

Abstract
Background. Dysfunction of polymorphonuclear leucocytes (PMNLs) in end-stage renal disease (ESRD) patients contributes to a diminished immune defence. The serum levels of leptin are elevated in patients with ESRD. We analysed in vitro effects of leptin on PMNLs from healthy subjects (HS; n = 12) and haemodialysis (HD) patients (n = 15) before and after HD.
Methods. PMNL oxidative burst and phagocytosis were tested by flow cytometry in whole blood. Chemotaxis of isolated PMNLs was assessed by the under-agarose method. To assess the involvement of leptin in PMNL signalling pathways, signal transduction inhibitors were used and the activity of intracellular kinases was investigated by western blotting, in vitro kinase assays and the Luminex technology.
Results. Increasing the leptin level in the blood of HS leads to a reduced activation of the oxidative burst by Escherichia coli and phorbol 12-myristate 13-acetate. Activation of the oxidative burst is reduced in the blood of HD patients and the addition of leptin does not lead to further PMNL inhibition. Leptin at a concentration measured in HD patients significantly reduces the chemotaxis of PMNLs from HS but had no effect on PMNLs from ESRD patients before and also after HD treatment with high-flux dialysers. The phosphoinositide 3-kinase/Akt pathway is involved in the inhibitory effects of leptin.
Conclusions. In the presence of leptin, PMNLs from HS and HD patients respond differently to stimuli. The lack of response to leptin in PMNLs from HD patients cannot be influenced by HD.
Keywords: haemodialysis, leptin, polymorphonuclear leucocyte functions, signal transduction
Introduction
Patients with chronic kidney disease (CKD) have an increased risk of morbidity and mortality. After cardiovascular disease, bacterial infections are the second major risk factor [1]. The disturbed non-specific immune system in CKD is mainly a consequence of disturbed polymorphonuclear leucocyte (PMNL) functions [2–4]. PMNLs, cells of the first-line non-specific immune defence, migrate to the site of infection along a chemotactic gradient; they ingest the invading microorganisms by phagocytosis and kill them with proteolytic enzymes and toxic oxygen radicals produced during the oxidative burst. Disturbances of any of those essential PMNL functions give rise to an increased risk for bacterial infections. For instance, PMNLs from haemodialysis (HD) patients have a lower intracellular killing capacity compared with controls [5, 6]. This may be attributed to a decreased oxidative burst upon stimulation [7, 8].
Various uraemic toxins, solutes that accumulate in patients with CKD and adversely affect essential biologic functions in general and the immune response in particular, have been previously reported [9–12]. Serum levels of adipokines, proteins secreted by adipocytes, are increased in uraemia [13]. We recently found that resistin attenuates chemotaxis and decreases the activation of the oxidative burst in PMNLs from healthy subjects (HS) [14]. The effect of the adipocytokine leptin, a signal of satiety to the brain and regulator of insulin and glucose metabolism, on PMNLs has not been clarified so far [15–18].
We tested the influence of leptin on essential PMNL functions and investigated the leptin effect on PMNL signalling. Because cells of CKD patients are exposed to the uraemic milieu and may respond differently compared with cells from HS, we included PMNLs from HD patients in our study.
Materials and methods
Materials
Human recombinant leptin was from BioVendor Laboratory Medicine (Brno, Czech Republic), saline solution (0.9% NaCl) from Fresenius Kabi (Graz, Austria), Giemsa stain from Merck (Darmstadt, Germany), polystyrene conical tubes and culture tissue dishes for chemotaxis assays from Becton Dickinson (Franklin Lakes, NJ), Ficoll-Hypaque from GE Healthcare (Uppsala, Sweden) and Hank's buffered saline solution from Gibco (Paisley, UK). Unless otherwise specified, all other chemicals were from Sigma-Aldrich, St Louis, MO.
Leptin serum concentrations
Leptin levels were measured by a sandwich enzyme immunoassay. A human leptin immunoassay (Quantakine) was used according to the instructions of the supplier (R&D Systems, Inc., Minneapolis, MN). The optical density was measured in a microplate reader (Anthos Reader HT 3; Anthos Labtec Instruments, Wals, Austria) at 450 nm.
Isolation of PMNLs
PMNLs were isolated from heparinized blood of 12 HS and 15 maintenance HD patients. Informed consent was given by all donors. For leucocyte function analysis, smokers and people taking medication or having infection or inflammation were excluded. Patients undergoing regular HD treatment with high-flux membranes (nitrocellulose triacetate from Nipro, Osaka, Japan, or polysulphone from Fresenius, Oberursel, Germany) had a Kt/V of 1.33 ± 0.05. Underlying diseases of the HD patients were diabetic, hypertensive or analgesics nephropathy, chronic glomerulonephritis and end-stage renal disease (ESRD) after acute renal failure induced by contrast medium. Four patients had ESRD of unknown origin. As previously described [9], discontinuous Ficoll-Hypaque density gradient centrifugation and hypotonic lysis of erythrocytes were used. The viability of the PMNLs obtained by this protocol was >95% as determined under the fluorescence microscope.
Chemotaxis
Chemotaxis was assessed by the under-agarose method as previously described [9]. The distance the PMNLs migrated under the agarose was determined under a microscope. To test the reversibility of the leptin effect, PMNLs were incubated in the presence of leptin at 37°C for 30 min. After washing the PMNLs twice, they were re-suspended in phosphate-buffered saline (PBS) and used for the chemotaxis assay.
Oxidative burst
PMNL oxidative burst was measured after adding 10 μL of leptin stock solutions or 10 μL PBS to 90 μL heparinized blood. Dihydrorhodamine 123 was used as fluorogenic substrate (Bursttest; Opregen Pharma, Heidelberg, Germany). After lysis of erythrocytes and fixation, the conversion of dihydrorhodamine 123 to rhodamine 123 by intracellularly produced H2O2 was quantified by flow cytometry (Epics XL-MCL; Coulter, Hialeah, FL). The mean fluorescence intensity without stimulus and leptin was set as 100%.
