

Abstract
We report the design, synthesis, biological evaluation, and the X-ray crystal structure of a novel inhibitor-bound HIV-1 protease. Various C3-functionalized cyclopentanyltetrahydrofurans (Cp-THF) were designed to interact with the flap Gly48 carbonyl or amide NH in the S2-subsite of the HIV-1 protease. We investigated the potential of those functionalized ligands in combination with hydroxyethyl sulfonamide isosteres. Inhibitor 26 containing a 3-(R)-hydroxyl group on the Cp-THF core, displayed the most potent enzyme inhibitory and antiviral activity. Our studies revealed a preference for the 3-(R)-configuration over the corresponding 3-(S)-derivative. Inhibitor 26 exhibited potent activity against a panel of multidrug-resistant HIV-1 variants. A high resolution X-ray structure of 26-bound HIV-1 protease revealed important molecular insight into the ligand-binding site interactions.
Introduction
Human immunodeficiency virus 1 (HIV-1) protease inhibitors are critical components of antiretroviral therapies.1,2 However, the emergence of drug resistance has raised serious questions about long-term treatment options.3,4 Our structure-based design of inhibitors targeting the protein-backbone has led to the discovery of a variety of novel HIV-1 protease inhibitors (PIs) with broad-spectrum activity against multidrug-resistant HIV-1 variants.5,6 One of these inhibitors, darunavir (1, Figure 1) was approved by the FDA for the treatment of HIV/AIDS patients.7–9 In an effort to address drug resistance, our inhibitor design strategy focused on maximizing active site interactions with the protease, particularly by promoting extensive hydrogen bonding interactions with backbone atoms throughout the active site.5,6
Figure 1.
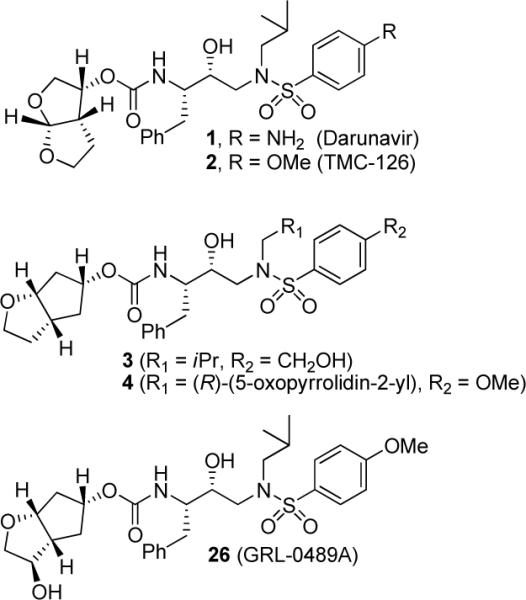
Structures of potent HIV-1 protease inhibitors 1–4, and 26.
We have recently reported a number of potent inhibitors incorporating a stereochemically defined (3aS,5R,6aR)-hexahydro-2H-cyclopenta[b]furan-5yl (Cp-THF) as the P2-ligand with a modified hydroxyethylsulfonamide isostere in inhibitor 3.10 The X-ray crystal structure of 3-bound HIV-1 protease revealed the formation of an extensive hydrogen-bonding network between the inhibitor and the active site. Based upon this molecular insight, we subsequently incorporated a stereochemically defined lactam at the P1'-position to further enhance backbone interactions.11 Interestingly, the resulting inhibitor 4 retained full potency against a range of multidrug-resistant HIV-1 variants.12 The X-ray structural studies of 4-bound HIV-1 protease evidenced enhanced backbone interactions with the Gly27' carbonyl at the S1'-subsite. The Cp-THF ligand appeared to fit within the S2-subsite and the cyclic ether oxygen has involved in a close hydrogen bonding interaction with the backbone NH of Asp29 (2.8 Å). Based upon this molecular insight, we have now investigated structural modifications of the Cp-THF ligand to further optimize ligand binding, particularly hydrogen bonding ability, in the S2-subsite. The X-ray structure of 3-bound HIV-1 protease indicated that the C3 methylene of the Cp-THF is in close proximity to the protease flap region. In fact, the X-ray data suggested a weak C3-H⋯O interaction with the Gly48 backbone carbonyl group.10 We therefore envisioned that introduction of a polar substituent at the C3 position may lead to additional interactions of the Cp-THF ligand with the protease flap residues. Furthermore, an inhibitor which makes tight interactions with the protease flap region could conceivably delay its dissociation via opening of the flaps. Herein, we report the design, synthesis, and biological evaluation of a series of protease inhibitors that incorporate a stereochemically defined functionality at the C3 position of the Cp-THF-ligand. Inhibitor 26, incorporating a 3-(R)-hydroxyl group was the most potent PI (Ki = 5 pM; antiviral IC50 = 2.9 nM). This inhibitor also maintained excellent potency against a range of multidrug-resistant HIV-1 variants. The protein ligand X-ray structure of 26-bound HIV-1 protease revealed important insights into the ligand-binding site interactions of the inhibitor with the flap region as well as other regions of the HIV-1 protease active site.
Chemistry
The synthesis of 3-keto and 3-(S)-methoxy Cp-THF ligands is shown in Scheme 1. Optically active alcohol 5 was prepared in multigram quantities as described previously.13,14 This was efficiently converted to ketone 6 as reported by us.14 The removal of TBS-ether by exposure to HF-pyridine afforded keto alcohol 7 in 94% yield. Ketone 6 was converted to 3-(S)-methoxy derivative 8 in a three-step sequence involving (1) reduction of the ketone with NaBH4 in ethanol at −23 °C to provide the corresponding alcohol as a single diastereomer; (2) methylation of the resulting alcohol with MeI in the presence of Ag2O in acetonitrile; and (3) removal of the silyl group with TBAF in THF to provide 8 in 66% yield, in 3 steps.
Scheme 1.
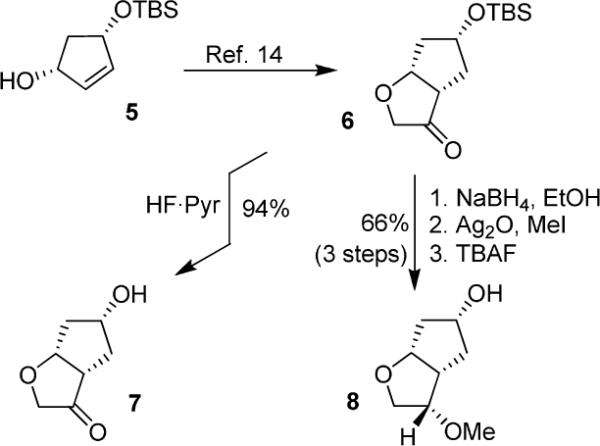
Syntheses of optically pure P2 ligands 7 and 8.
The syntheses of 3-(R)-acetoxy and 3-(R)-methoxy ligands 11 and 12 are outlined in Scheme 2. Treatment of alcohol 5 with NaH and 2-bromoacetic acid in THF provided the corresponding alkylated acid. The resulting acid was reacted with methyl iodide in the presence of NaHCO3 to provide methyl ester 9 in 53% yield (2 steps). Dibal-H reduction of ester 9 followed by radical cyclization15 of the resulting alkene using a catalytic amount of (nBu3Sn)2O, Ph2SiH2 and ethanol (2 equiv) in the presence of a catalytic amount of AIBN in benzene provided 3-(R)-hydroxy derivative 10 in 64% yield, in 2 steps. The 1H-NMR analysis showed a diastereomeric ratio of 10:1. The major isomer was separated by silica gel chromatography and used for the subsequent reactions. Reaction of alcohol 10 with acetic anhydride and triethylamine in the presence of a catalytic amount of DMAP afforded the corresponding acetate. The removal of the silyl group with TBAF in THF provided ligand 11 in 73% yield, in 2 steps. Alcohol 10 was converted to methoxy derivative 12 by alkylation with NaH and MeI in THF followed by removal of the silyl group in 71% yield (2 steps).
Scheme 2.
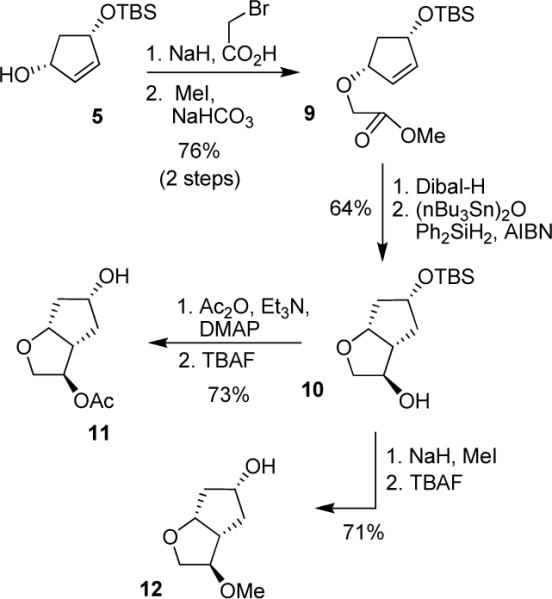
Syntheses of ligands 11 and 12.
We also planned to synthesize stereochemically defined 3-methyl derivatives to compare the effects of alkoxy and hydroxy groups. Towards this goal, we have carried out stereoselective syntheses of 3-(S) and 3-(R)-methyl derivatives and the synthetic route is shown in Scheme 3. Optically active olefin 13 was synthesized as described previously.14 Catalytic hydrogenation of 13 in the presence of Wilkinson's catalyst under a hydrogen filled balloon at 23° C for 3 h, followed by removal of the silyl group using TBAF afforded 3-(S)-methyl derivative 14.16 For the synthesis of the 3-(R)-methyl derivative, commercially available optically active lactone (+)-15 was methylated using LDA and MeI at −78 °C to provide methyl derivative 16 with high diastereoselectivity (dr = 20:1) and in 95% yield. Olefin 16 was then subjected to oxymercuration condition with Hg(OAc)2 and HClO4. The resulting organomercurial derivative was treated with aqueous sodium hydroxide solution followed by NaBH4 reduction to afford endo-alcohol 17 in 64% yield. The lactone was then reduced to the corresponding lactol with Dibal-H. Further reduction of the resulting lactol using Et3SiH and TiCl4 furnished ligand 3-(R)-methyl derivative 18 in 68% yield. The 1H-NMR NOESY experiments fully corroborated the assignment of 3-(S)- and 3-(R)-stereochemistry of methyl derivatives 14 and 18, respectively.
Scheme 3.
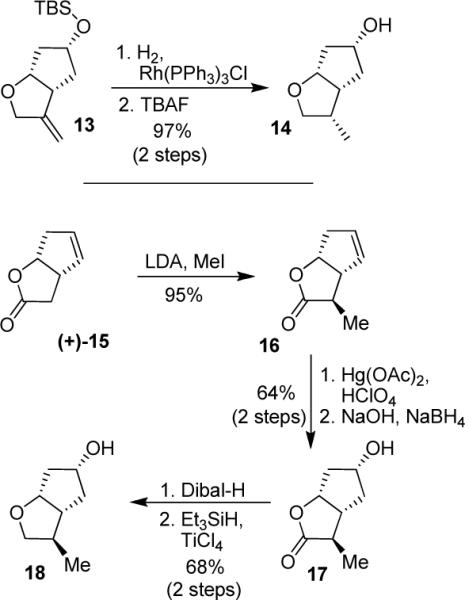
Syntheses of C3-methyl-substituted ligands 14 and 18.
