parental bands, appearing later in infection, are the wild type and the double-deletion recombinants, respectively (Fig. 2B, lane e). These bands did not arise when the parental phages were injected separately (see below). Probes specific for deleted regions detect either one or the other parent as well as the wild-type recombinant (Fig. 2B, lanes h–j and m–o); the double-deleted recombinant is invisible with both of these deletion-specific probes (Fig. 2B, lanes j and o vs. lane e).
To model double-strand end repair with such substrates, we have cut them in vivo at unique XhoI restriction sites and then followed the fate of one of the two resulting ends. In either phage, the unique restriction site was positioned roughly opposite the deletion in the other phage (Figs. 2A and 3A). If a spontaneous double-strand break occurs in the interval between the two deletions, its recombinational repair should yield both wild-type and double-deletion recombinants. However, if double-strand breaks are delivered at the unique restriction sites, only the double-deletion recombinant is expected, as the ends without homology in the interval between the deletions would have to invade outside, forming recombinants indistinguishable from one or the other parent (Fig. 3A).
Figure 3.


The influence of various combinations of cutting and repression on phage replication and recombinant yield. (A) Cutting at the unique restriction sites precludes formation of the wild-type recombinant. As in Fig. 2A, spi6 and nin5 regions are shown as open rectangles when they are present in the phages, whereas the short vertical lines mark the corresponding deletions. Chromosomes with double-strand breaks at the unique XhoI sites are marked cut, whereas unbroken chromosomes aremarked uncut. Exchanges (left) are indicated by diagonal lines leading to particular outcomes (right). (Top cross) Neither parent is cut, so both the wild-type (WT) and the double-deletion recombinants are possible. (Second from top cross) Both parents are cut, so only the double-deletion recombinant is possible; (two bottom crosses) one of the parents is cut, so two recombinants are formed, the double-deletion one and a recombinant which is not different from one of the parents. (B) The two parental phages were injected into either Rec+ or ΔrecA host cells containing various combinations of cutting and repression functions. Total DNA was extracted at the indicated postinjection times, cut with NcoI, and analyzed by blot hybridization with probe 1. (Top) Crosses in Rec+ cells; (bottom) crosses in ΔrecA mutant cells. Rec+ strain is WA800, ΔrecA mutant is AK24. Phages are MMS2660 (Δnin5) and MMS2663 (Δspi6). Plasmids supplying the XhoI cutting and cI repression functions are pPaoRM3.8 (XhoI), pK116 (cI), and pK107 (XhoI + cI); when no function is supplied, cells are plasmid-free.
To avoid excessive DNA degradation, we inactivated ExoV, the most powerful exonuclease of E. coli, with recD mutations. recD mutant cells do not degrade linear DNA as fast as recD+ cells do but are fully recombination proficient (Chaudhury and Smith 1984; Amundsen et al. 1986); we refer to recD cells as Rec+ cells. Homologous recombination in E. coli requires recA gene function (for review, see Roca and Cox 1997); recA recD mutants are deficient in all reactions involving strand exchange (Chaudhury and Smith 1984; Lovett et al. 1988), therefore, we used recA mutations when we wished to inactivate the host recombination machinery. The cI gene, coding for the repressor of phage λ, and a XhoI-specific restriction-modification system were provided on plasmids. Whenever phage expression was allowed, phages not only replicated freely but were also assumed to suppress the host RecBC recombinational pathway, substituting their own Red pathway in its place. Conversely, when phage expression was disallowed in the presence of cI repressor, phages could not replicate and had to use the host RecBC pathway to recombine.
Double-strand ends stimulate recombination
In the first experimental set, we compared the effects of phage expression and restriction cutting on recombination in Rec+ cells. When phages were neither repressed nor cut, they replicated freely, producing a small fraction of complementary recombinants (Fig. 3B, Rec+, lanes a–c). When phages were cut but not repressed, replication was limited, and, as explained in Figure 3A, only the double-deletion recombinant formed (Fig. 3B, Rec+, lanes g–i). In reciprocal conditions, when phages were repressed but not cut, limited replication still occurred, with formation of both complementary recombinants (Fig. 3B, Rec+, lanes j–l), perhaps resulting from random DNA breakage in the interval between the deletions. The apparent leakiness of phage repression was surprising, because the copy number of the plasmid-born repressor gene should have been some 50-fold higher than when the repressor resided in its usual place on the chromosome. We offer an explanation for this DNA replication later. Finally, when phages were both repressed and cut, there was little increase of parental DNA, and yet the double-deletion recombinant was produced in substantial amounts (Fig. 3B, Rec+, lanes m–o). These four crosses demonstrated that recombination in our system is stimulated either by DNA replication or by restriction-induced double-strand ends, as in other systems (Stahl et al. 1985; Thaler et al. 1987a 1989), supporting the idea that the mechanism by which DNA replication stimulates recombination is by generating double-strand ends (Skalka 1974; Wilkins and Mistry 1974; Stahl et al. 1985; Thaler et al. 1987b).
We then conducted crosses in recA mutant cells, deficient in host homologous recombination. When phages were neither repressed nor cut, the absence of RecA made little difference—phages were freely replicating, whereas the same low quantities of both recombinants were formed (Fig. 3B, ΔrecA, lanes a–c). This was expected, because neither phage λ replication nor its recombination in freely replicating crosses are dependent on the general recombination functions of the host (Takano 1966; van de Putte et al. 1966; Brooks and Clark 1967). When phages were cut, but not repressed, some DNA replication still occurred, but the amount of the double-deletion recombinant was lower than it was in recA+ cells (Fig. 3B, ΔrecA, lanes g–i). This was consistent with the earlier finding that, when both parental phages were cut at nonallelic locations, the Red system alone could catalyze a low level of recombination between them (Stahl et al. 1997). The last configuration, in which phages were both repressed and cut, yielded neither replication nor recombinant formation (Fig. 3B, ΔrecA, lanes m–o), which is explained by the absolute dependence of the host recombination system on RecA. The unexpected result came with the configuration in which phages were repressed but not cut. In contrast to RecA+ cells, there was no DNA replication and no recombinant formation (Fig. 3B, ΔrecA, lanes j–l). At face value, this implied two things: (1) cI repression was in fact complete; (2) phage DNA replication in Rec+ cells was due to homologous recombination.