Inhibition experiments
The signal transduction inhibitors SB203580, PD98059 and LY294002 were purchased from Calbiochem (EMD Biosciences, Inc., Darmstadt, Germany) and used at final concentrations of 30, 50 and 10 μM, respectively. For the stock solutions, inhibitors were dissolved in dimethyl sulphoxide (DMSO). The end concentration of DMSO of 0.1 w/v % did not influence the results and was used as control.
Activity of intracellular kinases
Incubations.
PMNLs (3 × 106 in 100 μl) were incubated in the presence of leptin (70 ng/mL) or PBS alone at 37°C for 5 min. Twenty microlitres of PBS or 20 μL solution containing the stimulus was added and incubated for 5 min. For phorbol 12-myristate 13-acetate (PMA), the final concentration was 1.35 μM and for E. coli (from the Bursttest kit) 1.2–2.4 × 108 cells/mL.
Preparation of extracts.
PMNLs were taken up in 125 μL lysis buffer [20 mM Tris, pH 7.5; 150 mM NaCl; 1 mM ethylenediaminetetraacetic acid; 1 mM ethyleneglycol-bis(aminoethylether)-tetraacetic acid, 1% (v/v) Triton X-100; 2.5 mM sodium pyrophosphate; 1 mM Na3VO4; 1 μg/mL leupeptin; 1 mM phenylmethylsulfonyl fluoride]. Protease inhibitor cocktail (6.25 μL) was added, and cells were vortexed for 30 s, incubated on ice for 5 min and sonicated four times for 5 s each. PMNL extracts were centrifuged (4°C, 10 s, 10 000 g), and 15 μL supernatant was used for western blotting and 100 μL for the in vitro kinase assay.
In vitro kinase assay.
The Akt Kinase Assay Kit (Cell Signaling Technology, Beverly, MA) was used to measure the activity of Akt, a downstream effector of phosphoinositide 3-kinase (PI3K). Ten microlitres of immobilised anti-Akt antibody bead slurry was added to 100 μL cell lysate and incubated over night at 4°C. After centrifugation (4°C, 30 s, 14 000 g), the beads were washed twice in lysis buffer and twice in 250 μL kinase buffer (2.5 mM Tris, pH 7.5; 500 μM β-glycerolphosphate; 200 μM dithiothreitol; 10 μM Na3VO4; 1 mM MgCl2). The pellet was suspended in 10 μL kinase buffer containing 200 μM adenosine triphosphate and 1 μg kinase substrate [glycogen synthase kinase 3 (GSK-3) fusion protein] and incubated at 30°C for 30 min.
Western blotting.
After the preparation of the cell extracts, respectively after the in vitro kinase reaction, ½ volume of 3× sample buffer [187 mM Tris, pH 6.8; 6% (v/v) sodium dodecyl sulphate ; 0.03% (w/v) bromophenol blue; 7.5% (v/v) β-mercaptoethanol] was added to the lysate. Proteins were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (GE Healthcare) and electrotransferred to a nitrocellulose membrane. Polyclonal rabbit anti-p38 mitogen-activated protein kinase (MAPK), anti-phospho-p38 MAPK, anti-p44/42 MAPK, anti-phospho-p44/42 MAPK [anti-extracellular signal-regulated kinase (ERK)] and anti-phospho-GSK-3 antibodies (Cell Signaling Technologies) were used as primary antibodies and detected by a horseradish peroxidase-labelled goat-anti-mouse antibody and the enhanced chemiluminescence detection system (GE Healthcare). The bands were quantified with the Personal Densitometer SI (Molecular Dynamics) and the image master 1D software (Amersham Biosciences).
Luminex technology.
Total and phosphorylated kinases were also measured using the MILLIPLEX (Luminex) system (Millipore Corporation, Billerica, MA) according to the manufacturer's protocol. This flow cytometric assay uses monoclonal antibodies linked to microspheres labelled with different proportions of two fluorescent dyes allowing the detection of different kinases in one sample.
Glucose uptake
The basal and N-formyl-methionyl-leucyl-phenylalanine (fMLP)-stimulated glucose uptake by isolated PMNLs in the absence and presence of leptin was measured as previously described [11, 19].
Phagocytosis
The phagocytotic activity of PMNL was quantified in heparinized whole blood. Flow cytometry (Epics XL-MCL) was used to determine the percentage of PMNL that had taken up fluorescein isothiocyanate-labelled opsonized E. coli (Phagotest; Opregen Pharma) and the amount of ingested E. coli per PMNL in the absence and presence of leptin was determined by flow cytometry.
Statistical analysis
Wilcoxon matched-pair signed-rank statistic was used for data analysis of assays testing the PMNL functions influenced by leptin. Data are presented as mean values ± SEM. For non-paired data, we used the t-test.
Results
Leptin serum concentrations
Serum leptin levels were significantly elevated in HD patients as compared with HS (Figure 1). Whereas the leptin concentrations in healthy women were significantly higher than in healthy men, no gender difference was observed in HD patients (Figure 1). Of note, results gained in the PMNL functional assays did not differ for cells obtained from healthy females or males.
Fig. 1.

Serum levels of leptin. Leptin serum levels were measured in HS (A; n = 35, 16 males, 19 females) and in HD patients (B; n = 27, 14 males, 13 females). Mean values ± SEM. **P < 0.01 male versus female. $$P < 0.01 total HS versus HD.
PMNL oxidative burst
The basal levels of PMNL oxidative burst were not different in the blood of HS compared with HD patients before and after an HD session (data not shown). However, the E. coli- and PMA-stimulated oxidative burst was significantly lower in HD patients before HD treatment (Table 1). HD did not improve PMNL reactivity towards PMA, whereas E. coli stimulation was increased after HD but did not reach levels of HS (Table 1).
Table 1.
Escherichia coli- and PMA-stimulated oxidative burst of PMNLs from HS (n = 12) and patients before (PRE; n = 15) and after (POST; n = 15) an HD sessiona
Basal values are set as 1. Mean values ± SEM.
P < 0.01 versus HS.
P < 0.01 versus PRE.