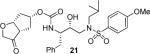
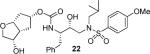
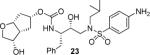
Various optically active ligand alcohols 7, 8, 11, 12, 14, and 18 were converted to the respective mixed activated carbonates. As shown in Scheme 4, reactions of ligand alcohols with 4-nitrophenyl chloroformate in the presence of pyridine in CH2Cl2 provided activated carbonates 19a–f in 50–96% yield.17 The synthesis of designed inhibitors was carried out by coupling these activated carbonates with various hydroxyethylsulfonamide isosteres containing functionalized P2'-phenylsulfonamide ligands. As shown in Scheme 5, amines 20a–c were readily prepared as described previously.10,17 Reaction of amine 20a with carbonate 19a provided inhibitor 21. Inhibitors 22–24 were prepared by reaction of carbonate 19a with respective amines 20a–c followed by NaBH4 reduction of the resulting ketone derivatives. The inhibitor structures are shown in Table 1. Inhibitors 25 and 28–30 were prepared by reactions of amine 20a with mixed carbonates 19b and 19d–f, respectively. The synthesis of inhibitors 26 and 27 was carried out by reactions of mixed carbonate 19c with amines 20a and 20b followed by removal of the acetyl group with K2CO3 in methanol. All inhibitors were prepared in good to excellent overall (41–96%) yields. The synthesis of inhibitor 31 containing a dimethylamine functionality was carried out by reductive amination of ketone 21 with Me2NH2+OAc− in the presence of NaHB(OAc)3. We have also attempted to prepare the corresponding methylamine derivative by reductive amination with MeNH3+OAc−. However, the resulting 3-(S)-methylamine derivative 32 turned out to be unstable.
Scheme 4.
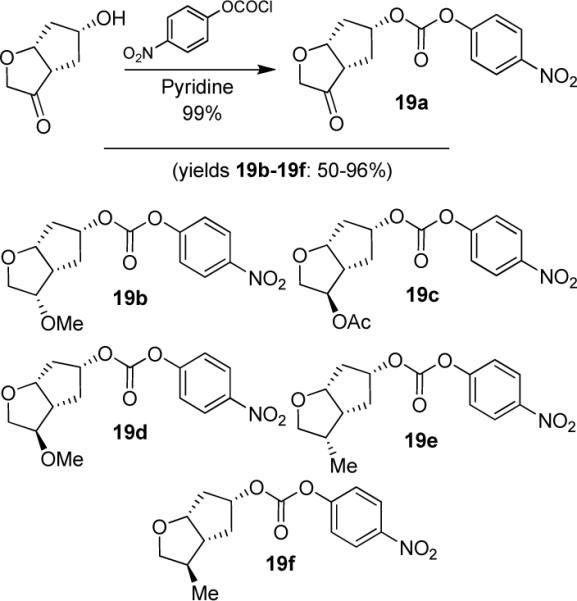
Syntheses of activated mixed carbonates 19a–f.
Scheme 5.

Syntheses of inhibitors 21–32.
Table 1.
Enzymatic inhibitory and antiviral activity of inhibitors 21–31.
| Entry | Inhibitor | Ki (nM) | IC50 (nM)a |
|---|---|---|---|
| 1 |
|
0.95 | 14 |
| 2 |
|
0.077 | 7 |
| 3 |
|
0.079 | 25 |
| 4 |
|
0.06 | 19 |
| 5 |
|
0.39 | 37 |
| 6 |
|
0.005 | 2.9 |
| 7 |
|
0.006 | 36 |
| 8 |
|
0.006 | 3.4 |
| 9 |
|
0.16 | 25 |
| 10 |
|
0.017 | --- |
| 11 |
|
12.5 | --- |
Values are means of at least two experiments. Human T-lymphoid (MT-2) cells (2×10 3) were exposed to 100 TCID50s of HIV-1LAIand cultured in the presence of each PI, and IC50 values were determined using the MTT assay. The IC50 values of amprenavir (APV), saquinavir (SQV), indinavir (IDV), and darunavir (DRV) were 0.03 (μM, 0.015 μM, 0.03 μM, and 0.003 μM respectively.
Results and Discussions
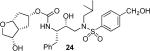
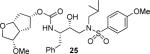
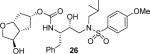
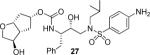
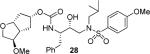
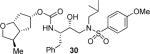
As mentioned previously, inhibitors were designed to make additional interactions in the S2-subsite of the protease, especially with the Gly48 backbone atoms in the flap region of the enzyme. All inhibitors in Table 1 were first evaluated in enzyme inhibitory assay developed by Toth and Marshall.18 Inhibitors that exhibited potent Ki values were subsequently evaluated for in vitro antiviral assays. As can be seen in the Table 1, all inhibitors displayed sub-nanomolar to low picomolar inhibitory potencies. Inhibitor 22, with a 3-(S)-hydroxy group on the Cp-THF, was significantly more potent than the keto derivative 21 (entries 1 and 2). The 3-(S)-hydroxy Cp-THF ligand was also investigated in combination with other sulfonamide substituents. Inhibitor 23 with a p-amino sulfonamide as the P2'-ligand displayed impressive inhibitory potency, however, its antiviral activity was 3-fold lower than 22. Inhibitor 24 with a p-hydroxymethyl sulfonamide as the P2'-ligand has shown a reduction in potency. Inhibitor 25 which contains a 3-(S)-methoxy substituent, exhibited a significant loss of potency and a near 5-fold loss of antiviral activity compared to 22. Interestingly, inhibitor 26 with 3-(R) configuration displayed an impressive enzyme inhibitory and antiviral activity. Inhibitor 27 with the 4-amino sulfonamide isostere also showed comparable enzyme inhibitory activity. Inhibitor 28, with a 3-(R)-methoxy group, also exhibited comparable inhibitory potency.
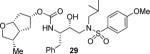
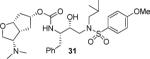
In order to probe the importance of the C3-hydroxy oxygen on the Cp-THF ring, inhibitors 29 and 30, with a methyl group in place of a C3-oxygen were synthesized. Inhibitor 29, which contains a 3-(S)-methyl group showed a significant reduction in potency compared to 22. Similarly, inhibitor 30 with a 3-(R)-methyl group has shown a reduction in enzyme Ki value compared to the corresponding hydroxy derivative 26. We have also investigated an amine substitution on the Cp-THF ligand. Inhibitor 31 with C3-dimethylamine exhibited a substantially lower enzyme inhibitory potency compared to the corresponding hydroxy or methoxy derivatives.
Inhibitors 22 with a 3-(S)-hydroxyl group and 26 with a 3-(R)-hydroxyl group on the Cp-THF ligand were tested against a panel of multidrug-resistant HIV-1 variants. Their antiviral activity was compared against other clinically available PIs including APV and DRV. The results are shown in Table 2. All inhibitors in Table 2 exhibited high antiviral activity against the wild-type HIV-1 laboratory strain, HIV-1ERS104pre, isolated from a drug-naïve patient.7 Compound 26 provided the most potent activity with an IC50 of 2.9 nM, comparable to that of DRV. When tested against various multidrug-resistant HIV-1 strains, the IC50 values of inhibitor 26 remained in the low nanomolar values (2.9–29 nM), and fold-change in IC50 did not exceed 10. Interestingly, isomeric inhibitor 22 displayed lower activity against the wild-type viral strain (IC50 = 20 nM). It also exhibited a much larger IC50 fold-change, and in some cases, only marginal activity against multidrug-resistant HIV-1 variants. Such a stark contrast in antiviral activity of 22 compared to 26 underlines the importance of the stereochemistry at the 3-position of the Cp-THF ligand. Inhibitor 26 displayed a superior profile compared to another approved PI, APV. Overall, inhibitor 26 maintained impressive potency against all tested multidrug-resistant HIV-1 strains. It compared favorably with DRV, which is the leading PI for the treatment of multidrug resistant HIV infection.
Table 2.
Comparison of the antiviral activity of 22, 26, and other PIs against multidrug resistant HIV-1 variants.
| IC50 (μM) ± SDs, (fold change)b |
||||
|---|---|---|---|---|
| Virusa | APV | DRV | 22 | 26 |
| HIV-1ERS104pre(wt) | 0.030 ± 0.006 | 0.0037 ± 0.0001 | 0.020 ± 0.004 | 0.0029 ± 0.0008 |
| HIV-1MDR/B | 0.93 ± 0.28 (31) | 0.036 ±0.013 (10) | >1 (>50) | 0.029 ± 0.007 (10) |
| HIV-1MDR/C | 0.26 ± 0.03 (9) | 0.013 ± 0.0004 (4) | >1 (>50) | 0.022 ± 0.003 (7) |
| HIV-1MDR/G | 0.38 ± 0.03 (12) | 0.0023±0.0006(1) | 0.27 ± 0.02 (13) | 0.0045±0.0007(2) |
| HIV-1MDR/TM | 0.19 ± 0.06 (6) | 0.0019±0.0003 (1) | 0.041 ± 0.004 (2) | 0.0031 ± 0.002(1) |
Amino acid substitutions identified in the protease-encoding region compared to the consensus type B sequence cited from the Los Alamos database; L10I, L33I, M36I, M46I, F53L, K55R, I62V, L63P, A71V, G73S, V82A, L90M, and I93L in HIV-1MDR/B; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q, V82A, and L89M in HIV-1MDR/C; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, and L90M in HIV-1MDR/G; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M, I93L in HIV-1MDR/TM. HIV-1ERS104pre served as a source of wild-type HIV-1. IC50S were determined by using PHA-PBMs as target cells, and inhibition of p24 Gag protein production by each drug was used as an end point. Numbers in parentheses represent n-fold changes of IC50S for each isolate compared to IC50S for the wild-type HIV-1ERS104pre. All assays were conducted in duplicate or triplicate, and data shown represent mean values (± 1 standard deviation) derived from results of three independent experiments. PHA-PBMs were derived from a single donor in each independent experiment. DRV (Darunavir), APV (Amprenavir).
In order to gain molecular insight into the ligand/binding-site interactions responsible for the activity of inhibitor 26, we have determined the X-ray crystal structure of the inhibitor-bound wild-type HIV-1 protease that was refined to a 1.45 Å resolution. The protease dimer binds with the inhibitor in two orientations related by a 180° rotation with a 0.55/0.45 ratio. The protease backbone structure showed a very low RMS deviation of 0.15 Å for all Cα atoms compared to protease complexes of 2-bound inhibitor or darunavir.19, 20 The inhibitor makes extensive interactions from the P2 to P2' ligands with the protease atoms, and most notably displays favorable polar interactions including hydrogen bonds, weaker C-H…O and C-H…pi interactions, as shown in Figure 2. The central hydroxyl group forms hydrogen bonds with the side chain carboxylate oxygen atoms of the catalytic Asp25 and Asp25' residues. The inhibitor hydrogen bonds with the protease backbone atoms of the amide of Asp30', the carbonyl oxygen of Gly27, and forms water-mediated interactions with the amides of Ile50 and 50', which are generally conserved in the majority of protease complexes with inhibitors21 or substrate analogs.22, 23 The inhibitor interactions with atoms in the binding cavity resemble those of darunavir and 2 (TMC-126) with the exception of the interactions of the new P2-ligand that replaces the bis-THF group. The 3-(R)-hydroxyl of the Cp-THF ligand extends towards the flap region and forms a new water-mediated hydrogen bond interaction with the backbone amide NH of Gly48, with interatomic distances of 2.5 Å, 3.1 Å for the major inhibitor orientation or 2.7 Å, 3.1 Å for the minor orientation, respectively. Also, the Cp-THF ether oxygen forms a strong hydrogen bond with the backbone amide NH of Asp29. These new interactions with the backbone atoms of Gly48 are likely to be responsible for the impressive antiviral activity and drug resistance properties of this inhibitor. The C3-functionality on the Cp-THF appears to enhance the affinity of the inhibitor. The new water-mediated interaction with the backbone NH of Gly48 on the protease flap may promote thermodynamic stabilization of the closed conformation of the protease-ligand complex. This may slow the kinetics of dissociation of the inhibitor through flexible opening of the protease flap.