Recombination stimulates DNA replication
Thus, we obtained conflicting results when phages were repressed and had to recombine using the E. coli recombination pathways. When phages were uncut, both their recombination and replication were RecA dependent (Fig. 3B, lanes j–l, cf. Rec+ to ΔrecA), suggesting a cause–effect relationship (break–copy model). On the other hand, the robust recombination without much DNA replication when phages were cut in the RecA+ cells (Fig. 3B, Rec+, lanes m–o) showed that host recombination does not need to entail extensive DNA replication (break–join model). However, when both phages are cut, the resulting recombination may not allow much DNA synthesis simply because cutting would limit its extent. From this point on, we did only the repressed crosses, to concentrate on E. coli’s recombination.
To distinguish between the break–copy and the break–join models of recombination, we cut one of the parents to stimulate recombination in the interval while leaving the other parent intact to provide the opportunity for DNA synthesis, should it be primed by recombination. To this end, two additional phages were used; they also carried either one or the other deletion, but lacked the restriction sites. Thus, for each deletion we had either cuttable or uncuttable phages, the final matrix comprising four crosses. The cut-by-cut and uncut-by-uncut configurations (Fig. 4A, lanes d–f and m–o) were similar to the previous crosses and served as controls for the two new configurations, in which one of the parents was cut, whereas the other was intact (Fig. 4A, lanes g–i and j–l).
Figure 4.


RecA-catalyzed double-strand end repair primes DNA synthesis. (A) The four parental phages in four pairwise combinations were injected into either Rec+ or ΔrecA host cells harboring pK107 (cutting + repression). The total DNA was extracted at the indicated postinjection times, cut with NcoI, and analyzed by blot hybridization with probe 1. (Top) Crosses in Rec+ cells; (Bottom) crosses in ΔrecA mutant cells. Rec+ strain is AK3, ΔrecA mutant is AK24. Phages are MMS2660 (Δnin5 cut), MMS2661 (Δnin5 uncut), MMS2662 (Δspi6 uncut) and MMS2663 (Δspi6 cut). Judging by 10-min time points, in vivo cutting at XhoI sites seems to be inefficient, as if the result of a rapid methylation of the injected DNA. However, at least 90% of the cuttable, yet uncut, parental DNA is likely to come from the adsorbed phages that failed to inject, as revealed by cutting the 10-min samples with XhoI in vitro. (B) The scheme of XhoI-stimulated formation of double-deletion recombinant with the simultaneous replication of the uncut parent. The phage duplex chromosomes are shown as single lines; boxes designate the presence of spi6 and nin5 DNA regions; short vertical lines mark the position of the corresponding deletions; cos sites are shown as chevrons. (step 1) Two parental phages aligned by the regions between the two deletions; (step 2) Δspi6 phage opened at the unique XhoI site; (step 3) RecA-catalyzed end invasion created a replication fork between the cut Δspi6 phage and the uncut Δnin5 phage; (step 4) replication fork copying the uncut Δnin5 phage. Note the formation of double-deletion DNA at the site of recombination.
First, we conducted the four crosses in Rec+ cells. The control crosses behaved as before. When only one of the parents was cut, however, recombinant formation was similar to that seen when both parents were cut, but DNA of the uncut parent was increasing independently of the configuration of the cuts and deletions (Fig. 4A, Rec+, lanes g–l). This was the result expected if recombination between a linear and a circular DNA initiates replication of the circular participant (Fig. 4B). However, other possible explanations must also be considered.
It seemed possible that cutting phages relieved repression, allowing replication of any uncut DNA. We addressed this possibility by repeating the same four crosses in a recA mutant strain. We saw that inactivation of RecA not only precluded all the recombination but also blocked DNA replication of the uncut parents (Fig. 4A, ΔrecA), arguing against the suspected derepression and supporting the view that RecA-catalyzed invasion of a double-strand end into an intact parent primed DNA replication (Fig. 4B).
However, this experiment failed to rule out the more specific possibility that the restriction cut activated RecA coprotease activity, which then degraded λ repressor (Roberts and Roberts 1975; Craig and Roberts 1980). To challenge this possibility, we did the cut-by-uncut crosses in the presence of plasmids expressing the uncleavable cI repressor (Roberts and Roberts 1975; see Materials and Methods for plasmid specifications). The results were not different from those obtained with wild-type cI repressor (not shown), allowing us to conclude that RecA-dependent inactivation of the phage repressor was not the reason for the replication of the uncut parent.
To make sure that the double-deletion recombinant was not a pre-existing low-level contaminant of the parental phage stocks, gaining replicative advantage because of the absence of XhoI sites, we monitored single infections of the four parental phages (Fig. 5). Monoparental infections yielded no additional bands, attesting to the purity of the parental phage stocks (Fig. 5, lanes b,d,f,h). In the same cells, the biparental cut-by-uncut crosses reproduced the previous result, that is, the formation of the double-deletion recombinant and replication of the uncut parent (Fig. 5, lanes i–l). Some DNA replication was also apparent in the monoparental infections (Fig. 5, cf. the 60 min lanes with the 10 min lanes), apparently the consequence of cryptic recombination between genetically identical molecules, as this DNA increase was not observed in recA mutant cells (not shown).
Figure 5.

Monoparental infections do not yield recombinant bands. The four parental phages were injected alone or in two cut-by-uncut combinations (controls) into Rec+ cells harboring pK107 (cutting + repression). The total DNA was extracted at the indicated postinjection times, cut with NcoI, and analyzed by blot hybridization with probe 1. The host strain is WA800; phages are MMS2660 (Δnin5 cuttable), MMS2661 (Δnin5 uncuttable), MMS2662 (Δspi6 uncuttable) and MMS2663 (Δspi6 cuttable).