Addition of leptin to blood from HS did not change the basal oxidative burst (data not shown) but attenuated the stimulation of the PMNL oxidative burst by E. coli (Figure 2A) or PMA (Figure 2B). Already, the lowest added amount of leptin (10 ng/mL) significantly reduced the stimulation by both E. coli and PMA. There was no stronger inhibition when the leptin levels were further increased. Increasing the leptin concentration in the blood of HD patients did not lead to further inhibition (Figures 2C and 2D).
Fig. 2.

Effect of leptin on PMNL oxidative burst. The oxidative burst was measured in PMNLs from HS (A and B) and from HD patients (C and D) stimulated by Escherichia coli (A and C) or by PMA (B and D). For (C) and (D): Values before (black bars) and after (grey bars) an HD session. Leptin was added to whole blood and thereby the leptin concentration increased by 10, 70 and 500 ng/mL. A total of 6–15 independent experiments were performed. Mean values ± SEM. *P < 0.05 versus 0 ng/mL.
The activation of the oxidative burst of PMNLs from HS (Figure 3A) and HD patients pre- and post-HD (Figure 3C) by E. coli was significantly diminished by SB203580 (an inhibitor of p38 MAPK) and LY294002 (an inhibitor of PI3K) alone indicating an involvement of p38 MAPK and PI3K in oxidative burst activation, whereas PD98059 had no impact. Inhibition of PI3K led to a strongly diminished stimulation of the oxidative burst by PMA in HS and HD patients pre- and post-HD (Figure 3B and D) pointing to a prominent role of the PI3K pathway in the stimulation of PMNLs with PMA. SB203580 and PD98059 decreased the PMA-stimulated oxidative burst of PMNLs obtained from HS. In contrast, this effect did not reach significance for HD patients before and after HD treatment (Figure 3C and D). Except for the inhibition of the PMA-stimulated oxidative burst by SB203580, leptin did not show a significant additional inhibitory effect.
Fig. 3.

Effect of signal transduction inhibitors on the PMNL oxidative burst in the absence and presence of leptin. The oxidative burst was measured in PMNLs from HS (A and B) and from patients on HD treatment isolated before (PRE) and after (POST) an HD session (C and D) stimulated by Escherichia coli (A and C) or by PMA (B and D) without (grey bars) or with (black bars) the addition of 70 ng/mL leptin and the signal transduction inhibitors SB203580 (SB; 30 μM), PD98059 (PD; 50 μM) and LY294002 (LY; 10 μM). Co: control in the absence of inhibitor. A total of four to seven independent experiments in the presence of inhibitors were performed. Mean values ± SEM. *P < 0.05 and **P < 0.01 versus control in the absence of leptin; $P < 0.05 and $$P < 0.01 versus the absence (grey bars) of leptin.
Activation of intracellular kinases
Escherichia coli and PMA induced a significant activation (phosphorylation) of p38 MAPK and ERK (p44/42 MAPK) in PMNLs of both HS and HD patients (Figures 4 and 5). Leptin did not change the degree of phosphorylation indicating that leptin has no effect on or upstream of p38 MAPK and ERK (p44/42 MAPK). These results obtained by western blotting were confirmed by using the Luminex technology (Figures 6 and 7).
Fig. 4.

Effect of leptin on p38 MAPK activation (western blotting). p38 MAPK (p38) and phospho-p38 MAPK (P-p38) were detected in extracts of PMNLs from HS (A and B) and from patients on HD treatment (C and D) stimulated by Escherichia coli (A and C) or by PMA (B and D) incubated in the presence of 70 ng/mL leptin or buffer alone. Representative western blots and the densitometric evaluation of the blots are shown. Mean values ± SEM. n = 3–7; *P < 0.05 and **P < 0.01 versus the absence of stimulus.
Fig. 6.

Effect of leptin on p38 MAPK activation (Luminex): p38 MAPK (p38) and phospho-p38 MAPK (P-p38) were detected in extracts of PMNLs from HS (A and B) and from patients on HD treatment (C and D) stimulated by Escherichia coli (A and C) or by PMA (B and D) incubated in the presence of 70 ng/mL leptin or buffer alone. The ratio between P-p38 and p38 was calculated and set as 1 for the absence of stimulus and leptin. Mean values ± SEM. n = 4; *P < 0.05 and **P < 0.01 versus the absence of stimulus.
Fig. 5.

Effect of leptin on p44/42 MAPK activation. p44/42 MAPK (p44/42) and phospho-p44/42 MAPK (P-p44/42) were detected in extracts of PMNLs from HS (A and B) and from patients on HD treatment (C and D) stimulated by Escherichia coli (A and C) or by PMA (B and D) incubated in the presence of 70 ng/mL leptin or buffer alone. Representative western blots and the densitometric evaluation of the blots are shown. Mean values ± SEM. n = 4–7; *P < 0.05 versus the absence of stimulus.
Fig. 7.

Effect of leptin on p44/42 MAPK activation (Luminex): p44/42 MAPK (p44/42) and phospho-p44/42 MAPK (P-p44/42) were detected in extracts of PMNLs from HS (A and B) and from patients on HD treatment (C and D) stimulated by Escherichia coli (A and C) or by PMA (B and D) incubated in the presence of 70 ng/mL leptin or buffer alone. The ratio between P-p44/42 and p44/42 was calculated and set as 1 for the absence of stimulus and leptin. Mean values ± SEM. n = 4; *P < 0.05 versus the absence of stimulus.
We determined the activity of Akt, a downstream effector of PI3K, by measuring the phosphorylation of GSK-3, a substrate of AKT. Leptin decreased the basal Akt activity in PMNLs of HS and HD patients (Figure 8). PMA did not change this inhibitory effect of leptin. However, in the absence of leptin, PMA significantly increased the Akt activity in cells from HS but not from HD patients suggesting that Akt phosphorylation is blocked in PMNLs from HD patients. Escherichia coli reduced the Akt activity in both PMNLs from HS and HD patients as a result of phosphatases activated during the incubation of PMNLs with E. coli (data not shown).