Figure 2.

Stereoview of the X-ray structure of inhibitor 26-bound to the active site of the wild-type HIV-1 protease.
Conclusion
In summary, we have designed a number of C3-substituted hexahydrocyclopentafuranyl urethanes as P2-ligands to enhance interactions with the protein backbone in the S2-subsite. The ligands were stereoselectively synthesized in optically active form. Incorporation of these ligands in (R)-hydroxyethylsulfonamide isosteres resulted in a series of novel and highly potent HIV-1 protease inhibitors. In particular, inhibitor 26 displayed remarkable enzyme inhibitory and antiviral potency. Also, inhibitor 26 has shown excellent activity against multi-PI-resistant variants compared to other FDA approved inhibitors. A protein-ligand X-ray structure of 26-bound HIV-1 protease was determined at 1.45 Å resolution. The inhibitor appeared to make extensive interactions throughout the active site. Of particular interest, the 3-(R)-hydroxyl of the Cp-THF ligand formed a new water-mediated hydrogen bond interaction with the backbone amide NH of Gly48 and the Cp-THF ether oxygen formed a strong hydrogen bond with backbone amide NH of Asp29. The extensive interactions with the protein backbone may be responsible for inhibitor 26's impressive antiviral activity and drug resistance profiles. The design of inhibitors targeting the protein backbone has led us to develop inhibitors characterized by high potency against both wild-type and multi-drug-resistant HIV-1 strains. Further design and optimization of inhibitors utilizing this molecular insight are in progress.
Experimental Section
General
All anhydrous solvents were obtained according to the following procedures: diethyl ether and tetrahydrofuran (THF) were distilled from sodium/benzophenone under argon; toluene, methanol, acetonitrile, and dichloromethane from calcium hydride, and benzene from sodium. Other solvents were used without purification. All moisture-sensitive reactions were carried out in flame-dried flasks under argon atmosphere. Reactions were monitored by thin layer chromatography (TLC) using Silicycle 60A-F254 silica gel pre-coated plates. Flash column chromatography was performed using Silicycle 230–400 mesh silica gel. Yields refer to chromatographically and spectroscopically pure compounds. Optical rotations were recorded on a Perkin Elmer 341 polarimeter. 1H NMR and 13C NMR spectra were recorded on a Varian Inova-300 (300 MHz and 75 MHz, respectively), Bruker Avance ARX- 400 (400 MHz and 100 MHz), and Bruker Avance ARX-500 (500 MHz and 125 MHz). High and low resolution mass spectra were carried out by the Mass Spectroscopy Center at Purdue University. The purity of all test compounds was determined by HRMS and HPLC analysis in the different solvent systems. All test compounds showed ≥95% purity.
(3aS,5R,6aR)-5-Hydroxytetrahydro-2H-cyclopenta[b]furan-3(3aH)-one (7)
A solution of ketone 6 (23 mg, 0.09 mmol) in CH3CN (0.5 mL) was cooled to 0 °C under argon. Pyridine (50 μL) was added followed by dropwise addition of HF. Pyridine (0.18 mL). The solution was stirred at 0 °C for 4 h. The reaction was quenched by addition of saturated aqueous NaHCO3 solution followed by solid NaHCO3. The aqueous phase was extracted multiple times with EtOAc and the combined organic layer was dried over Na2SO4. Following careful evaporation of the solvent, the residue was purified by column chromatography on silica gel using hexanes:EtOAc (1:1 then 1:2) as the eluent to yield the desired ketone 7 (12 mg, 94%) as a colorless oil. TLC: Rf = 0.32 (hexanes:EtOAc = 1:2); (c 0.86, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 5.04 (t, J = 6.6 Hz, 1H), 4.38 (t, J = 4.0 Hz, 1H), 4.20 (d, J = 17.0 Hz, 1H), 3.97 (d, J = 17.0 Hz, 1H), 2.85 (t, J = 8.4 Hz, 1H), 2.21 (d, J = 13.9 Hz, 1H), 2.15 (d, J = 15.2 Hz, 1H), 2.01 (ddd, J = 3.9, 10.5, 14.1 Hz, 1H), 1.95 (ddd, J = 4.5, 6.0, 15.1 Hz, 1H), 1.90 (br. s., 1H); 13C NMR (CDCl3, 100 MHz) δ 217.7, 84.0, 72.9, 70.8, 48.8, 43.4, 40.7.
(3S,3aS,5R,6aR)-3-Methoxyhexahydro-2H-cyclopenta[b]furan-5-ol (8)
Ketone 6 (35.6 mg, 0.14 mmol) was dissolved in EtOH (1 mL) under argon and cooled to −25 °C. To the solution was added NaBH4 (10 mg, 0.26 mmol) in one protion, and the resulting mixture was stirred at this temperature for 20 min. Saturated aqueous NH4Cl solution was added and the volume of solvent reduced under vacuum. Additional water was added and the aqueous phase was extracted several times with EtOAc. The combined organic layer was washed with brine, dried (Na2SO4), and evaporated in vacuo. Purification of the crude alcohol by column chromatography on silica gel using hexanes/EtOAc (5:1) as the eluent provided the corresponding alcohol (32.2 mg, 90%) as a colorless oil. TLC: Rf = 0.22 (hexanes/EtOAc = 5:1); + 21.5 (c 1.05, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 4.55 (br. s, 1H), 4.42 (t, J = 7.0 Hz, 1H), 4.34 (t, J = 4.5 Hz, 1H), 4.11 (m, 1H), 3.87 (d, J = 9.9 Hz, 1H), 3.48 (dd, J = 3.6, 9.9 Hz, 1H), 2.89 (dt, J = 7.8, 10.2 Hz, 1 H), 2.13 (dd, J = 2.4, 14.7 Hz, 1H), 1.99 (dd, J = 2.4, 14.7 Hz, 1H), 1.78–1.65 (m, 2 H), 0.89 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 85.3, 77.3, 74.9, 72.9, 48.1, 41.8, 34.8, 25.7, 18.0, −4.7, −5.2; HRMS-CI (m/z): [M + H]+ calcd for C13H27O3Si 259.1729, found 259.1731. To a solution of the above alcohol (27 mg, 0.1 mmol) in CH3CN (1 mL) was added MeI (0.3 mL, excess) followed by Ag2O (50 mg, 0.2 mmol). The mixture was gently refluxed for 12 h under argon then additional MeI (0.3 mL) and Ag2O (50 mg) were added. Reflux was continued for an additional 12 h until all SM disappeared. The resulting mixture was diluted in Et2O and filtered on celite. After evaporation of the solvent, the residue was purified by column chromatography on silica gel using hexanes/EtOAc (8:1) as the eluent to yield the corresponding 3-(S)-methoxy intermediate as a clear oil (23 mg, 85%). TLC: Rf = 0.38 (hexanes:EtOAc = 5:1); + 42.6 (c 1.02, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 4.41 (dt, J = 5.1, 7.2 Hz, 1H), 4.12 (m, 1H), 3.93 (m, 1H), 3.87–3.70 (m, 2H), 3.32 (s, 3H), 2.61 (m, 1H), 2.18 (m, 1H), 1.90–1.75 (m, 2H), 1.63 (m, 1H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (CDCl3, 75 MHz) δ 82.5, 81.1, 73.5, 69.0, 57.8, 42.6, 33.5, 25.8, 18.1, −4.8. To a solution of the above intermediate (16 mg, 0.059 mmol) in THF (1 mL) was added TBAF (1M solution in THF, 0.1 mmol, 100 μL). The solution was stirred at room temperature for 2 h. The solvent was evaporated and the residue purified by flash column chromatography on silica gel using hexanes/EtOAc (1:1 then 1:2.5) to furnish 8 as a colorless oil (8 mg, 86%). TLC: Rf = 0.32 (CHCl3/3% EtOH); 1H NMR (CDCl3, 500 MHz) δ 4.41 (t, J = 4.4 Hz, 1H), 4.21 (dt, J = 5.1, 10.1 Hz, 1H), 4.08 (d, J = 10.3 Hz, 1H), 3.85 (dd, J = 3.0, 7.3 Hz, 1H), 3.83 (d, J = 10.3 Hz, 1H), 3.42 (s, 3H), 2.94–2.86 (m, 1H), 2.13 (d, J = 14.5 Hz, 1H), 2.08 (dd, J = 2.4, 15.1 Hz, 1H), 1.81 (ddd, J = 5.3, 10.6, 14.5 Hz, 1H), 1.74 (ddd, J = 5.3, 6.0, 15.0 Hz, 1H); 13C NMR (CDCl3; 125 MHz) δ 86.0, 82.1, 73.6, 71.6, 57.4, 47.2, 42.0, 34.0.
Methyl 2-{(1R,4S)-4-[(tert-Butyldimethylsilyl)oxy]cyclopent-2-en-1-yloxy}-acetate (9)
To an ice-cold suspension of NaH (60% susp. oil, 360 mg, 9 mmol) in dry THF (5 mL) under argon was slowly added a solution of 2-bromoacetic acid (520 mg, 3.75 mmol) in dry THF (3 mL + 3 mL rinse). The resulting mixture was stirred at room temperature for 30 min then cooled back to 0 °C. Desymmetrized meso-cyclopentenediol 5 (642 mg, 3 mmol) in dry DMF (10 mL) was slowly added to this solution and the reaction was let warming to room temperature and stirred for 24 h. A pH 4 phosphate buffer solution was added at 0 °C and the aqueous phase was extracted with EtOAc (× 4). The combined organic phase was dried (Na2SO4), filtered, and evaporated in vacuo to give the corresponding crude carboxylic acid along with residual DMF. The acid was diluted with additional DMF (10 mL), the solution was cooled to 0 °C and NaHCO3 (630 g, 7.5 mmol) was added at once followed by MeI (14 mmol, 1.98 g, 871 μL). After stirring at room temperature for 12 h, saturated aqueous NH4Cl solution was added (10 mL), then water (10 mL), and the aqueous phase was successively extracted with Hexanes:Et2O 1:1 (× 3). The combined organic phase was dried (Na2SO4), filtered, and evaporated. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc (20:1 then 15:1) to give methyl ester 9 as a colorless oil (654 mg, 76%, 2 steps). TLC: Rf = 0.45 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 300 MHz) δ 5.92 (s, 2H), 4.63 (dd, J = 4.8, 7.0 Hz, 1H), 4.53 (dd, J = 5.1, 7.2 Hz, 1H), 4.10 (s, 2H), 3.74 (s, 3H), 2.64 (dt, J = 7.2, 13.8 Hz, 1H), 1.60 (dt, J = 4.9, 13.7 Hz, 1H), 0.87 (s, 9H), 0.065 (s, 3H), 0.06 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 171.2, 138.4, 131.9, 82.4, 74.6, 64.7, 51.8, 40.5, 25.8, 18.0, −4.7.