Recombination and replication are not region, phage, or plasmid specific
The observed increase of uncut repressed parents (Fig. 4A) could be due to local DNA synthesis rather than replication of the entire chromosome. To see what happens to other regions of the parental chromosomes, we probed for a DNA segment located >10 kb upstream of the invading Δspi6 end (25-kb downstream around the circular chromosome; Fig. 2A). We found that this DNA segment undergoes a similar increase (not shown), confirming that what we observe is replication of the entire chromosome.
One of the parental phages was deleted for the phage recombination functions, but the other was wild type for them. Although phage genes were repressed, low level expression of phage recombination functions might have contributed to the observed phenomena. To verify that we were dealing with E. coli’s recombination system only, we repeated the experiment with both parents mutant for the phage recombination system (Red− Gam−) and obtained the same results (not shown).
The plasmid harboring the cI repressor carried a 1.2-kb homology to the recombinational interval, offering an opportunity for recombination with the phage. If the plasmid recombined into the phage chromosome, it would have driven phage replication. This possibility was unlikely, as such a cointegrate would have had a distinct mobility in our gels, whereas we observed no increasing species other than the uncut parent and recombinants, even 2 hr after the injection. However, to control against such a possibility, we constructed recD mutant strains with the genes coding for the restrictase and the phage repressor residing in the host chromosome instead of in a plasmid. These strains gave essentially the same results (A. Kuzminov and F.W. Stahl, unpubl.).
To rule out the possibility that the cI region on the plasmid influenced recombination between phages by interacting homologously with the cut phage, we used a pair of substrate phages with XhoI sites opposite a different pair of deletions delineating an interval away from cI (Fig. 6A). This configuration yielded the same pattern of DNA replication dependence on recombination—in Rec+ cells, the double-deletion recombinant was formed and the uncut parent was replicating (Fig. 6B, lanes a–f), whereas in recA mutant cells, there was no recombinant formation and the parental DNA slowly disappeared (Fig. 6B, lanes j–o).
Figure 6.


Double-strand end repair far from the cI-containing interval primes DNA synthesis. (A) A schematic diagram (not to scale) of the two alternative substrate DNAs, the two complementary recombinants, and the probe (2) used in this alternative setup. The b519 and spi6 regions are boxed in the DNAs that retain them; boxes are absent in the DNAs having the corresponding deletions; the nin5 region, present throughout, serves as the position reference. Also shown are the unique XhoI restriction sites for in vivo cutting and the EagI and BsiWI restriction sites used in the subsequent in vitro analysis. The sizes of the fragments generated as a result of in vivo and subsequent in vitro cutting are indicated. (B) The three parental phages in two pairwise combinations were injected into either Rec+ or ΔrecA host cells harboring pK107 (cutting + repression). The total DNA was extracted at the indicated postinjection times, cut with BsiWI + EagI and analyzed by blot hybridization with probe 2. Lanes a-f are from crosses in Rec+ cells; lanes j-o are from crosses in ΔrecA mutant cells. Rec+ strain is AK3, ΔrecA mutant is AK24. Phages are IS4 (Δb519 cut), λAK1 (Δspi6 cut) and MMS2662 (Δspi6 uncut).
Genetic requirements of the recombinant formation
As already demonstrated (Figs. 3, 4, and 6), one genetic requirement of the recombinant formation and associated DNA replication is RecA protein, which catalyzes strand invasion. Another genetic requirement is inactivation of ExoV nuclease, the enzyme that attacks and destroys linear DNA in E. coli (Telander-Muskavitch and Linn 1981; Taylor 1988). ExoV is embodied in a heterotrimeric RecBCD enzyme, and its degradation activities can be either down-regulated by recD mutations, which leave recombinational activities of the enzyme intact, or completely inactivated by recBC mutations, which also make cells Rec−. Throughout this work, we used recD mutations to prevent rapid degradation of the cut DNA by ExoV and at the same time to preserve the recombination proficiency of the cells (Fig. 7, lanes a, b). When the same cut-by-uncut cross is repeated in wild-type cells (ExoV+), there is no recombinant formation and no parental phage replication (Fig. 7, lanes c, d), apparently due to the rapid and complete degradation of the cut DNA. To recombine well in wild-type cells, λ mutant for its own recombination genes needs χ sites (for review, see Myers and Stahl 1994), which our λ substrates are lacking.
Figure 7.

Genetic requirements of the recombinant formation. Two parental phages in a single cut-by-uncut combination were injected into various host strains harboring pK107 (cutting + repression). The total DNA was extracted at the indicated postinjection times, cut with NcoI, and analyzed by blot hybridization with probe 1. recD strain is WA800, wild-type strain is AB1157, recBC mutant is JC5519, recD recF mutant is AK61, recD dnaE mutant is AK64. Phages are MMS2660 (Δnin5 cut) and MMS2662 (Δspi6 uncut). Incubation was at 42°C to inactivate DnaEts protein.
Under physiological conditions, RecA filament assembly on the SSB-complexed ssDNA is a slow reaction; in vivo, it has to be accelerated by two different activities, defining the two pathways of recombinational repair in E. coli (for review, see Kuzminov 1995b, 1996a). In double-strand end repair, RecA polymerization is promoted by RecBCD enzyme (Anderson and Kowalczykowski 1997), whereas in daughter-strand gap repair, RecA polymerization is controlled by RecF protein (Webb et al. 1997 and references therein). Because our recombination was the result of double-strand end repair, we expected it to be blocked in recBC mutants but to be unaffected in recF mutants.
As already mentioned, besides being incapacitated in double-strand end repair, recBC mutants are also deficient in linear duplex DNA degradation. This is illustrated by the relative stability of the cut parental DNA in recBC cells (Fig. 7, cf. lanes e and f with a and b). As expected, no recombinants are formed in the recBC mutant, and no replication of the uncut parent is observed. In contrast, recD recF mutant is proficient in both recombinant formation and replication of the uncut parent (Fig. 7, lanes g,h), confirming that the phenomenon in question follows the RecBC, rather than the RecF, recombinational pathway. Both the extent of DNA replication of the uncut parent and the amount of the recombinant band in recD recF mutant are less than in recD recF+ cells (Fig. 7, cf. lanes b and h), suggesting that RecF has a role in this recombination and stressing the need for quantification in further genetic characterization of the phenomenon.