Fig. 8.
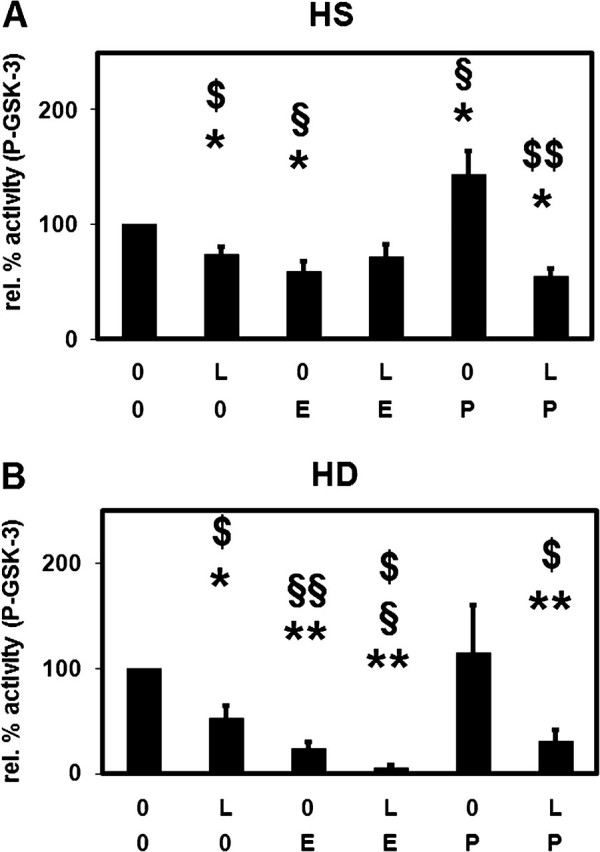
Effect of leptin on the stimulation of Akt activity. Phospho-GSK-3 (P-GSK-3) was detected in extracts of PMNLs from HS (A) and from patients on HD treatment (B) stimulated by Escherichia coli (E) or by PMA (P) incubated in the presence of 70 ng/mL leptin (L) or buffer alone (0). Mean values ± SEM. n = 3–6; *P < 0.05 and **P < 0.01 versus the absence of both stimulus and leptin (0; 0). $P < 0.05 and $$P < 0.01 versus the absence of leptin. §P < 0.05 and §§P < 0.01 versus the absence of stimulus.
Phosphorylation of IκBα
Nuclear factor-κB (NF-κB) is another important downstream effector of the PI3K/Akt pathway. Phosphorylation of NF-κB-bound IκBα, the major inhibitor of NF-κB, results in IκBα degradation and thereby activates NF-κB by allowing its nuclear translocation [20]. Therefore, the phosphorylation of IκBα is used as a measure for NF-κB activation. PMA induced the phosphorylation of IκBα in PMNLs from healthy donors but not from HD patients (Figure 9). This effect was abolished in the presence of leptin. These results are consistent with an involvement of Akt in NF-κB activation.
Fig. 9.
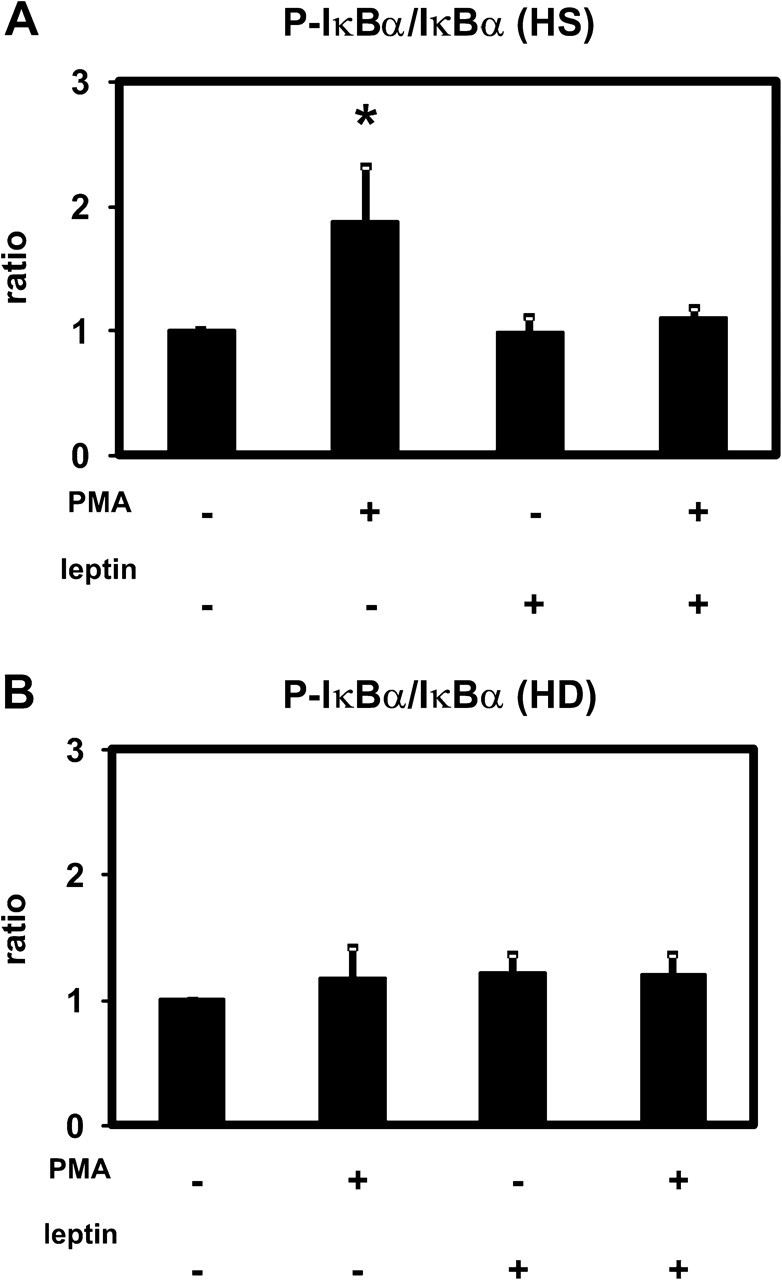
Effect of leptin on the phosphorylation of IκBα as a measure for NF-κB activation. IκBα and phospho-IκBα (P-IκBα) were detected by the Luminex technology in extracts of PMNLs from HS (A) and from patients on HD treatment (B) stimulated by PMA incubated in the presence of 70 ng/mL leptin or buffer alone. The ratio between P-p44/42 and p44/42 was calculated and set as 1 for the absence of stimulus and leptin. Mean values ± SEM. n = 4; *P < 0.05 versus the absence of stimulus.