(3R,3aR,5R,6aR)-5-[(tert-Butyldimethylsilyl)oxy]hexahydro-2H-cyclopenta[b]furan-3-ol (10)
A solution of methyl ester 9 (87.2 mg, 0.304 mmol) in dry CH2Cl2 (8 mL) was cooled to − 78 °C under argon. DIBAL-H (1M sol. in hexanes, 0.40 mL) was added slowly and the reaction mixture was stirred for 1 h at this temperature. The reaction was quenched by addition of MeOH (100 μL) and the mixture was warmed to rt. A pH 7 phosphate buffer (0.5M solution, 2 mL) was added. The aqueous phase was extracted with CH2Cl2 (× 3) and the combined organic phase washed with brine, dried (Na2SO4), filtered, and evaporated in vacuo. The crude aldehyde solution was passed through a short plug of silica gel, prealably deactivated with 1% Et3N using hexanes/EtOAc (5:1) as the eluent. After evaporation, the crude product was directly submitted to the next step. The residue was dissolved in dry benzene (degassed, 2 mL) under argon and the solution transferred into a sealable tube. (nBu3Sn)2O (22.5 μL, 26.3 mg, 44 μmol), Ph2SiH2 (42 μL, 41.6 mg, 0.22 mmol), EtOH (45 μL), and AIBN (10 mg) were successively added. The sealed tube was placed with stirring in an oil bath at 80 °C for 6 h. After cooling to room temperature, the reaction mixture was diluted with Et2O (2 mL) and aqueous 0.5 M HCl solution (2 mL) was added. After stirring at room temperature for 15 min, the aqueous phase was extracted with Et2O (× 3). The combined organic phase was washed with saturated aqueous NaHCO3 solution, brine, dried (MgSO4), filtered, and evaporated. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc (6:1 to 2:1) to afford the (R)-alcohol 10 (50 mg, 64%, over 2 steps) as a white solid; 5 mg of the other 3-(S)-diastereoisomer was also isolated and only traces of 1,2 reduction product were observed. TLC: Rf = 0.46 (hexanes/EtOAc = 3:1); + 22.3 (c 0.67, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 4.69 (dt, J = 3.8, 7.2 Hz, 1H), 4.19–4.02 (m, 3H), 3.72 (d, J = 9.8 Hz, 1H), 2.48 (q, J = 7.5 Hz, 1H), 2.13–1.96 (m, 2H), 1.70–1.55 (m, 1H), 1.47–1.25 (m, 1H), 0.86 (s, 9H), 0.03 (s, 6H); 13C NMR (CDCl3, 75 MHz) δ 82.2, 78.3, 73.1, 72.7, 50.9, 42.4, 39.0, 25.8, 17.5, −4.8, −4.9.
(3R,3aS,5R,6aR)-5-Hydroxyhexahydro-2H-cyclopenta[b]furan-3-yl Acetate (11)
Alcohol 10 (45 mg, 0.175 mmol) was diluted in CH2Cl2 (5 mL), and the solution cooled to 0 °C under argon. Et3N (0.525 mmol, 53 mg, 75 μL) and DMAP (1 crystal) were added followed by acetic anhydride (25 μL, 0.23 mmol). The mixture was warmed to rt and stirred overnight. The solution was evaporated to dryness and purification of the residue by flash column chromatography on silica gel using hexanes/EtOAc (20:1) furnished the corresponding acetate derivative (38 mg, 73%). TLC: Rf = 0.24 (hexanes/EtOAc = 10:1); 1H NMR (CDCl3, 300 MHz) δ 5.02 (d, J = 3.8 Hz, 1H), 4.69 (dt, J = 3.2, 7.2 Hz, 1H), 4.22 (dd, J = 4.0, 10.4 Hz, 1H), 4.11 (m, 1H), 3.80 (d, J = 10.4 Hz, 1H), 2.56 (m, 1H), 2.09–1.97 (m, 2H), 2.04 (s, 3H), 1.71 (m, 1H), 1.68–1.54 (m, 1H), 0.86 (s, 9H), 0.03 (s, 6H); 13C NMR (CDCl3, 75 MHz) δ 170.8, 82.9, 81.2, 72.8, 70.9, 48.5, 42.7, 39.2, 25.8, 21.2, 18.0, −4.9. The acetate (28 mg, 0.093 mmol) was diluted in THF (1.5 mL), TBAF (1M sol. THF, 200 μL, 0.2 mmol) was added at 0 °C and the mixture was stirred for 2.5 h while warming to 23 °C. The solvent was evaporated and the residue purified by flash column chromatography on silica gel using hexanes/EtOAc (3:1 to 1:1) to give pure alcohol 11 as a colorless oil (17.5 mg, quant.). TLC: Rf = 0.18 (EtOAc 100%); 1H NMR (CDCl3, 300 MHz) δ 5.51 (m, 1H), 4.70–4.62 (m, 1H), 4.32 (dd, J = 5.0, 10.4 Hz, 1H), 4.28–4.20 (m, 1H), 3.71 (dd, J = 3.2, 10.4 Hz, 1H), 2.65–2.56 (m, 1H), 2.30–2.13 (m, 2H), 2.06 (s, 3H), 1.99–1.88 (m, 1H), 1.77–1.68 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 170.8, 84.8, 81.1, 73.4, 71.9, 48.9, 41.9, 39.5, 21.1.
(3R,3aS,5R,6aR)-3-Methoxyhexahydro-2H-cyclopenta[b]furan-5-ol (12)
A solution of alcohol 10 (30 mg, 0.116 mmol) in dry DMF (1 mL) was cooled to 0 °C under argon and NaH (60% suspension in oil, 15 mg, 0.348 mmol) was added in one portion. The mixture was warmed to r.t., stirred for 30 min then cooled to 0 °C. MeI (22 μL, 50 mg, 0.35 mmol) was added to the solution and the reaction was warmed to 23 °C and stirred for 3.5 h. Saturated aqueous NH4Cl solution was added and the aqueous phase was extracted with Et2O/hexanes (1:1). The combined organic phase was washed with brine, dried (Mg2SO4), filtered, and evaporated in vacuo. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc (10:1 then 8:1) to afford the corresponding TBS-protected methoxy compound (28.6 mg, 86%) as a clear oil. TLC: Rf = 0.61 (hexanes/EtOAc = 3:1); + 20.6 (c 1.52, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 4.63 (dt, J = 3.6, 7.2 Hz, 1H), 4.11 (m, 1H), 4.05 (dd, J = 4.1, 9.9 Hz, 1H), 3.81 (dd, J = 1.3, 9.9 Hz, 1H), 3.74 (m, 1H), 3.31 (s, 3H), 2.54 (td, J = 7.7, 8.3 Hz, 1H), 2.10–1.96 (m, 2H), 1.70–1.60 (m, 1H), 1.47 (m, 1H), 0.87 (s, 9H), 0.04 (6H), 13C NMR (CDCl3, 100 MHz) δ 87.7, 82.5, 72.9, 70.3, 56.6, 47.4, 42.5, 39.5, 25.8, 18.0, −4.8, −4.9. To an ice-cold solution of the above methoxy intermediate (27 mg, 0.1 mmol) in THF (2 mL) under argon was added TBAF (1M solution in THF, 0.2 mL, 0.2 mmol). The reaction was stirred for 1.5 h. The solvent was evaporated and the crude residue was purified by flash column chromatography on silica gel using hexanes/EtOAc (1:1) as the eluent to furnish alcohol 12 as a clear oil (13 mg, 82%). TLC: Rf = 0.17 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 300 MHz) δ 4.63 (t, J = 5.5 Hz, 1H), 4.26 (m, 1H), 4.20 (dd, J = 5.3, 9.7 Hz, 1H), 3.89 (dt, J = 2.0, 4.8 Hz, 1H), 3.65 (dd, J = 4.4, 9.7 Hz, 1H), 3.34 (s, 3H), 2.64–2.54 (m, 1H), 2.40 (br. s, 1H), 2.16 (ddd, J = 5.5, 10.6, 14.3 Hz, 1H), 2.02 (dd, J = 1.7, 14.7 Hz, 1H), 1.87 (dt, J = 5.1, 14.7 Hz, 1H), 1.77–1.61 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 88.4, 85.1, 74.0, 71.9, 57.0, 48.4, 41.8, 40.2.
(3S,3aR,5R,6aR)-3-Methylhexahydro-2H-cyclopenta[b]furan-5-ol (14)
To a solution of olefin 13 (29.7 mg, 0.12 mmol) in toluene (3 mL) was added Wilkinson's catalyst, RhCl(PPh3)3 (18 μmol, 17 mg). The resulting solution was then placed under a hydrogen atmosphere and stirred for 3 h. After dilution with additional toluene, the solution was filtered on a celite pad, and the pad was rinsed with toluene. Evaporation of the solvent and purification of the residue on silica gel using hexanes:EtOAc (100:1 then 50:1) as the eluent furnished the corresponding 3-(S)-methyl compound (29 mg, 97%). TLC: Rf = 0.28 (hexanes/EtOAc = 20:1); + 22.7 (c 1.01, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 4.43 (m, 1H), 4.03 (m, 1H), 3.81 (dd, J = 7.4, 8.0 Hz, 1H), 3.42 (dd, J = 8.0, 10.7 Hz, 1H), 2.43 (m, 1H), 2.32–2.18 (m, 2H), 1.72 (m, 1H), 1.51 (ddd, J = 4.8, 8.6, 13.5 Hz, 1H), 1.44 (m, 1H), 0.95 (d, J = 6.8 Hz, 3H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 82.6, 73.2, 71.6, 44.5, 43.2, 36.2, 34.7, 25.9, 18.2, 11.3, −4.7, −4.8; HRMS-CI (m/z): [M + H]+ calcd for C14H29O2Si 257.1937, found 257.1936. A solution of methyl-based ligand (28 mg, 0.1 mmol) in THF (2 mL) was treated with TBAF (1 M sol. in THF, 150 μL) and stirred at room temperature for 2.5 h. The reaction mixture was diluted with Et2O and filtered on a short silica pad. The solvent containing the alcohol was carefully reduced and essentially pure alcohol 14 was directly used to the next step without purification (>99%). TLC: Rf = 0.26 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 4.46 (dt, J = 2.7, 5.5 Hz, 1H), 4.20 (m, 1H), 3.88 (t, J = 7.8 Hz, 1H), 3.48 (t, J = 9.2 Hz, 1H), 2.59–2.40 (m, 2H), 2.22 (d, J = 6.9 Hz, 1H), 2.08 (dt, J = 5.9, 14.1 Hz), 1.87 (ddd, J = 6.0, 9.5, 13.7 Hz, 1H), 1.83 (m, 1H), 1.68–1.54 (m, 1H), 1.01 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 85.3, 73.8, 72.9, 46.3, 42.4, 36.5, 34.5, 12.7.
(3R,3aR,6aR)-3-Methyl-3,3a,6,6a-tetrahydro-2H-cyclopenta[b]furan-2-one (16)
To a solution of lithium diisopropylamide, prepared by adding nBuLi (2.5 M sol. in hexanes, 1.36 mL, 3.39 mmol) to diisopropylamine (477 μL, 3.39 mmol) in THF (15 mL) at 0 °C, was added a pre-cooled solution of known (3aS,6aR)-3,3a,6,6a-tetrahydro-2H-cyclopenta[b]furan-2-one (+)-15 (350 mg, 2.82 mmol) in THF (5 mL + 3 mL rinse) at −78 °C, dropwise. The reaction mixture was stirred at this temperature for 30 min, then methyl iodide (352 μL, 5.65 mmol) was added dropwise and the reaction mixture was stirred at −78 °C for 6 h. The reaction mixture was quenched with 2 M aqueous HCl. The aqueous phase was extracted with Et2O (× 3). The combined organic layer was dried (Na2SO4), filtered, and concentrated under vacuum. The residue was purified by column chromatography on silica gel using hexanes/Et2O (5:1) to yield lactone 16 (369 mg, 95%) as a brown oil. TLC: Rf = 0.54 (hexanes/EtOAc = 2:1); + 54.2 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 5.74 (m, 1H), 5.59 (m, 1H), 5.12 (t, J = 5.6 Hz, 1H), 3.13 (dd, J = 3.8, 1.8 Hz, 1H), 2.65 (m, 2H), 2.52 (q, J = 7.6 Hz, 1H), 1.33 (d, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 180.0, 130.9, 129.4, 81.3, 53.8, 39.9, 39.2, 17.4.