We interpret the observed connection between recombinant formation and uncut parent replication (Figs. 4–6) to mean that double-strand end recombination primes a full-fledged replication fork (Fig. 4B). This means that recombination intermediates are often resolved by DNA replication rather than by cutting strands and predicts that blocking DNA replication would reduce the recombination reaction. dnaEts mutation allows rapid inactivation of DNA synthesis at the nonpermissive temperature (Wechsler and Gross 1971; Kuzminov et al. 1994), due to the defect in the main replicative DNA polymerase of E. coli (for review, see Baker and Wickner 1992). Incubation of a recD dnaEts mutant at the nonpermissive temperature severely reduces recombinant formation (Fig. 7, lanes i,j), supporting the idea that recombination intermediates in our system are resolved by DNA synthesis.
Discussion
Using the λ chromosome as an in vivo substrate for the E. coli RecBC pathway of recombination, we inquired whether homologous interactions between a double-strand end and a circular DNA would stimulate replication of the circular DNA (break–copy) or would result in exchange without replication (break–join). We obtained the following evidence: (1) In our system, homologous recombination is stimulated by either ordinary phage chromosome replication or by introduction of double-strand ends; (2) when the parental phages are repressed but not cut, their limited DNA replication is dependent on recombination between them, which, in its turn, is likely to be due to random double-strand ends; (3) when one of the repressed parental chromosomes is cut, replication of the uncut parental chromosome is dependent on recombination between the cut and uncut chromosomes; (4) both the recombination and the replication require RecA, RecBC, and the replicative DNA polymerase. We conclude that DNA replication in E. coli gives rise to double-strand ends that recombine with an intact homologous DNA via the RecBC pathway to generate replication forks.
Double-strand end repair was proposed to prime DNA replication in E. coli (Smith 1991; Asai et al. 1993; Kuzminov 1995a), but the only direct demonstration that has been offered (Asai et al. 1994) has shortcomings (see the Introduction). Our results offer direct, compelling evidence that double-strand end repair catalyzed by the E. coli recombination system promotes DNA replication. They also bring back the ideological pluralism of the early recombination principles—although E. coli is apparently able to perform break–join recombination when DNA synthesis is inhibited, it prefers to go break–copy and, whenever the break is itself the consequence of DNA replication, copy–break–copy (reminiscent of copy–choice).
Skalka may have been the first to advance the idea of interdependence of DNA replication and recombinational repair, when trying to explain the stimulating effect of the recombination functions on phage λ DNA synthesis (Skalka 1974). An even tighter relationship between homologous recombination and DNA replication exists in phage T4, which depends on strand invasion to prime its late DNA replication (for review, see Mosig 1998). The recent finding that E. coli priA mutants, affecting the principal primosome-assembly protein, are deficient in homologous recombination is the strongest genetic evidence that double-strand end repair in this bacterium cannot be completed without initiation of DNA replication (Kogoma et al. 1996; Sandler et al. 1996). However, E. coli is not the only free-living organism with the copy–break–copy cycle—genetic data suggesting that double-strand end repair primes DNA replication in yeast have been reported (Voelkel-Meiman and Roeder 1990; Malkova et al. 1996; Morrow et al. 1997).
The demonstration that homologous recombination between a double-strand end and an intact duplex is associated with DNA synthesis has broad implications. Historically, the efforts to understand the recombination apparatus were driven by efforts to understand meiotic recombination rather than DNA repair, with the consequence that dominant models of homologous recombination emphasize genetic exchange (Clark and Sandler 1994; Kowalczykowski et al. 1994; Taylor and Smith 1995; Eggleston and West 1996) (Fig. 8A–D). However, the conclusion that recombination is linked with DNA replication in a copy–break–copy cycle suggests that the recombinational apparatus was selected primarily because it served the needs of DNA replication.
Figure 8.