Leptin effect on PMNL chemotaxis
Leptin at a uraemic concentration significantly attenuated chemotaxis of PMNLs isolated from HS (Figure 10A). When PMNLs were pre-incubated with leptin and the protein was subsequently removed, no inhibition of chemotaxis was observed (Figure 10A). The chemotaxis of PMNLs isolated from HD patients before or after dialysis was also not affected by leptin (Figure 10B). Because the absolute distances migrated by PMNLs from HS and HD patients were not different (data not shown), the absence of an effect of leptin on chemotaxis of PMNLs from HD patients cannot be explained by an already diminished migration of PMNLs from HD patients.
Fig. 10.

Effect of leptin on PMNL chemotaxis. (A) Chemotactic movement of PMNLs from HS in the presence of 70 ng/mL leptin (L) or after pre-incubation with 70 ng/mL leptin and subsequent removal (L r). Mean values ± SEM. n = 6. *P < 0.05 versus the absence of leptin (0). (B) Chemotactic movement of PMNLs from HD patients before (black bars) and after (grey bars) an HD session in the presence of 0, 10, 70 and 500 ng/mL leptin. Mean values ± SEM. n = 15.
Effects of leptin on PMNL glucose uptake and phagocytosis
fMLP stimulated the glucose uptake, a measurement of PMNL metabolic activity, 2.63-fold above basal level. Leptin had no effect on the glucose uptake of PMNLs from HS stimulated by fMLP (data not shown).
Approximately 90% of PMNLs from healthy donors took up opsonized E. coli bacteria in whole blood. Increasing the leptin concentration did not alter this value (data not shown). The number of E. coli ingested per PMNL did not differ when leptin levels were elevated (data not shown).
Discussion
Addition of leptin to the blood of HS leads to a diminished activation of the oxidative burst by E. coli and PMA. Increasing the leptin concentration in the blood of HD patients with an already reduced PMNL response to stimulation does not lead to further inhibition (Figures 2C and 2D). Leptin at a concentration measured in HD patients significantly reduces the chemotaxis of PMNLs from HS in a reversible manner but had no effect on PMNLs from HD patients (Figure 10). The PI3K/Akt pathway is involved in the leptin effect.
In the present study, we tested the effect of leptin on functions of PMNLs, cells of the first-line non-specific immune defence. Whereas low serum levels of leptin correlate to an increased incidence of infections related to malnutrition [21], leptin concentrations are elevated in the blood of ESRD patients, who have a high risk of bacterial infections [22]. Serum leptin levels correlate negatively with the glomerular filtration rate [23] but are also increased as a result of increased production in uraemia [24]. Hence, leptin is a potential uraemic toxin contributing to the disturbed immune response by interfering with PMNL functions presumably via interaction with a previously identified leptin receptor [15]. Obese people who have high leptin serum levels [25] have also an increased susceptibility to infections [26, 27]. Conversely, leptin production is acutely increased during infection and inflammation [28].
For the in vitro experiments, leptin levels were chosen according to those previously described [22] and determined in this study. We measured leptin serum concentrations of 17.8 ± 3.7 ng/mL (mean value ± standard deviation) for HS and 64.4 ± 12.1 ng/mL for HD patients (Figure 1). In agreement with the literature [29, 30], we found that leptin levels are higher in healthy females (29.5 ± 5.5 ng/mL) than males (4.0 ± 0.9 ng/mL), but no gender dependency in HD patients. Considering the circadian rhythm of leptin secretion in humans [31], it is important to state that all serum samples were obtained between 7 AM and 9 AM.
In our study, the oxidative burst stimulated by E. coli or PMA was significantly lower in HD patients compared with healthy controls. This is consistent with some previous reports [7, 8, 32], whereas others did not observe a different PMNL reactivity [33, 34]. There are also contradictory results regarding the basal levels of PMNL oxidative burst. Our group and others [8, 35] observed no difference between cells from HS and HD patients, whereas older studies found higher levels of basal reactive oxygen species (ROS) production in ESRD [36, 37]. Part of this discrepancy may be explained by the different methods used to assess the oxidative burst detecting different ROS intra- or extracellular in whole blood or in isolated PMNLs. Interestingly, Patruta et al. [32] showed that basal oxidative burst activity is significantly increased only in HD patients ‘over treated’ with iron or with iron overload.
There are a few inconsistent reports on the influence of leptin on PMNL oxidative burst. Ottonello et al. [17] and Montecucco et al. [18] found that leptin had no effect on the basal or fMLP-triggered oxidative burst. Caldefie-Chezet et al. [15] detected a significant increase in oxidative burst activity of PMNLs stimulated by PMA by leptin. In another study of the same group [16], no significant variation was seen for PMA-stimulated PMNLs incubated with leptin. We found that the stimulation of the oxidative burst was significantly attenuated when the concentration of leptin was increased in the blood of HS but not in the blood of HD patients. Already, the smallest increase in leptin levels significantly reduced the activation by both E. coli and PMA in HS. A further increase in leptin concentration did not result in a stronger inhibition. This seems plausible since the pathway affected by leptin is not the only one involved in the activation of the oxidative burst, i.e. other kinases (originally termed ‘novel kinases’) besides Akt are stimulated by PI3K during the activation of the oxidative burst [38]. As expected from the same resting levels of PMNL oxidative burst in HS and HD patients, increasing the leptin concentration did not change the basal cell activity.