(3R,3aR,5R,6aR)-5-Hydroxy-3-methylhexahydro-2H-cyclopenta[b]furan-2-one (17)
To a ice-cold yellow solution of Hg(OAc)2 (1.45 g, 4.57 mmol) in THF:H2O (2.5:1 ratio, 23 mL) was added perchloric acid (~ 0.7 mL) until the solution became colorless. A solution of lactone 16 (350 mg, 2.54 mmol) in THF (7 mL) was then added at 0 °C and the reaction was stirred for 1 h. Additional Hg(OAc)2 (646 mg, 2.03 mmol) similarly pretreated with perchloric acid in THF:H2O (2.5:1, 10 mL) was added and stirring was continued for 2 h at 0 °C. The pH of the mixture was then adjusted to ~10 by addition of a 1M aqueous NaOH solution. Stirring was then continued for 1 h at room temperature. The solution was cooled to 0 °C and NaBH4 (145 mg, 3.81 mmol) was added in small portions. After 1 h, the reaction mixture was acidified to pH = 2 with concentrated HCl and stirring was continued for 1 h. The reaction solution was saturated with NaCl, and the aqueous phase extracted several times with EtOAc. The combined organic phase was dried (Na2SO4), filtered, and concentrated under vacuum. The residue was purified by column chromatography on silica gel using 2% MeOH in CH2Cl2 to yield alcohol 17 (252 mg, 64%) as a colorless oil. TLC: Rf = 0.31 (CHCl3/MeOH = 9/1); + 53.5 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 4.98 (dt, J = 8.0, 14.9 Hz, 1H), 4.55 (m, 1H), 2.69 (qd, J = 3.9, 7.6 Hz, 1H), 2.58 (m, 1H), 2.55 (m, 1H), 2.09 (d, J = 15.0 Hz, 1H), 1.94 (m, 2H), 1.83 (m, 1H), 1.26 (d, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 181.0, 83.8, 74.6, 51.2, 45.5, 43.9, 41.3, 18.4.
(3R,3aR,5R,6aR)-3-Methylhexahydro-2H-cyclopenta[b]furan-5-ol (18)
To a solution of lactone 17 (205 mg, 1.31 mmol) in CH2Cl2 (9 mL) was added DIBAL-H (1M sol. in CH2Cl2, 1.45 mL, 1.45 mmol) dropwise at −78 °C. The reaction was stirred for 3 h then quenched with saturated Rochelle's salt solution. After stirring overnight at room temperature, the phases were separated and the aquoues pahse was extracted with EtOAc (× 3). The combined organic phase was dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using 2% MeOH in CH2Cl2 to yield the corresponding lactol (150 mg, 72%). To a solution of this lactol (143 mg, 0.9 mmol) in CH2Cl2 (9 mL) at −78 °C was added TiCl4 (100 μL, 0.9 mmol) dropwise and the reaction was stirred for 20 min. Et3SiH (289 μL, 1.81 mmol) was then added and the solution was stirred for an additional 1 h. The reaction was quenched with saturated NaHCO3 solution once completion was reached as observed by TLC. The aqueous phase was extracted Et2O, dried (Na2SO4), filtered, and concentrated under reduced pressure at 25 °C. The residue was purified by column chromatography on silica gel using 2% MeOH in CH2Cl2 to yield alcohol 18 (122 mg, 95%) as a colorless oil. TLC: Rf = 0.38 (CHCl3/MeOH = 9/1); + 12.6 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 4.47 (t, J = 5.7 Hz, 1H), 4.22 (m, 1H), 4.06 (dd, J = 6.5, 8.7 Hz, 1H), 3.18 (t, J = 8.4 Hz, 1H), 2.60 (d, J = 7.2 Hz, 1H), 2.27–2.18 (m, 1H), 2.18–2.12 (m, 1H), 2.00 (dd, J = 1.5, 16.2 Hz, 1H), 1.98–1.91 (m, 1H), 1.80 (dt, J = 5.2, 14.6 Hz, 1H), 1.69 (ddd, J = 2.3, 5.4, 13.9 Hz, 1H), 1.03 (d, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 85.4, 75.1, 74.7, 50.3, 43.2, 41.6, 41.5, 17.6.
Synthesis of activated mixed carbonates
(3aS,5R,6aR)-3-Oxohexahydro-2H-cyclopenta[b]furan-5-yl (4-Nitrophenyl) Carbonate (19a)
To a solution of ketone 7 (20 mg, 0.14 mmol) in CH2Cl2 (1 mL) was added pyridine (30 μL). The solution was cooled to 0 °C under argon and 4 nitrophenyl chloroformate (42 mg, 0.21 mmol) was added at once. A white suspension formed. The solution was stirred at this temperature and slowly warmed to r.t., until all starting material was consumed. The reaction mixture was evaporated to dryness and the residue purified by column chromatography on silica gel using hexanes/EtOAc (3:1 then 2:1) as eluent to furnish pure mixed carbonate 19a (39 mg, 92%) as a white solid. TLC: Rf = 0.25 (hexanes/EtOAc = 2:1); 1H NMR (CDCl3, 400 MHz) δ 8.27 (d, J = 9.2 Hz, 2H), 7.34 (d, J = 9.2 Hz, 2H), 5.23 (t, J = 4.2 Hz, 1H), 5.13 (t, J = 6.9 Hz, 1H), 4.19 (d, J = 17.1 Hz, 1H), 4.06 (d, J = 17.1 Hz, 1H), 2.99 (dd, J = 8.7, 8.8 Hz, 1H), 2.54–2.41 (m, 2H), 2.22–2.08 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 216.8, 155.3, 151.5, 145.4, 125.3, 121.9, 82.4, 81.0, 70.6, 48.5, 41.2, 37.1.
(3S,3aS,5R,6aR)-3-Methoxyhexahydro-2H-cyclopenta[b]furan-5-yl (4-Nitrophenyl) Carbonate (19b)
The title compound was obtained from 8 in 82% yield as described for 7 after purification by column chromatography on silica gel using hexanes/EtOAc (3:1 to 2:1) as the eluent (white solid). TLC: Rf = 0.25 (hexanes/EtOAc = 2:1); 1H NMR (CDCl3, 300 MHz) δ 8.27 (d, J = 9.2 Hz, 2H), 7.38 (d, J = 9.2 Hz, 2H), 5.15 (tt, J = 4.5, 6.2 Hz, 1H), 4.56 (dt, J = 2.8, 6.1 Hz, 1H), 4.04 (dt, J = 6.3, 7.5 Hz, 1H), 3.87 (AB, dd, J = 5.2, 9.1 Hz, 1H), 3.86 (AB, dd, J = 6.6, 9.1 Hz, 1H), 3.34 (s, 3H), 2.83 (m, 1H), 2.38–2.02 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 155.6, 152.1, 145.2, 125.2, 121.8, 83.7, 81.4, 77.2, 70.3, 57.9, 44.2, 39.6; 30.2.
(3R,3aS,5R,6aR)-5-((4-Nitrophenoxy)carbonyloxy)hexahydro-2H-cyclopenta[b]furan-3-yl Acetate (19c)
The title compound was obtained from 11 in 96% yield as described for 7 after purification by column chromatography on silica gel using hexanes/EtOAc (6:1 then 4:1) as the eluent (white solid). TLC: Rf = 0.26 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 300 MHz) δ 8.27 (d, J = 9.1 Hz, 2H), 7.37 (d, J = 9.1 Hz, 2H), 5.20 5.12 (m, 1H), 5.12 5.08 (m, 1H), 4.76 (dt, J = 2.0, 6.0 Hz, 1H), 4.27 (dd, J = 4.6, 10.4 Hz, 1H), 3.80 (dd, J = 2.5, 10.4 Hz, 1H), 2.79–2.70 (m, 1H), 2.37 (ddd, J = 6.0, 10.2, 15.0 Hz, 1H), 2.29–2.19 (m, 1H), 2.15 (dt, J = 5.6, 15.6 Hz, 1H), 2.07 (s, 3H), 2.02–1.92 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 170.7, 155.4, 151.9, 145.3, 125.3, 121.8, 83.2, 81.0, 80.5, 71.7, 48.8, 39.6, 35.9, 21.1.
(3R,3aS,5R,6aR)-3-Methoxyhexahydro-2H-cyclopenta[b]furan-5-yl (4-Nitrophenyl) Carbonate (19d)
The title was obtained from 12 in 93% yield as described for 7 after purification by column chromatography on silica gel using hexanes/EtOAc (6:1 to 4:1) as the eluent (white solid). TLC: Rf = 0.40 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 400 MHz) δ 8.27 (d, J = 9.2 Hz, 2H), 7.37 (d, J = 9.2 Hz, 2H), 5.19–5.12 (m, 1H), 4.74–4.67 (m, 1H), 4.13 (dd, J = 5.4, 5.5 Hz, 1H), 3.84–3.78 (m, 2H), 3.35 (s, 3H), 2.75–2.68 (m, 1H), 2.36 (ddd, J = 6.1, 10.0, 14.8 Hz, 1H), 2.25–2.11 (m, 2H), 1.82 (dddd, J = 1.2, 3.8, 5.5, 14.7 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 155.5, 151.9, 145.3, 125.3, 121.8, 87.5, 83.0, 81.3, 71.3, 56.9, 48.2, 39.4, 36.2.
(3S,3aR,5R,6aR)-3-Methylhexahydro-2H-cyclopenta[b]furan-5-yl (4-Nitrophenyl) Carbonate (19e)
The title compound was obtained from 14 in 83% yield as described for 7 after purification by column chromatography on silica gel using hexanes/EtOAc (10:1 then 7:1) as the eluent (white solid). TLC: Rf = 0.25 (hexanes:EtOAc = 3:1); 1H NMR (CDCl3, 300 MHz) δ 8.27 (d, J = 9.0 Hz, 2H), 7.37 (d, J = 9.0 Hz, 2H), 5.09 (m, 1H), 4.57 (dt, J = 2.8, 6.2 Hz, 1H), 3.93 (t, J = 8.1 Hz, 1H), 3.51 (dd, J = 8.1, 10.4, 1H), 2.65 (m, 1H), 2.46 (m, 1H), 2.38 (m, 1H), 2.11 (m, 1H), 2.02 (ddd, J = 2.7, 5.5, 14.8 Hz, 1H), 1.81 (m, 1H), 1.02 (d, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ 155.5, 152.0, 145.3, 125.3, 121.8, 83.4, 80.8, 72.3, 45.8, 39.7, 36.5, 31.0, 11.8.
(3R,3aR,5R,6aR)-3-Methylhexahydro-2H-cyclopenta[b]furan-5-yl (4-Nitrophenyl) Carbonate (19f)
The title was obtained from 18 in 50% yield as described for 7 following column chromatography on silica gel using hexanes/EtOAc (10:1 then 7:1) as the eluent (white solid). TLC: Rf = 0.29 (hexanes/EtOAc = 3:1); ; 1H NMR (CDCl3, 400 MHz) δ 8.26 (d, J = 9.3 Hz, 2H), 7.37 (d, J = 9.3 Hz, 2H), 5.19–5.12 (m, 1H), 4.57 (dt, J = 2.0, 6.8 Hz, 1H), 4.07 (dd, J = 6.3, 8.6 Hz, 1H), 3.31 (dd, J = 7.1, 8.6 Hz, 1H), 2.35–2.25 (m, 1H), 2.23–2.17 (m, 2H), 2.17–2.07 (m, 2H), 1.88 (ddd, J = 3.8, 5.3, 14.5 Hz, 1H), 1.05 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 155.6, 151.9, 145.2, 125.2, 121.8, 83.3, 82.2, 74.9, 50.4, 41.8, 39.5, 37.3, 17.5.