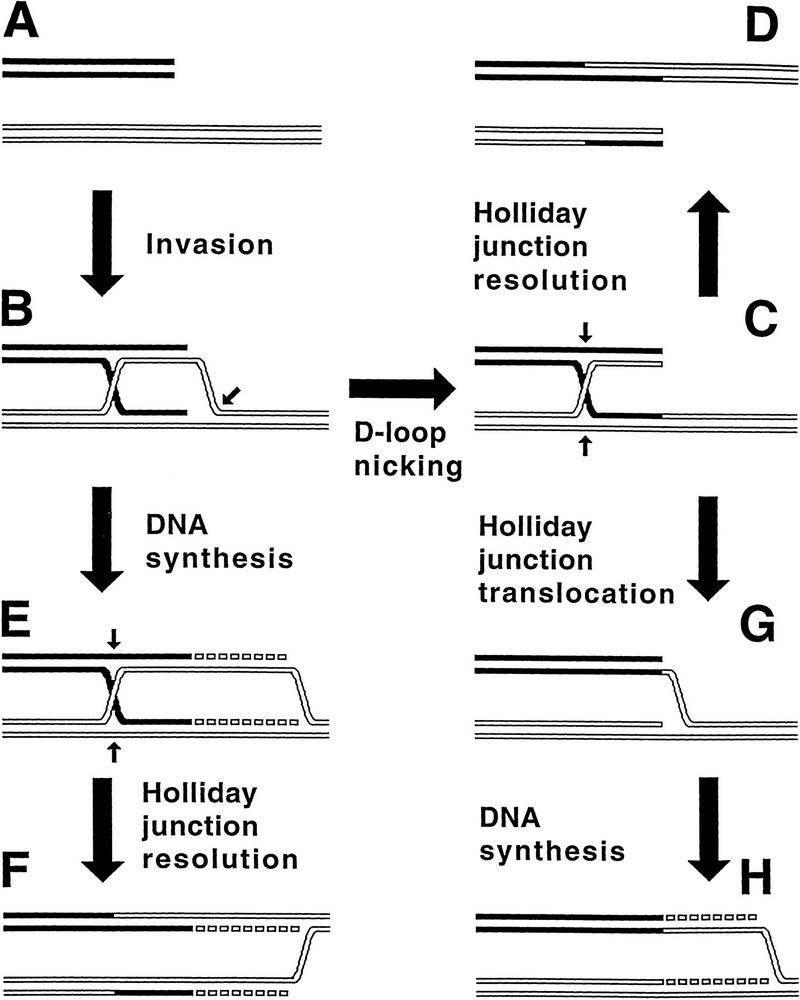
Exchange (break–join) versus repair (break–copy) in contemporary schemes of recombination. A–D represents the break–join, exchange-oriented pathway (Clark and Sandler 1994; Kowalczykowski et al. 1994; Taylor and Smith 1995; Eggleston and West 1996), whereas A, B, E, and F (Asai et al. 1993; Kuzminov 1995a; Cox 1998) or A–C, G, and H (Kuzminov 1996b) are the break–copy, repair-oriented pathways. DNA duplexes are depicted as double lines; the strands to be cut are indicated by small arrows. For simplicity, only one of the two possible orientations of Holliday junction resolution is shown. (A) Two homologous DNA duplexes, the black one with the double-strand end, the white one intact; (B) the double-strand end, with the help of homologous pairing activity of RecA protein, invades the intact duplex; (C) the intact duplex is nicked in the invasion loop, and one of the invading strands is linked with the nicked strand; (D) the Holliday junction is resolved, yielding two recombinant DNA molecules; (E) the invading end primes DNA synthesis; (F) the Holliday junction is resolved, completing the replication fork; (G) the Holliday junction is removed by sliding towards the double-strand end, which creates a replication fork framework; (H) DNA synthesis begins at the replication fork framework.
The role of homologous recombination in postreplicational DNA repair is reflected in several models (Smith 1991; Asai et al. 1993; Kuzminov 1995a; Cox 1998) (Fig. 8, A,B,E,F or A,B,C,G,H). In the more recent models, DNA replication is postulated to produce substrates for recombinational repair in the form of disrupted replication forks. The cell reverses the disruption via local repairing of the separated parental strands (discussed in Kuzminov 1996a). This pairing reaction is the same as the one that is central to most models of homologous recombination (Fig. 8B), so the restart of DNA replication is called recombinational repair.
During meiosis, when interactions between sister chromatids are specifically inhibited, most exchange occurs between homologs. Judging from the phenotype of recombination-deficient mutants, an immediate, physiological role for meiotic exchange is to promote segregation of homologs. Nevertheless, a role for exchange in the generation of hereditary diversity, especially in creatures with few chromosomes, is hard to dismiss. However, one suspects that meiotic exchange, so much a focus of investigation in recombination, reflects the usurpation, with modification, of a mechanism whose primary task was, and is, the restoration of broken replication forks arising during vegetative growth and reproduction.
Materials and methods
Bacterial strains, phages, and plasmids
Bacterial strains JC5519 (Willetts et al. 1969), WA800 (Rinken et al. 1992), BT125 (Rinken et al. 1992), AK3 (Kuzminov and Stahl 1997), AK24, AK61, and AK64 are all derivatives of AB1157 (Bachmann 1971). AK24 is ΔrecA304 recD1903; it was constructed by transducing JC10287 (Czonka and Clark 1979) harboring pBEU14 (Uhlin and Clark 1981) to tetracycline resistance with P1 lysate of AK3 (Kuzminov and Stahl 1997), followed by selection for T4 2 permissivity. The resulting strain was then grown at 40°C to eliminate the recA plasmid and was verified to be extremely UVs.
AK61 is recC1010 recF400::Tn5; it was constructed by transducing WA800 to kanamycin resistance with P1 lysate of WA576 (Thoms and Wackernagel 1987) with subsequent selection for moderate UV sensitivity. AK64 is recC1010 dnaE486::mini-Kan; it was constructed by transducing WA800 to kanamycin resistance with P1 lysate of AK12 (AB1157 dnaE486ts zae3095::mini-Kan) with the following selection for inability to grow at 42°C.
All λ phages are from the laboratory collection. The full genotypes of MMS2660–MMS2663 are described (Stahl et al. 1997). MMS207 is cI Ind−; MMS718 is int4 red3 gam210 cI857 Psus80 Δnin5; IS4 is Δb519 int4 Psus80; λAK1, constructed for this work, is SR1::XhoI int4 Δspi6. Phages were purified from plate lysates by centrifugation in a CsCl gradient (Stahl et al. 1997) or by PEG precipitation (Arber et al. 1983).
The plasmid pPaoRM3.8, carrying PaeR7I RM system (a XhoI isoschizomer), is described (Gingeras and Brooks 1983). Plasmids pK107, pK110, pK111, and pK116 were constructed for this work. pK107 is pPaoRM3.8, into the unique BamHI site of which the cI-containing (BamHI–BglII 1550 bp) fragment from pKB252 (Backman et al. 1976) has been inserted so that the restored BamHI site is proximal to the restriction-modification genes. The coordinates of the λ chromosome piece in this plasmid are 36895–38108.
Plasmid pK110 is pPaoRM3.8, into the unique BamHI site of which the cI(Ind−)-containing (2392-bp BglII–BglII) fragment from λ MMS207 has been inserted so that the λ genes are transcribed away from the restriction-modification genes. The coordinates of the λ chromosome piece in this plasmid are 35711–38108. Plasmid pK111 has been constructed from pK110 by deleting the (1600-bp ClaI–ClaI) fragment containing rexB and most of rexA gene. The presence of the cI Ind mutation (Daniels et al. 1983) in pK111 has been confirmed by sequencing. The coordinates of the λ chromosome piece in this plasmid are 36967–38108. pK116 was constructed from pK107, first by cutting with BstXI, then treating with S1 nuclease, then cutting with NruI, then ligating. The deleted 1350-bp piece removes two-thirds of the XhoI restrictase gene.