In the hypothalamus, the appetite-controlling action of leptin is mediated by the long isoform of the leptin receptor (Ob-Rb) activating PI3K, MAPKs and the principal signal transduction pathway induced by leptin, activation of signal transducer and activator of transcription 3 (STAT 3) [39]. Only Ob-Rb mediates intracellular signalling via phosphorylation of STAT3 [40, 41]. PMNLs express only the short form of the leptin receptor (Ob-Ra) mediating signals into PMNLs via PI3K and p38 MAPK [42]. Since PI3K and MAPKs participate in several PMNL functions [43], we measured the activities of p38 MAPK, ERK (p44/42 MAPK) and Akt, a kinase that mediates PI3K-dependent phosphorylation of p47phox, a nicotinamide adenine dinucleotide phosphate oxidase component, and contributes to the activation of the oxidative burst in PMNLs [44].
Inhibition of PI3K led to a strongly diminished stimulation of the oxidative burst by PMA pointing to a prominent role of the PI3K pathway. Previous reports [38, 45] state that PI3K inhibition does not interfere with the stimulation of the oxidative burst. However, more recent publications show that LY294002 inhibits the PMA-stimulated oxidative burst [46] and protein kinase C activates PI3K via phosphorylation of the tyrosine kinase Syk [47]. Furthermore, protein kinase C activation contributes to PI3K/Akt activation in T cells [48].
In a following set of experiments, we directly tested whether leptin interferes with the activation of intracellular kinases. Escherichia coli and PMA induced a significant activation of p38 MAPK and ERK (p44/42 MAPK). The presence of leptin did not change the degree of the basal and E. coli- or PMA-induced phosphorylation of p38 MAPK and ERK (p44/42 MAPK) in PMNLs of HS and HD patients indicating that leptin has no effect on or upstream of p38 MAPK and ERK (p44/42 MAPK).
Figure 11 shows a simplified chart of signalling pathways compatible with the results obtained in inhibition and kinase activity experiments. In contrast to PMNLs, leptin activates the PI3K/Akt signalling pathway in several other cell types such as in human dendritic cells [49] and macrophages [50]. However, the absence of a functional Ob-Rb in these cells leads to a down-regulated activity of the PI3K/Akt pathway. This may explain the different signalling in PMNLs lacking Ob-Rb. Consistent with this explanation, leptin activates NF-κB, another important downstream effector of the PI3K/Akt pathway, in dendritic cells [51] but not in PMNLs (Figure 9). Escherichia coli reduced the Akt activity as a result of phosphatases activated during the incubation with PMNLs [14]. Therefore, the mechanism of E. coli stimulation could not be clearly defined at present. PMA significantly increased the Akt activity only in cells from healthy donors suggesting that Akt phosphorylation is hampered in patients' PMNLs and other kinases may mediate the PMA stimulatory effect (Figure 8). These differences in the activation of Akt by PMA (in the absence of leptin) point to differences in intracellular signalling between PMNLs from HS and HD patients. This further suggests that the lack of effect of increasing the leptin concentration in the blood of HD patients may be caused by an intrinsic defect of PMNLs rather than the already increased leptin level. Many of the observed effects may be explained by a different leptin receptor expression or activation. However, the fact that the major effect of leptin in our study, i.e. the inhibition of Akt activity, is the same for HS and HD patients argues against this explanation.
Fig. 11.

Oxidative burst induced by PMA in PMNLs: Simplified chart of signalling pathways compatible with the results obtained in inhibition and kinase activity experiments. SB: SB203580; PD: PD98059; LY: LY294002; MEK1,2: MAPK kinases.
Several partly contradictory reports describing the effect of leptin on PMNL chemotaxis have been published so far. Caldefie-Chezet et al. [15] and Ottonello et al. [17] both found that leptin per se acts as a chemoattractant for PMNLs. Whereas the former group showed that leptin increases PMNL migration as expressed by the chemotactic index, the latter described a dose-dependent inhibitory effect of leptin on PMNL chemotaxis applying the leading front method using blind well chambers. We found that leptin at a concentration measured in HD patients significantly reduces the chemotaxis of PMNLs from HS in a reversible manner. The absolute distances migrated by PMNLs from HS and HD patients were not different. Therefore, the absence of a leptin effect on chemotaxis of PMNLs from HD patients cannot be explained by an already diminished migration of patients' cells but rather by an insensitivity of patients' cells. Hence, in the case of chemotaxis, data from cells of HS may be representative for the immediate but not for the chronic response of PMNLs to leptin. In agreement with our present data, we previously showed that the uraemic retention solute para-hydroxy-hippuric acid affects PMNLs isolated from healthy donors and from HD patients differently [52].
In conclusion, our data suggest as a consequence of continuous exposure to the uraemic milieu, a difference in signal transduction between PMNLs from HS and HD patients. Leptin reduces the activity of Akt in both groups but had no effect on PMNLs from ESRD patients before and after effective HD treatment with high-flux membranes. Similar results have been obtained with para-hydroxy-hippuric acid [52] and resistin (Gerald Cohen, Jana Raupachova, Walter H. Hörl, unpublished observation). Therefore, results obtained with PMNLs from HS may reflect the response to an acute (local) exposure of PMNLs to uraemic toxins but cannot necessarily be extrapolated to the chronic effect of uraemic retention solutes. This has to be considered when interpreting data from in vitro experiments. Nevertheless, since leptin has been shown to be a mediator of the inflammatory response by activating other immune cells [53] that express the long isoform of the leptin receptor, decreasing the leptin concentration to normal levels remains a challenge in clinical practice.
Acknowledgments
Acknowledgments. This study was made possible by an unrestricted grant from Baxter Healthcare GmbH. The authors acknowledge the European Uraemic Toxin (EUTox) Work Group, a group of European researchers involved in studies and reviews related to uraemic toxicity. EUTox was created under the auspices of the European Society for Artificial Organs and is composed by 20 research groups throughout Europe.
Conflict of interest statement. None declared.