General method for the synthesis of HIV-protease inhibitors
(3aS,5R,6aR)-3-Oxohexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (21)
The N-Boc-protected isostere 20a (82 mg, 0.16 mmol) was dissolved in a 2:1 mixture CH2Cl2/TFA and stirred at r.t. for 2 h. The solution was then evaporated and dried in vacuo. The residue was re-dissolved in CH3CN (1 mL) and the solution cooled down to 0 °C under argon. Et3N (100 μL) was added followed by a solution of activated carbonate 19a (40 mg, 0.13 mmol) in THF (1 mL). The solution was stirred for 36 h and the solution evaporated in vacuo. The residue was purified by column chromatography on silica gel using hexanes/EtOAc (3:1 to 1.5:1) as the eluent to furnish inhibitor 21 (46 mg, 61%) as a white solid. TLC: Rf = 0.23 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 500 MHz) δ 7.70 (d, J = 8.8 Hz, 2H), 7.34–7.28 (m, 2H), 7.25–7.19 (m, 3H), 6.98 (d, J = 8.8 Hz, 2H), 5.03 (t, J = 6.9 Hz, 1H), 4.99 (m, 1H), 4.71 (d, J = 8.7 Hz, 1H), 3.97 (s, 2H), 3.87 (s, 3H), 3.89–3.85 (m, 1H), 3.83–3.70 (m, 2H), 3.09 (dd, J = 8.0, 15.2 Hz, 1H), 3.00 (dd, J = 3.0, 15.1 Hz, 1H), 2.99–2.89 (m, 3H), 2.89–2.82 (m, 1H), 2.79 (dd, J = 6.8, 13.4 Hz, 1H), 2.24–2.12 (m, 2H), 2.06–1.94 (m, 2H), 1.81 (m, 1H), 0.90 (d, J = 6.4 Hz, 3H), 0.86 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 216.2, 163.0, 155.4, 137.5, 129.8, 129.6, 129.5, 128.5, 126.6, 114.3, 82.9, 76.3, 72.2, 70.6, 58.8, 55.6, 55.1, 53.7, 48.7, 41.2, 37.3, 35.1, 27.3, 20.1, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C29H38N2O8SNa 597.2247, found 597.2251.
(3S,3aR,5R,6aR)-3-Hydroxyhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-iso-butyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (22)
A solution of keto-inhibitor 21 (10 mg, 17 μmol) in EtOH was cooled to −25 °C and NaBH4 (6 mg) was added at once. The solution was stirred for 30 min then saturated aqueous NH4Cl solution was added. The aqueous phase was extracted with EtOAc (× 4). The combined organic layer was dried, filtered, and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using hexanes:EtOAc (2:1, 1:1 then 1:2) as the eluent to afford the desired inhibitor 22 (8 mg, 80%) as a white solid. TLC: Rf = 0.39 (hexanes/EtOAc = 1:5); 1H NMR (CDCl3, 500 MHz) δ 7.70 (d, J = 8.8 Hz, 2H), 7.32–7.19 (m, 5H), 6.97 (d, J = 8.8 Hz, 2H), 5.07 (m, 1H), 4.89 (d, J = 7.2 Hz, 1H), 4.38 (t, J = 6.8 Hz, 1H), 4.21 (m, 1H), 3.87 (s, 3H), 3.88–3.74 (m, 4H), 3.54 (dd, J = 3.2, 9.6 Hz, 1H), 3.10 (dd, J = 7.8, 15.0 Hz, 1H), 3.05–2.82 (m, 5H), 2.78 (dd, J = 6.8, 13.4 Hz, 1H), 2.42 (br.s, 1H), 2.18–2.04 (m, 2H), 1.94–1.76 (m, 3H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 163.0, 155.3, 137.6, 129.9, 129.6, 129.5, 128.6, 126.5, 114.3, 84.9, 78.1, 76.3, 73.0, 72.2, 58.7, 55.6, 55.3, 53.7, 47.3, 39.1, 35.4, 31.3, 27.2, 20.1, 19.9; LRMS-ESI (m/z): [M + Na]+ 599.3.
(3S,3aR,5R,6aR)-3-Hydroxyhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-4-(4-Amino-N-isobutylphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-yl]carbamate (23)
The ketone intermediate was first synthesized from the coupling of 19a with isostere 20b as described for 21 after stirring 48 h at r.t. in a CH2Cl2/THF (1:1) mixture. Purification of the crude compound by column chromatography on silica gel using Chloroform with 1% to 3% EtOH as the eluent afforded the corresponding ketone inhibitor (60%, white solid). TLC: Rf = 0.18 (Chloroform/3% EtOH); 1H NMR (CDCl3, 500 MHz) δ 7.54 (d, J = 8.6 Hz, 2H), 7.34–7.28 (m, 2H), 7.25–7.18 (m, 3H), 6.68 (d, J = 8.6 Hz, 2H), 5.02 (t, J = 6.9 Hz, 1H), 4.99 (m, 1H), 4.68 (d, J = 8.8 Hz, 1H), 4.14 (br. s, 2H), 3.98 (s, 2H), 3.91 (m, 1H), 3.84–3.72 (m, 2H), 3.08 (dd, J = 8.4, 15.1 Hz, 1H), 3.00–2.83 (m, 5H), 2.76 (dd, J = 8.7, 13.4 Hz, 1H), 2.22–2.15 (m, 2H), 2.05–1.97 (m, 2H), 1.80 (m, 1H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 216.9, 155.4, 150.6, 137.5, 129.6, 129.5, 128.5, 126.5, 126.3, 114.1, 82.9, 76.3, 72.2, 70.6, 58.9, 55.0, 53.8, 48.7, 41.2, 37.3, 35.2, 27.3, 20.2, 19.9; HRMS-ESI (m/z): [M+Na+] calcd for C28H37N3O7SNa 582.2250, found 582.2246. The title compound was obtained in 82% yield by reduction of the above ketone intermediate as described for 22, followed by column chromatography on silica gel using hexanes/EtOAc (1:2 to 1:5) as the eluent. Off-white solid. TLC: Rf = 0.12 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 500 MHz) δ 7.54 (d, J = 8.8 Hz, 2H), 7.34–7.18 (m, 5H), 6.68 (d, J = 8.8 Hz, 2H), 5.07 (m, 1H), 4.87 (d, J = 7.2 Hz, 1H), 4.38 (t, J = 6.8 Hz, 1H), 4.24–4.19 (m, 1H), 4.14 (s, 2H), 3.86 (br.s, 1H), 3.85–3.75 (m, 3H), 3.55 (dd, J = 3.6, 9.6 Hz, 1H), 3.10 (dd, J = 8.2, 15.0 Hz, 1H), 3.04–2.79 (m, 5H), 2.76 (dd, J = 6.8, 13.2 Hz, 1H), 2.37 (d, J = 11.6 Hz, 1H), 2.10 (t, J = 14.4 Hz, 2H), 1.94–1.75 (m, 3H), 0.90 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 155.3, 150.6, 137.6, 129.6, 129.5, 128.5, 126.5, 126.4, 114.1, 84.9, 78.0, 70.3, 73.0, 72.2, 58.8, 55.2, 53.7, 47.3, 39.1, 35.5, 31.3, 27.3, 20.2, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C28H39N3O7SNa 584.2406, found 584.2410.
(3S,3aR,5R,6aR)-3-Hydroxyhexahydro-2H-cyclopenta[b]furan-5-yl {(2S,3R)-3-Hydroxy-4-[4-(hydroxymethyl)-N-iso-butylphenylsulfonamido]-1-phenylbutan-2-yl}carbamate (24)
The ketone intermediate was first synthesized from the coupling of 19a with isostere 20c as described for 21. Purification of the crude compound by column chromatography on silica gel using hexanes/EtOAc (2:1 to 1:2) as the eluent afforded the corresponding ketone inhibitor in 49% yield as a white solid. TLC: Rf = 0.38 (hexanes/EtOAc = 1:3); 1H NMR (CDCl3, 500 MHz) δ 7.76 (d, J = 8.2 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.34–7.28 (m, 2H), 7.25–7.20 (m, 3H), 5.02 (t, J = 6.9 Hz, 1H), 4.96 (m, 1H), 4.79 (s, 2H), 4.67 (d, J = 8.4 Hz, 1H), 3.98–3.93 (m, 2H), 3.84–3.75 (m, 2H), 3.75–3.67 (m, 1H), 3.10 (dd, J = 8.2, 15.1 Hz, 1H), 3.07–2.80 (m, 5H), 2.83 (dd, J = 6.9, 13.5 Hz, 1H), 2.23–2.15 (m, 2H), 2.04–1.96 (m, 2H), 1.88–1.76 (m, 1H), 0.90 (dd, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H). The title compound was obtained in 83% yield by reduction of the above ketone intermediate as described for 22, followed by column chromatography on silica gel using hexanes/EtOAc (1:1 to 1:3 then 100% EtOAc) as the eluent. White solid. TLC: Rf = 0.23 (hexanes/EtOAc = 1:3); 1H NMR (CDCl3, 400 MHz) δ 7.76 (d, J = 8.2 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.34–7.27 (m, 2H), 7.24–7.20 (m, 3H), 5.06 (m, 1H), 4.82 (d, J = 7.8 Hz, 1H), 4.76 (s, 2H), 4.38 (t, J = 7.0 Hz, 1H), 4.21 (m, 1H), 3.85–3.68 (m, 4H), 3.55 (dd, J = 3.5, 9.8 Hz, 1H), 3.11 (dd, J = 8.3, 15.1 Hz, 1H), 3.05–2.79 (m, 6H), 2.38 (d, J = 8.3 Hz, 1H), 2.15–1.99 (m, 2H), 1.94–1.80 (m, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); HRMS-ESI (m/z): [M + Na]+ calcd for C29H40N2O8SNa 599.2403, found 599.2401.
(3S,3aS,5R,6aR)-3-Methoxyhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-iso-butyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (25)
The title compound was obtained in 73% yield from 19b and 20a as described for 21 following column chromatography on silica gel using hexanes/EtOAc (1:1) as the eluent. White solid. TLC: Rf = 0.21 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 500 MHz) δ 7.70 (d, J = 8.9 Hz, 2H), 7.33–7.19 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 4.97–4.90 (m, 1H), 4.78 (d, J = 7.5 Hz, 1H), 4.47–4.42 (m, 1H), 3.99 (q, J = 6.9 Hz, 1H), 3.87 (s, 3H), 3.88–3.82 (m, 2H), 3.81–3.76 (m, 2H), 3.70 (dd, J = 6.9, 9.0 Hz, 1H), 3.30 (s, 3H), 3.14–3.06 (dd, J = 7.8, 14.9 Hz, 1H), 3.04–2.88 (m, 4H), 2.78 (dd, J = 6.7, 13.4 Hz, 1H), 2.70 (m, 1H), 2.14 (m, 1H), 1.93 (dd, J = 6.3, 8.6 Hz, 2H), 1.86–1.76 (m, 2H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 163.0, 156.3, 137.5, 129.9, 129.6, 129.5, 128.5, 126.5, 114.3, 83.5, 81.3, 76.3, 72.3, 70.0, 58.7, 57.8, 55.6, 54.9, 53.7, 43.8, 39.5, 35.5, 30.4, 27.2, 20.1, 19.9; HRMS-ESI (m/z): [M + H]+ calcd for C30H43N2O8S 591.2740, found 591.2742.