Tryptone Broth (TB) and LB broth, as well as TM buffer (TMG without gelatin) are described (Arber et al. 1983). When cells carried plasmids, the medium was supplemented with 100 μg/ml ampicillin.
The standard cross
A fresh single colony was used to inoculate an overnight culture in LB, which was then incubated with shaking at 28°C. The next morning, 200–400 μl of the O/N culture were used to inoculate 20 ml of TB, supplemented with 0.1% maltose, and the diluted culture was grown with shaking at 28°C to mid-log phase. Cells were collected by centrifugation, resuspended in 20 ml of TM, and shaken at 28°C for 1 hr. Cells were again collected by centrifugation, resuspended in 1 ml of TM and counted. (N+1) × 2 × 108 cells (where N is the number of expected time points in the experiment) were brought to 200 μl with TM and chilled on ice. An appropriate phage mixture, both phages at moi = 3, was added in 40 μl of TM, the adsorption mixture was vortexed gently and chilled on ice for 15 min. The tube with the mixture was transferred to 37°C for 1 min, and then the mixture was introduced into (N+1) ml of prewarmed LB culture. Incubation was at 37°C unless indicated otherwise. At indicated time points, 1-ml aliquots were taken for DNA isolation.
DNA isolation, labeling, gel electrophoresis, and blot hybridization
DNA was isolated, restricted with indicated enzymes, and separated in 0.7% agarose gels as before (Stahl et al. 1997). As all of our experiments are, in essence, time courses with live cultures in which DNA content increases with time, it was undesirable to normalize loading of lanes by DNA content. Instead, we started all the experiments with the same number of cells and phages, used the same volumes during DNA isolation, and took the same fraction of the DNA preparations (30% of total) for restriction analysis. This protocol allows one to see the real changes in intracellular phage DNA. Southerns and DNA labeling for Southerns were as before (Stahl et al. 1997). Membranes to be reprobed were washed three times for 15 min each in 100 mm sodium phosphate buffer (pH 7.0–7.5) containing 1% SDS and were exposed wet. The probe was later stripped in 0.4 n NaOH at 65°C for 30 min. Film exposure was at −70°C with intensifying screens.
Probe 1, specific for the interval between Δspi6 and Δnin5, is a 1489-bp StyI–StyI λ fragment with coordinates 35016–36505. The probe away from the interval is a 4572-bp NcoI–NcoI λ fragment with coordinates 19329–23901. The spi6+-specific probe is a 2932-bp EcoRI–NheI λ fragment with coordinates 31747–34679. The nin5+-specific probe is a 2014-bp StuI–AflII λ fragment with coordinates 40616–42630. Probe 2, specific for the interval between Δb519 and Δspi6, is a 3003-bp SphI–SphI λ fragment with coordinates 24375–27378.
Acknowledgments
We thank Wilfried Wackernagel for bacterial strains, Gerry Smith and Lenna Kuzminova for helpful comments on the manuscript, and an anonymous reviewer for proposing an insightful experiment. This work was supported by National Science Foundation grant MCB-9402695.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kuzminov@molbio.uoregon.edu; FAX (541) 346-5891.
References
- Amundsen SK, Taylor AF, Chaudhury AM, Smith GR. recD: The gene for an essential third subunit of exonuclease V. Proc Natl Acad Sci. 1986;83:5558–5562. doi: 10.1073/pnas.83.15.5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DG, Kowalczykowski SC. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a χ-regulated manner. Cell. 1997;90:77–86. doi: 10.1016/s0092-8674(00)80315-3. [DOI] [PubMed] [Google Scholar]
- Arber W, Enquist L, Hohn B, Murray N, Murray K. Experimental methods for use with lambda. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 433–466. [Google Scholar]
- Asai T, Sommer S, Bailone A, Kogoma T. Homologous recombination-dependent initiation of DNA replication from damage-inducible origins in Escherichia coli. EMBO J. 1993;12:3287–3295. doi: 10.1002/j.1460-2075.1993.tb05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Bates DB, Kogoma T. DNA replication triggered by double-strand breaks in E. coli: Dependence on homologous recombination functions. Cell. 1994;78:1051–1061. doi: 10.1016/0092-8674(94)90279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann BJ. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol Rev. 1971;36:525–557. doi: 10.1128/br.36.4.525-557.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman K, Ptashne M, Gilbert W. Construction of plasmids carrying the cI gene of bacteriophage λ. Proc Natl Acad Sci. 1976;73:4174–4178. doi: 10.1073/pnas.73.11.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Wickner SH. Genetics and enzymology of DNA replication in Escherichia coli. Annu Rev Genet. 1992;26:447–477. doi: 10.1146/annurev.ge.26.120192.002311. [DOI] [PubMed] [Google Scholar]
- Belling J. Chiasmas in flowering plants. Univ Calif Publ Bot. 1931a;16:311–338. [Google Scholar]
- ————— Chromomeres of liliaceous plants. Univ Calif Publ Bot. 1931b;16:153–170. [Google Scholar]
- Brooks K, Clark AJ. Behavior of λ bacteriophage in a recombination deficient strain of Escherichia coli. J Virol. 1967;1:283–293. doi: 10.1128/jvi.1.2.283-293.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury AM, Smith GR. A new class of Escherichia coli recBC mutants: Implications for the role of RecBC enzyme in homologous recombination. Proc Natl Acad Sci. 1984;81:7850–7854. doi: 10.1073/pnas.81.24.7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ, Sandler SJ. Homologous recombination: The pieces begin to fall into place. Crit Rev Microbiol. 1994;20:125–142. doi: 10.3109/10408419409113552. [DOI] [PubMed] [Google Scholar]