References
- 1.Hauser AB, Stinghen AE, Kato S, et al. Characteristics and causes of immune dysfunction related to uremia and dialysis. Perit Dial Int. 2008;28(Suppl 3):S183–S187. [PubMed] [Google Scholar]
- 2.Chonchol M. Neutrophil dysfunction and infection risk in end-stage renal disease. Semin Dial. 2006;19:291–296. doi: 10.1111/j.1525-139X.2006.00175.x. [DOI] [PubMed] [Google Scholar]
- 3.Mahajan S, Kalra OP, Asit KT, et al. Phagocytic polymorphonuclear function in patients with progressive uremia and the effect of acute hemodialysis. Ren Fail. 2005;27:357–360. [PubMed] [Google Scholar]
- 4.Sardenberg C, Suassuna P, Andreoli MC, et al. Effects of uraemia and dialysis modality on polymorphonuclear cell apoptosis and function. Nephrol Dial Transplant. 2006;21:160–165. doi: 10.1093/ndt/gfi095. [DOI] [PubMed] [Google Scholar]
- 5.Anding K, Gross P, Rost JM, et al. The influence of uraemia and haemodialysis on neutrophil phagocytosis and antimicrobial killing. Nephrol Dial Transplant. 2003;18:2067–2073. doi: 10.1093/ndt/gfg330. [DOI] [PubMed] [Google Scholar]
- 6.Jin H, Xu Q, Zhou P, et al. Impaired gp-91phox gene expression and dysfunction of peripheral blood neutrophils in patients maintaining hemodialysis. Chin Med J (Engl) 2000;113:120–123. [PubMed] [Google Scholar]
- 7.Shimazu T, Ominato M, Toyama K, et al. Effects of a vitamin E-modified dialysis membrane on neutrophil superoxide anion radical production. Kidney Int Suppl. 2001;78:S137–S143. doi: 10.1046/j.1523-1755.2001.59780137.x. [DOI] [PubMed] [Google Scholar]
- 8.Ritchey EE, Wallin JD, Shah SV. Chemiluminescence and superoxide anion production by leukocytes from chronic hemodialysis patients. Kidney Int. 1981;19:349–358. doi: 10.1038/ki.1981.26. [DOI] [PubMed] [Google Scholar]
- 9.Cohen G, Hörl WH. Retinol binding protein isolated from acute renal failure patients inhibits polymorphonuclear leucocyte functions. Eur J Clin Invest. 2004;34:774–781. doi: 10.1111/j.1365-2362.2004.01418.x. [DOI] [PubMed] [Google Scholar]
- 10.Cohen G, Rudnicki M, Deicher R, et al. Immunoglobulin light chains modulate polymorphonuclear leucocyte apoptosis. Eur J Clin Invest. 2003;33:669–676. doi: 10.1046/j.1365-2362.2003.01191.x. [DOI] [PubMed] [Google Scholar]
- 11.Cohen G, Rudnicki M, Walter F, et al. Glucose-modified proteins modulate essential functions and apoptosis of polymorphonuclear leukocytes. J Am Soc Nephrol. 2001;12:1264–1271. doi: 10.1681/ASN.V1261264. [DOI] [PubMed] [Google Scholar]
- 12.Cohen G, Rudnicki M, Hörl WH. Uremic toxins modulate the spontaneous apoptotic cell death and essential functions of neutrophils. Kidney Int Suppl. 2001;78:S48–S52. doi: 10.1046/j.1523-1755.2001.59780048.x. [DOI] [PubMed] [Google Scholar]
- 13.Diez JJ, Iglesias P, Fernandez-Reyes MJ, et al. Serum concentrations of leptin, adiponectin and resistin, and their relationship with cardiovascular disease in patients with end-stage renal disease. Clin Endocrinol (Oxf) 2005;62:242–249. doi: 10.1111/j.1365-2265.2005.02207.x. [DOI] [PubMed] [Google Scholar]
- 14.Cohen G, Ilic D, Raupachova J, et al. Resistin inhibits essential functions of polymorphonuclear leukocytes. J Immunol. 2008;181:3761–3768. doi: 10.4049/jimmunol.181.6.3761. [DOI] [PubMed] [Google Scholar]
- 15.Caldefie-Chezet F, Poulin A, Tridon A, et al. Leptin: a potential regulator of polymorphonuclear neutrophil bactericidal action? J Leukoc Biol. 2001;69:414–418. [PubMed] [Google Scholar]
- 16.Caldefie-Chezet F, Poulin A, Vasson MP. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic Res. 2003;37:809–814. doi: 10.1080/1071576031000097526. [DOI] [PubMed] [Google Scholar]
- 17.Ottonello L, Gnerre P, Bertolotto M, et al. Leptin as a uremic toxin interferes with neutrophil chemotaxis. J Am Soc Nephrol. 2004;15:2366–2372. doi: 10.1097/01.ASN.0000139321.98029.40. [DOI] [PubMed] [Google Scholar]
- 18.Montecucco F, Bianchi G, Gnerre P, et al. Induction of neutrophil chemotaxis by leptin: crucial role for p38 and Src kinases. Ann N Y Acad Sci. 2006;1069:463–471. doi: 10.1196/annals.1351.045. [DOI] [PubMed] [Google Scholar]
- 19.McCall AL, Gould JB, Ruderman NB. Diabetes-induced alterations of glucose metabolism in rat cerebral microvessels. Am J Physiol. 1984;247:E462–E467. doi: 10.1152/ajpendo.1984.247.4.E462. [DOI] [PubMed] [Google Scholar]
- 20.Ferreiro DU, Komives EA. Molecular mechanisms of system control of NF-kappaB signaling by IkappaBalpha. Biochemistry. 2010;49:1560–1567. doi: 10.1021/bi901948j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matarese G, La Cava A. The intricate interface between immune system and metabolism. Trends Immunol. 2004;25:193–200. doi: 10.1016/j.it.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Vanholder R, De Smet R, Glorieux G, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63:1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 23.Axelsson J, Bergsten A, Qureshi AR, et al. Elevated resistin levels in chronic kidney disease are associated with decreased glomerular filtration rate and inflammation, but not with insulin resistance. Kidney Int. 2006;69:596–604. doi: 10.1038/sj.ki.5000089. [DOI] [PubMed] [Google Scholar]