(3R,3aR,5R,6aR)-3-Hydroxyhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-iso-butyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (26)
The acetyl intermediate was first synthesized in 84% yield by coupling of 19c with 20a as described for 21 followed by purification by column chromatography on silica gel using hexanes/EtOAc (3:1 to 1:1) as the eluent. White solid. TLC: Rf = 0.23 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 300 MHz) δ 7.71 (d, J = 8.9 Hz, 2H), 7.34–7.19 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 4.91 (m, 1H), 4.88 (m, 1H), 4.75 (d, J = 8.2 Hz, 1H), 4.67 (m, 1H), 3.98 (dd, J = 4.1, 10.4 Hz, 1H), 3.87 (s, 3H), 3.85–3.77 (m, 3H), 3.73 (dd, J = 1.5, 10.4 Hz, 1H), 3.19–2.92 (m, 4H), 2.90–2.72 (m, 2H), 2.67–2.55 (m, 1H), 2.23–2.09 (m, 1H), 2.06 (s, 3H), 2.04–1.76 (m, 3H), 1.50–1.43 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ 170.6, 163.0, 156.0, 137.6, 129.8, 129.5, 128.5, 126.6, 114.3, 83.5, 80.6, 76.1, 72.6, 71.6, 58.8, 55.6, 54.9, 53.7, 48.5, 39.6, 36.2, 35.7, 27.2, 21.1, 20.1, 19.9. The acetate intermediate (18 mg, 0.029 mmol) was diluted in MeOH (1.5 mL) at 0 °C. K2CO3 (5 mg, 0.04 mmol) was added and the solution was stirred for 6 h. Saturated aqueous NH4Cl solution (1 mL) and the solvent was reduced under vacuum. The aqueous phase was diluted and extracted with EtOAc (× 4). The combined organic phase was dried (MgSO4), filtered, and evaporated. The residue was purified by column chromatography on silica gel using chloroform/0.5% to 3% EtOH as the eluent to provide inhibitor 26 (15.9 mg, 94%) as a white solid. TLC: Rf = 0.26 (hexanes/EtOAc = 1:5); 1H NMR (CDCl3, 500 MHz) δ 7.71 (d, J = 8.9 Hz, 2H), 7.33–7.25 (m, 2H), 7.24–7.19 (m, 3H), 6.98 (d, J = 8.9 Hz, 2H), 4.86 (m, 1H), 4.81 (d, J = 8.3 Hz, 1H), 4.69 (t, J = 5.4 Hz, 1H), 4.01 (m, 1H), 3.91–3.76 (m, 4H), 3.87 (s, 3H), 3.64 (dd, J = 2.0, 9.7 Hz, 1H), 3.12 (dd, J = 8.2, 15.1 Hz, 1H), 3.11–3.05 (m, 1H), 3.03 (dd, J = 2.7, 15.2 Hz, 1H), 2.95 (dd, J = 8.3, 13.4 Hz, 1H), 2.85–2.75 (m, 2H), 2.52 (m, 1H), 2.12 (ddd, J = 6.1, 10.0, 14.7 Hz, 1H), 1.99 (dt, J = 6.1, 15.0 Hz, 1H), 1.93–1.79 (m, 3H), 1.35 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 163.0, 156.0, 137.8, 129.8, 129.5, 128.4, 126.4, 114.3, 83.1, 78.3, 76.3, 73.8, 72.7, 58.8, 55.6, 54.8, 53.7, 51.3, 39.5, 36.1, 35.8, 27.2, 20.1, 19.9; HRMS-ESI (m/z): [M + H]+ calcd for C29H41N2O8S 577.2584, found 577.2572.
(3R,3aR,5R,6aR)-3-Hydroxyhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-4-(4-Amino-N-iso-butylphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-yl]carbamate (27)
The acetyl intermediate was first synthesized in 63% yield by coupling of 19c with 20b as described for 21 followed by purification by column chromatography on silica gel using CHCl3/0.25% to 1.5 % EtOH as the eluent. White solid. TLC: Rf = 0.44 (hexanes/EtOAc = 1:3); 1H NMR (CDCl3, 500 MHz) δ 7.54 (d, J = 8.7 Hz, 2H), 7.32–7.27 (m, 2H), 7.25–7.19 (m, 3H), 6.67 (d, J = 8.7 Hz, 2H), 4.94 (m, 1H), 4.90–4.85 (m, 1H), 4.75 (d, J = 8.7 Hz, 1H), 4.66 (t, J = 5.4 Hz, 1H), 4.16 (br.s, 2H), 3.99 (dd, J = 4.1, 10.4 Hz, 1H), 3.88–3.77 (m, 3H), 3.74 (d, J = 10.3 Hz, 1H), 3.11 (dd, J = 8.5, 15.1 Hz, 1H), 3.05 (dd, J = 4.0, 14.1 Hz, 1H), 2.98 (dd, J = 1.5, 15.2 Hz, 1H), 2.93 (dd, J = 8.4, 13.2 Hz, 1H), 2.84 (dd, J = 8.8, 13.9 Hz, 1H), 2.77 (dd, J = 6.7, 13.3 Hz, 1H), 2.64–2.56 (m, 1H), 2.20–2.12 (m, 1H), 2.07 (s, 3H), 2.00 (dt, J = 6.1, 15.2 Hz, 1H), 1.95–1.88 (m, 1H), 1.81 (m, 1H), 1.51–1.44 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.6, 155.9, 150.6, 137.7, 129.5, 128.5, 126.5, 126.2, 114.1, 83.5, 80.6, 76.0, 72.6, 71.5, 58.9, 54.9, 53.8, 48.5, 39.6, 36.2, 35.7, 27.3, 21.1, 20.2, 19.9. The title compound was obtained from the above acetate intermediate in 88% yield as described for 26 following purification by column chromatography on silica gel using a gradient, 1% to 5% EtOH in CHCl3, as the eluent. White solid. TLC: Rf = 0.4 (CHCl3/10% EtOH); 1H NMR (CDCl3, 500 MHz) δ 7.55 (d, J = 8.7 Hz, 2H), 7.31–7.26 (m, 2H), 7.24–7.19 (m, 3H), 6.67 (d, J = 8.7 Hz, 2H), 4.89–4.83 (m, 1H), 4.79 (d, J = 8.9 Hz, 1H), 4.69 (t, J = 5.4 Hz, 1H), 4.16 (br.s, 2H), 4.01 (m, 1H), 3.88 (dd, J = 3.7, 9.9 Hz, 1H), 3.87–3.76 (m, 3H), 3.65 (dd, J = 1.4, 9.8 Hz, 1H), 3.14–3.04 (m, 2H), 2.99 (dd, J = 2.9, 15.1 Hz, 1H), 2.92 (dd, J = 8.3, 13.3 Hz, 1H), 2.81 (dd, J = 9.1, 14.0 Hz, 1H), 2.78 (dd, J = 6.8, 13.3 Hz, 1H), 2.54–2.47 (m, 1H), 2.11 (ddd, J = 6.2, 10.2, 14.6 Hz, 1H), 2.00 (dt, J = 6.1, 15.1 Hz, 1H), 1.93–1.85 (m, 1H), 1.85–1.76 (m, 2H), 1.35 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 156.0, 150.7, 137.8, 129.5, 128.4, 126.4, 126.2, 114.1, 83.1, 78.3, 76.2, 73.7, 72.7, 58.9, 54.8, 53.8, 51.3, 39.5, 36.0, 35.8, 27.3, 20.2, 19.9; HRMS-ESI (m/z): [M + H]+ calcd for C28H39N3O7S 584.2406, found 584.2398.
(3R,3aS,5R,6aR)-3-Methoxyhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-iso-butyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (28)
The title compound was obtained in 57% yield from 19d and 20a as described for 21 following purification by column chromatography on silica gel using hexanes/EtOAc (2:1 to 1:1) as the eluent. White solid. TLC: Rf = 0.34 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 500 MHz) δ 7.71 (d, J = 8.8 Hz, 2H), 7.33–7.25 (m, 2H), 7.26–7.20 (m, 3H), 6.98 (d, J = 8.8 Hz, 2H), 4.89 (m, 1H), 4.78 (d, J = 8.3 Hz, 1H), 4.61 (t, J = 5.7 Hz, 1H), 3.93–3.85 (m, 1H), 3.88 (s, 3H), 3.85–3.79 (m, 2H), 3.73 (d, J = 2.7, 9.8 Hz, 1H), 3.60 (m, 1H), 3.31 (s, 3H), 3.13 (dd, J = 8.3, 15.2 Hz, 1H), 3.10–3.00 (m, 2H), 2.95 (dd, J = 8.4, 13.4 Hz, 1H), 2.88–2.77 (m, 1H), 2.80 (dd, J = 7.0, 13.3 Hz, 1H), 2.62–2.53 (m, 1H), 2.19–2.09 (m, 1H), 1.99 (dt, J = 6.0, 15.1 Hz, 1H), 1.92 (m, 1H), 1.90–1.78 (m, 2H), 1.47–1.38 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 163.0, 156.1, 137.7, 129.9, 129.5, 128.5, 126.5, 114.3, 87.6, 83.3, 76.5, 72.7, 71.1, 58.8, 56.7, 55.6, 54.9, 53.7, 48.0, 39.5, 36.6, 35.8, 27.2, 20.1, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C30H42N2O8SNa 613.2560, found 613.2555.
(3S,3aR,5R,6aR)-3-Methylhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-iso-butyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (29)
The title compound was obtained from 19e and 20a in 78% yield as described for 21 following purification by column chromatography on silica gel using hexanes/EtOAc (3:1 to 2:1) as the eluent. White solid. TLC: Rf = 0.32 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 300 MHz) δ 7.71 (d, J = 8.9 Hz, 2H), 7.34–7.19 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 4.84 (m, 1H), 4.75 (d, J = 8.2 Hz, 1H), 4.49–4.40 (m, 1H), 3.87 (s, 3H), 3.86–3.76 (m, 4H), 3.32 (dd, J = 8.3, 10.5 Hz, 1H), 3.11 (dd, J = 7.8, 15.1 Hz, 1H), 3.06–2.84 (m, 4H), 2.79 (dd, J = 6.7, 13.4 Hz, 1H), 2.54–2.45 (m, 1H), 2.40–2.31 (m, 1H), 2.18 (dt, J = 6.5, 14.8 Hz, 1H), 1.90–1.77 (m, 2H), 1.74–1.66 (m, 1H), 1.50–1.40 (m, 1H), 0.91 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ 162.7, 156.3, 137.6, 129.8, 129.5, 128.5, 126.5, 114.3, 83.5, 76.0, 72.6, 72.2, 58.7, 55.6, 54.9, 53.7, 45.6, 39.8, 36.4, 35.6, 31.3, 27.2, 20.1, 19.8, 11.8. LRMS-ESI (m/z): [M + Na]+ 597.3, [M + H]+ 575.1.