- Cox MM. A broadening view of recombinational DNA repair in bacteria. Genes Cells. 1998;3:65–78. doi: 10.1046/j.1365-2443.1998.00175.x. [DOI] [PubMed] [Google Scholar]
- Craig NL, Roberts JW. E. coli RecA protein-directed cleavage of phage λ repressor requires polynucleotide. Nature. 1980;283:26–30. doi: 10.1038/283026a0. [DOI] [PubMed] [Google Scholar]
- Czonka LN, Clark AJ. Deletions generated by the transposon Tn10 in the srl-recA region of the Escherichia coli K-12 chromosome. Genetics. 1979;93:321–343. doi: 10.1093/genetics/93.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DL, Schroeder JL, Szybalski W, Sanger F, Blattner FR. A molecular map of coliphage lambda. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 469–517. [Google Scholar]
- Delbrück M, Stent GS. On the mechanism of DNA replication. In: McElroy WD, Glass B, editors. The chemical basis of heredity. Baltimore, MD: The Johns Hopkins Press; 1957. pp. 699–736. [Google Scholar]
- Eggleston AK, West SC. Exchanging partners: Recombination in E. coli. Trends Genet. 1996;12:20–26. doi: 10.1016/0168-9525(96)81384-9. [DOI] [PubMed] [Google Scholar]
- Enquist LW, Skalka A. Replication of bacteriophage λ DNA dependent on the function of host and viral genes. I. Interaction of red, gam, and rec. J Mol Biol. 1973;75:185–212. doi: 10.1016/0022-2836(73)90016-8. [DOI] [PubMed] [Google Scholar]
- Gingeras TR, Brooks JE. Cloned resriction/modification system from Pseudomonas aeruginosa. Proc Natl Acad Sci. 1983;80:402–406. doi: 10.1073/pnas.80.2.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey AD, Rotman R. Genetic recombination between host-range and plaque-type mutants of bacteriophage in single bacterial cells. Genetics. 1949;34:44–71. doi: 10.1093/genetics/34.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Wollman EL. Étude génétique d’un bactériophage tempéré d′Escherichia coli. I. Le systéme génétique du bactériophage λ. Ann Inst Pasteur (Paris) 1954;87:653–673. [PubMed] [Google Scholar]
- Janssens FA. La théorie de la chiasmatypie. Nouvelle interpretation des cinèses de maturation. Cellule. 1909;22:387–411. [Google Scholar]
- Kaiser AD. A genetic study of the temperate coliphage λ. Virology. 1955;1:424–443. doi: 10.1016/0042-6822(55)90036-2. [DOI] [PubMed] [Google Scholar]
- Kellenberger-Gujer G, Weisberg RA. Recombination in bacteriophage lambda. I. Exchange of DNA promoted by phage and bacterial recombination mechanisms. In: Hershey AD, editor. The bacteriophage lambda. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1971. pp. 407–415. [Google Scholar]
- Kogoma T, Cadwell GW, Barnard KG, Asai T. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J Bacteriol. 1996;178:1258–1264. doi: 10.1128/jb.178.5.1258-1264.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995a;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- ————— A mechanism for induction of the SOS response in E. coli: Insights into the regulation of reversible protein polymerization in vivo. J Theor Biol. 1995b;177:29–43. doi: 10.1006/jtbi.1995.0222. [DOI] [PubMed] [Google Scholar]
- ————— . Recombinational repair of DNA damage. Austin, TX: R.G. Landes Company; 1996a. [Google Scholar]
- ————— Unraveling the late stages of recombinational repair: Metabolism of DNA junctions in Escherichia coli. BioEssays. 1996b;18:757–765. doi: 10.1002/bies.950180911. [DOI] [PubMed] [Google Scholar]
- Kuzminov A, Stahl FW. Stability of linear DNA in recA mutant Escherichia coli cells reflects ongoing chromosomal DNA degradation. J Bacteriol. 1997;179:880–888. doi: 10.1128/jb.179.3.880-888.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A, Schabtach E, Stahl FW. χ-sites in combination with RecA protein increase the survival of linear DNA in E. coli by inactivating exoV activity of RecBCD nuclease. EMBO J. 1994;13:2764–2776. doi: 10.1002/j.1460-2075.1994.tb06570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J. Recombination mechanisms in bacteria. J Cell Comp Physiol (Suppl. 2) 1955;45:75–107. doi: 10.1002/jcp.1030450506. [DOI] [PubMed] [Google Scholar]
- Lovett ST, Luisi-DeLuca C, Kolodner RD. The genetic dependence of recombination in recD mutants of Escherichia coli. Genetics. 1988;120:37–45. doi: 10.1093/genetics/120.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee TR, Asai T, Malka D, Kogoma T. DNA damage-inducible origins of DNA replication in Escherichia coli. EMBO J. 1992;11:4219–4225. doi: 10.1002/j.1460-2075.1992.tb05516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Ivanov EL, Haber JE. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc Natl Acad Sci. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly KF, Signer ER, Radding CM. Nonessential functions of bacteriophage λ. Virology. 1969;37:177–188. doi: 10.1016/0042-6822(69)90197-4. [DOI] [PubMed] [Google Scholar]
- Meselson M. On the mechanism of genetic recombination. J Mol Biol. 1964;9:734–745. doi: 10.1016/s0022-2836(64)80178-9. [DOI] [PubMed] [Google Scholar]
- Meselson M, Weigle JJ. Chromosome breakage accompanying genetic recombination in bacteriophage. Proc Natl Acad Sci. 1961;47:857–868. doi: 10.1073/pnas.47.6.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, Hieter P. “Break copy” duplication: A model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics. 1997;147:371–382. doi: 10.1093/genetics/147.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig G. Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu Rev Genet. 1998;32:379–413. doi: 10.1146/annurev.genet.32.1.379. [DOI] [PubMed] [Google Scholar]
- Myers RS, Stahl FW. χ and the RecBCD enzyme of Escherichia coli. Annu Rev Genet. 1994;28:49–70. doi: 10.1146/annurev.ge.28.120194.000405. [DOI] [PubMed] [Google Scholar]
- Poteete AR, Fenton AC. Efficient double-strand break-stimulated recombination promoted by the general recombination systems of phages λ and P22. Genetics. 1993;134:1013–1021. doi: 10.1093/genetics/134.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinken R, Thoms B, Wackernagel W. Evidence that recBC-dependent degradation of duplex DNA in Escherichia coli recD mutants involves DNA unwinding. J Bacteriol. 1992;174:5424–5429. doi: 10.1128/jb.174.16.5424-5429.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JW, Roberts CW. Proteolytic cleavage of bacteriophage lambda repressor in induction. Proc Natl Acad Sci. 1975;72:147–151. doi: 10.1073/pnas.72.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca AI, Cox MM. RecA protein: Structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- Sandler SJ, Samra HS, Clark AJ. Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics. 1996;143:5–13. doi: 10.1093/genetics/143.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalka A. A replicator’s view of recombination (and repair) In: Grell RF, editor. Mechanisms in recombination. New York, NY: Plenum Press; 1974. pp. 421–432. [Google Scholar]
- Smith GR. Conjugational recombination in E. coli: Myths and mechanisms. Cell. 1991;64:19–27. doi: 10.1016/0092-8674(91)90205-d. [DOI] [PubMed] [Google Scholar]
- Stahl FW, Chung S, Crasemann J, Faulds D, Haemer J, Lam S, Malone RE, McMillin KD, Nozu Y, Siegel J, et al. Recombination, replication, and maturation in phage lambda. In: Fox CF, Robinson WS, editors. Virus research. New York, NY: Academic Press; 1973. pp. 487–503. [Google Scholar]
- Stahl FW, McMilin KD, Stahl MM, Crasemann JM, Lam S. The distribution of crossovers along unreplicated lambda bacteriophage chromosomes. Genetics. 1974;77:395–408. doi: 10.1093/genetics/77.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl FW, Kobayashi I, Stahl MM. In phage λ, cos is a recombinator in the Red pathway. J Mol Biol. 1985;181:199–209. doi: 10.1016/0022-2836(85)90085-3. [DOI] [PubMed] [Google Scholar]
- Stahl MM, Thomason L, Poteete AR, Tarkowski T, Kuzminov A, Stahl FW. Annealing vs. invasion in phage λ recombination. Genetics. 1997;147:961–977. doi: 10.1093/genetics/147.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T. Behavior of some episomal elements in a recombination-deficient mutant of Escherichia coli. Japan J Microbiol. 1966;10:201–210. [Google Scholar]
- Taylor AF. RecBCD enzyme of Escherichia coli. In: Kucherlapati R, Smith GR, editors. Genetic recombination. Washington, D.C.: American Society for Microbiology; 1988. pp. 231–263. [Google Scholar]
- Taylor AF, Smith GR. Strand specificity of nicking of DNA at Chi sites by RecBCD enzyme. Modulation by ATP and magnesium levels. J Biol Chem. 1995;270:24459–24467. doi: 10.1074/jbc.270.41.24459. [DOI] [PubMed] [Google Scholar]
- Telander-Muskavitch KM, Linn S. RecBC-like enzymes: Exonuclease V deoxyribonucleases. In: Boyer PD, editor. The enzymes. New York, NY: Academic Press; 1981. pp. 234–250. [Google Scholar]
- Thaler DS, Stahl MM, Stahl FW. Double-chain-cut sites are recombination hotspots in the Red pathway of phage λ. J Mol Biol. 1987a;195:75–87. doi: 10.1016/0022-2836(87)90328-7. [DOI] [PubMed] [Google Scholar]
- ————— Evidence that the normal route of replication-allowed Red-mediated recombination involves double-chain ends. EMBO J. 1987b;6:3171–3176. doi: 10.1002/j.1460-2075.1987.tb02628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler DS, Sampson E, Siddiqi I, Rosenberg SM, Thomason LC, Stahl FW, Stahl MM. Recombination of bacteriophage λ in recD mutants of Escherichia coli. Genome. 1989;31:53–67. doi: 10.1139/g89-013. [DOI] [PubMed] [Google Scholar]
- Thoms B, Wackernagel W. Regulatory role of recF in the SOS response of Escherichia coli: Impaired induction of SOS genes by UV irradiation and nalidixic acid in a recF mutant. J Bacteriol. 1987;169:1731–1736. doi: 10.1128/jb.169.4.1731-1736.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlin BE, Clark AJ. Overproduction of the Escherichia coli RecA protein without stimulation of its proteolytic activity. J Bacteriol. 1981;148:386–390. doi: 10.1128/jb.148.1.386-390.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Putte P, Zwenk H, Rörsch A. Properties of four mutants of Escherichia coli defective in genetic recombination. Mutat Res. 1966;3:381–392. doi: 10.1016/0027-5107(66)90048-0. [DOI] [PubMed] [Google Scholar]
- Voelkel-Meiman K, Roeder GS. Gene conversion tracts stimulated by HOT1-promoted transcription are long and continuous. Genetics. 1990;126:851–867. doi: 10.1093/genetics/126.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb BL, Cox MM, Inman RB. Recombinational DNA repair: The RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell. 1997;91:347–356. doi: 10.1016/s0092-8674(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Wechsler JA, Gross JD. Escherichia coli mutants temperature-sensitive for DNA synthesis. Mol & Gen Genet. 1971;113:273–284. doi: 10.1007/BF00339547. [DOI] [PubMed] [Google Scholar]
- Wilkins AS, Mistry J. Phage lambda’s generalized recombination system. Study of intracellular DNA pool during lytic infection. Mol & Gen Genet. 1974;129:275–293. doi: 10.1007/BF00265693. [DOI] [PubMed] [Google Scholar]
- Willetts NS, Clark AJ, Low KB. Genetic location of certain mutations conferring recombination deficiency in Escherichia coli. J Bacteriol. 1969;97:244–249. doi: 10.1128/jb.97.1.244-249.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]