(3R,3aR,5R,6aR)-3-Methylhexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (30)
The title compound was obtained from 19f and 20a in 96% yield as described for 21 following purification by column chromatography on silica gel using hexanes/EtOAc (5:1) as the eluent. White solid. TLC: Rf = 0.50 (hexanes/EtOAc = 1:1); (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 7.70 (d, J = 8.8 Hz, 2H), 7.30–7.16 (m, 5H), 6.96 (d, J = 8.8 Hz, 2H), 4.88 (m, 1H), 4.80 (d, J = 7.1 Hz, 1H), 4.46 (m, 1H), 3.91 (dd, J = 6.1, 8.5 Hz, 1H), 3.86 (s, 3H), 3.84 (m, 1H), 3.80 (m, 3H), 3.22 (dd, J = 7.5, 14.8 Hz, 1H), 3.15–2.99 (m, 3H), 2.94 (dd, J = 8.2, 13.4 Hz, 1H), 2.87–2.75 (m, 2H), 2.14 (m, 1H), 2.00–1.77 (m, 6H), 1.50 (d, J = 12.3 Hz, 1H), 0.98 (d, J = 6.6 Hz, 3H), 0.90 (d, J = 6.6 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 162.9, 156.2, 137.7, 129.8, 129.4, 128.4, 126.4, 114.3, 83.7, 77.2, 74.7, 72.5, 58.7, 55.6, 54.8, 53.7, 50.2, 41.8, 39.4, 37.7, 35.7, 27.2, 20.1, 19.8, 17.6.. LRMS-ESI (m/z): [M + Na]+ 597.1, [M + H]+ 575.3.
(3S,3aR,5R,6aR)-3-(Dimethylamino)hexahydro-2H-cyclopenta[b]furan-5-yl [(2S,3R)-3-Hydroxy-4-(N-iso-butyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl]carbamate (31)
To a solution of AcOH (~15 μL) in dichloromethane (1 mL), a slow stream of Me2NH gas was bubbled briefly at 0 °C for 5 min. After flushing the flask with argon, a solution of ketone inhibitor 21 (13 mg, 0.02 mmol) in dichloroethane (0.5 mL) was added over at 0 °C and after 15 min, NaBH(OAc)3 (10 mg, 0.05 mmol) was added. The resulting solution was warmed to room temperature. After 24 h, another 10 mg portion of NaBH(OAc)3 was added and the solution stirred for 48 h. The reaction was quenched by addition of saturated aqueous NaHCO3 solution adjusting the pH to 10 with 1M NaOH solution. The aqueous phase was extracted multiple times with EtOAc. The combined organic phase was dried (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using Ethanol (0.5 to 2 %) in CHCl3 to furnish the corresponding dimethylamine inhibitor 31 (11.3 mg, 82%) as a white solid. TLC: Rf = 0.35 (CHCl3/8% EtOH); 1H NMR (CDCl3, 500 MHz) δ 7.70 (d, J = 8.9 Hz, 2H), 7.32–7.26 (m, 2H), 7.25–7.19 (m, 3H), 6.97 (d, J = 8.9 Hz, 2H), 4.92 (m, 1H), 4.86 (m, 1H), 4.55 (m, 1H), 3.90–3.85 (m, 1H), 3.87 (s, 3H), 3.83–3.75 (m, 2H) 3.70–3.63 (m, 1H), 3.30–3.20 (m, 1H), 3.10 (dd, J = 3.14–3.06, 1H), 3.04–2.88 (m, 4H), 2.79 (dd, J = 6.9, 13.4 Hz, 1H), 2.64–2.55 (m, 1H), 2.23–2.18 (m, 1H), 2.24 (br.s., 6H), 2.18–2.10 (m, 1H), 2.06–1.97 (m, 1H), 1.88–1.75 (m, 2H), 0.90 (d, J = 6.6 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 163.0, 156.4, 137.6, 130.0, 129.6, 129.5, 128.5, 126.5, 114.3, 83.8, 77.2, 76.0, 72.3, 69.4, 58.7, 55.6, 55.0, 53.7, 45.4, 45.3, 40.1, 35.5, 31.4, 27.2, 20.1, 19.9; LRMS-ESI (m/z): [M + H]+ 604.3
Determination of X-ray Structure of HIV-1 Protease-Inhibitor Complex
The optimized HIV-1 protease was expressed and purified as described.23 The protease-inhibitor complex was crystallized by the hanging drop vapor diffusion method with well solutions of 1.2 M ammonium chloride, 0.1 M sodium acetate buffer (pH 4.8). Diffraction data were collected on a single crystal cooled to 90 K at the SER-CAT BM beamline 22, Advanced Photon Source, Argonne National Laboratory (Chicago, USA) with X-ray wavelength of 1.0 Å, and processed by HKL-200024 with Rmerge of 7.2%. The PR structure in was used in molecular replacement by PHASER25,26 in CCP4i Suite27,28 and refined to 1.45 Å resolution using SHELX-9729,30 and COOT31 for manual modification. PRODRG-232 was used to construct the inhibitor and the restraints for refinement. Alternative conformations were modeled, anisotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules, and hydrogen atoms were added in the final round of refinement. The final refined solvent structure comprised two Cl− ions and 142 water molecules. The crystallographic statistics are listed in Table 3 in the Supporting Information provided. The coordinates and structure factors of the PR with 26 complex have been deposited in Protein Data Bank33 with code 3ST5.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institutes of Health (GM53386, AKG and GM62920, ITW). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho.
Footnotes
† The PDB accession code for 26-bound HIV-1 protease X-ray structure is 3ST5.
aAbbreviations: bis-THF, bis-tetrahydrofuran; Cp-THF, hexahydrocyclopentafuran; PI, protease inhibitor; APV, amprenavir; DRV, darunavir; SQV, saquinavir; IDV, indinavir.
Supporting Information Available: HPLC, LRMS and HRMS data of inhibitors 21–31; crystallographic data collection and refinement statistics for inhibitor 26. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Palella FJ, Jr., Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N. Engl. J. Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- (2).Sepkowitz KA. AIDS — The First 20 Years. N. Engl. J. Med. 2001;344:1764–1772. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- (3).Gupta R, Hill A, Sawyer AW, Pillay D. Emergence of Drug Resistance in HIV Type 1-Infected Patients after Receipt of First-Line Highly Active Antiretroviral Therapy: A Systematic Review of Clinical Trials. Clin. Infect. Dis. 2008;47:712–722. doi: 10.1086/590943. [DOI] [PubMed] [Google Scholar]
- (4).Wainberg MA, Friedland G. Public Health Implications of Antiretroviral Therapy and HIV Drug Resistance. J. Am. Med. Assoc. 1998;279:1977–1983. doi: 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]
- (5).Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV Protease Inhibitors Targeting Protein Backbone: An Effective Strategy for Combating Drug Resistance. Acc. Chem. Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- (6).Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Bis-Tetrahydrofuran: a Privileged Ligand for Darunavir and a New Generation of HIV Protease Inhibitors that Combat Drug Resistance. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- (7).Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor (PI) UIC 94017 (TMC114) with Potent Activity against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob. Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ghosh A, Chapsal B, Mitsuya H. In: Aspartic Acid Proteases as Therapeutic Targets. Ghosh A, editor. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2010. pp. 245–262. [Google Scholar]
- (9).Ghosh AK, Dawson ZL, Mitsuya H. Darunavir, a Conceptually New HIV-1 Protease Inhibitor for the Treatment of Drug-resistant HIV Bioorg. Med. Chem. Lett. 2007;15:7576–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-Based Design of Novel HIV-1 Protease Inhibitors to Combat Drug Resistance. J. Med. Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- (11).Ghosh AK, Leshchenko-Yashchuk S, Anderson DD, Baldridge A, Noetzel M, Miller HB, Tie YF, Wang Y-F, Koh Y, Weber IT, Mitsuya H. Design of HIV-1 Protease Inhibitors with Pyrrolidinones and Oxazolidinones as Novel P1'-Ligands To Enhance Backbone-Binding Interactions with Protease: Synthesis, Biological Evaluation, and Protein-Ligand X-ray Studies. J. Med. Chem. 2009;52:3902–3914. doi: 10.1021/jm900303m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Koh Y, Das D, Leschenko S, Nakata H, Ogata-Aoki H, Amano M, Nakayama M, Ghosh AK, Mitsuya H. GRL-02031, a Novel Nonpeptidic Protease Inhibitor (PI) Containing a Stereochemically Defined Fused Cyclopentanyltetrahydrofuran Potent against Multi-PI-Resistant Human Immunodeficiency Virus Type 1 In Vitro. Antimicrob. Agents Chemother. 2009;53:997–1006. doi: 10.1128/AAC.00689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Deardorff DR, Windham CQ, Craney CL. Org. Synth., Coll. Vol. IX. 1998;9:487. [Google Scholar]
- (14).Ghosh AK, Chapsal BD, Baldridge A, Ide K, Koh Y, Mitsuya H. Design and Synthesis of Stereochemically Defined Novel Spirocyclic P2-Ligands for HIV-1 Protease Inhibitors. Org. Lett. 2008;10:5135–5138. doi: 10.1021/ol8020308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hays DS, Fu GC. Organotin Hydride Catalyzed Carbon-Carbon Bond Formation: Radical-Mediated Reductive Cyclization of Enals and Enones. J. Org. Chem. 1996;61:4–5. [Google Scholar]
- (16).Bueno JM, Coterón JM, Chiara JL, Fernández-Mayoralas A, Fiandor JM, Valle N. Stereoselective Synthesis of the Antifungal GM222712. Tetrahedron Lett. 2000;41:4379–4382. [Google Scholar]
- (17).Ghosh AK, Chapsal BD, Baldridge A, Steffey MP, Walters DE, Koh Y, Amano M, Mitsuya H. Design and Synthesis of Potent HIV-1 Protease Inhibitors Incorporating Hexahydrofuropyranol-Derived High Affinity P2 Ligands: Structure–Activity Studies and Biological Evaluation. J. Med. Chem. 2011;54:622–634. doi: 10.1021/jm1012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Toth MV, Marshall GR. A Simple Continuous Fluorometric Assay for HIV Protease. Int. J. Pept. Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- (19).Ghosh AK, Kulkarni S, Anderson DD, Hong L, Baldridge A, Wang Y-F, Chumanevich AA, Kovalevsky AY, Tojo Y, Amano M, Koh Y, Tang J, Weber IT, Mitsuya H. Design, Synthesis, Protein-Ligand X-ray Structure, and Biological Evaluation of a Series of Novel Macrocyclic Human Immunodeficiency Virus-1 Protease Inhibitors to Combat Drug Resistance. J. Med. Chem. 2009;52:7689–7705. doi: 10.1021/jm900695w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-High Resolution Crystal Structure of HIV-1 Protease Mutant Reveals Two Binding Sites for Clinical Inhibitor TMC114. J. Mol. Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gustchina A, Sansom C, Prevost M, Richelle J, Wodak SY, Wlodawer A, Weber IT. Energy Calculations and Analysis of HIV-1 Protease-Inhibitor Crystal Structures. Protein Eng. 1994;7:309–316. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- (22).Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular Basis for Substrate Recognition and Drug Resistance from 1.1 to 1.6 Å Resolution Crystal Structures of HIV-1 Protease Mutants with Substrate Analogs. FEBS J. 2005;272:5265–5277. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural Implications of Drug-Resistant Mutants of HIV-1 Protease: High Resolution Crystal Structures of the Mmutant Protease/ Substrate Analog Complexes. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- (24).Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr., Sweet RM, editors. Methods in Enzymology, 276: Macromolecular Crystallography, Part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- (25).Shen CH, Wang YF, Kovalevasky AY, Harrison RW, Weber IT. Amprenavir Complexes with HIV-1 Protease and its Drug-Resistant Mutants Altering Hydrophobic Clusters. FEBS J. 2010;277(18):3699–3714. doi: 10.1111/j.1742-4658.2010.07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Collaborative Computational Project Number 4 The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- (28).Potterton E, Briggs P, Turkenburg M, Dodson E. A Graphical User Interface to the CCP4 Program Suite. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- (29).Sheldrick GM. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- (30).Sheldrick GM, Schneider TR. SHELXL: High Resolution Refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- (31).Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (32).Schuettelkopf AW, van Aalten DMF. PRODRG; a Tool for High-Throughput Crystallography of Protein-Ligand Complexes. Acta Crystallogr, Sect. D: Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- (33).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.