- 24.Aminzadeh MA, Pahl MV, Barton CH, et al. Human uraemic plasma stimulates release of leptin and uptake of tumour necrosis factor-alpha in visceral adipocytes. Nephrol Dial Transplant. 2009;24:3626–3631. doi: 10.1093/ndt/gfp405. [DOI] [PubMed] [Google Scholar]
- 25.Magni P, Liuzzi A, Ruscica M, et al. Free and bound plasma leptin in normal weight and obese men and women: relationship with body composition, resting energy expenditure, insulin-sensitivity, lipid profile and macronutrient preference. Clin Endocrinol (Oxf) 2005;62:189–196. doi: 10.1111/j.1365-2265.2005.02195.x. [DOI] [PubMed] [Google Scholar]
- 26.Falagas ME, Kompoti M. Obesity and infection. Lancet Infect Dis. 2006;6:438–446. doi: 10.1016/S1473-3099(06)70523-0. [DOI] [PubMed] [Google Scholar]
- 27.Lamas O, Marti A, Martinez JA. Obesity and immunocompetence. Eur J Clin Nutr. 2002;56(Suppl 3):S42–S45. doi: 10.1038/sj.ejcn.1601484. [DOI] [PubMed] [Google Scholar]
- 28.Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol. 2000;68:437–446. [PubMed] [Google Scholar]
- 29.Bossola M, Muscaritoli M, Valenza V, et al. Anorexia and serum leptin levels in hemodialysis patients. Nephron Clin Pract. 2004;97:C76–C82. doi: 10.1159/000078634. [DOI] [PubMed] [Google Scholar]
- 30.Heimburger O, Lonnqvist F, Danielsson A, et al. Serum immunoreactive leptin concentration and its relation to the body fat content in chronic renal failure. J Am Soc Nephrol. 1997;8:1423–1430. doi: 10.1681/ASN.V891423. [DOI] [PubMed] [Google Scholar]
- 31.Sinha MK, Ohannesian JP, Heiman ML, et al. Nocturnal rise of leptin in lean, obese, and non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 1996;97:1344–1347. doi: 10.1172/JCI118551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patruta SI, Edlinger R, Sunder-Plassmann G, et al. Neutrophil impairment associated with iron therapy in hemodialysis patients with functional iron deficiency. J Am Soc Nephrol. 1998;9:655–663. doi: 10.1681/ASN.V94655. [DOI] [PubMed] [Google Scholar]
- 33.Dahlgren C. Polymorphonuclear leukocyte chemiluminescence induced by formylmethionyl-leucyl-phenylalanine and phorbol myristate acetate: effects of catalase and superoxide dismutase. Agents Actions. 1987;21:104–112. doi: 10.1007/BF01974930. [DOI] [PubMed] [Google Scholar]
- 34.Lewis SL, Van Epps DE. Neutrophil and monocyte alterations in chronic dialysis patients. Am J Kidney Dis. 1987;9:381–395. doi: 10.1016/s0272-6386(87)80141-5. [DOI] [PubMed] [Google Scholar]
- 35.Paul JL, Roch-Arveiller M, Man NK, et al. Influence of uremia on polymorphonuclear leukocytes oxidative metabolism in end-stage renal disease and dialyzed patients. Nephron. 1991;57:428–432. doi: 10.1159/000186308. [DOI] [PubMed] [Google Scholar]
- 36.Perianayagam MC, Balakrishnan VS, Guo D, et al. Quantification of Bax and Bcl2 in polymorphonuclear leukocytes from haemodialysis patients: relation to hydrogen peroxide. Eur J Clin Invest. 2003;33:905–911. doi: 10.1046/j.1365-2362.2003.01225.x. [DOI] [PubMed] [Google Scholar]
- 37.Chen MF, Chang CL, Liou SY. Increase in resting levels of superoxide anion in the whole blood of uremic patients on chronic hemodialysis. Blood Purif. 1998;16:290–300. doi: 10.1159/000014347. [DOI] [PubMed] [Google Scholar]
- 38.Ding J, Vlahos CJ, Liu R, et al. Antagonists of phosphatidylinositol 3-kinase block activation of several novel protein kinases in neutrophils. J Biol Chem. 1995;270:11684–11691. doi: 10.1074/jbc.270.19.11684. [DOI] [PubMed] [Google Scholar]
- 39.Morris DL, Rui L. Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab. 2009;297:E1247–E1259. doi: 10.1152/ajpendo.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers MG., Jr. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res. 2004;59:287–304. doi: 10.1210/rp.59.1.287. [DOI] [PubMed] [Google Scholar]
- 41.Bjorbaek C, Uotani S, da Silva B, et al. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 42.Bruno A, Conus S, Schmid I, et al. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J Immunol. 2005;174:8090–8096. doi: 10.4049/jimmunol.174.12.8090. [DOI] [PubMed] [Google Scholar]
- 43.Coffer PJ, Geijsen N, M'Rabet L, et al. Comparison of the roles of mitogen-activated protein kinase kinase and phosphatidylinositol 3-kinase signal transduction in neutrophil effector function. Biochem J. 1998;329:121–130. doi: 10.1042/bj3290121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Q, Powell DW, Rane MJ, et al. Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J Immunol. 2003;170:5302–5308. doi: 10.4049/jimmunol.170.10.5302. [DOI] [PubMed] [Google Scholar]
- 45.Vlahos CJ, Matter WF, Brown RF, et al. Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. J Immunol. 1995;154:2413–2422. [PubMed] [Google Scholar]
- 46.Wrann CD, Tabriz NA, Barkhausen T, et al. The phosphatidylinositol 3-kinase signaling pathway exerts protective effects during sepsis by controlling C5a-mediated activation of innate immune functions. J Immunol. 2007;178:5940–5948. doi: 10.4049/jimmunol.178.9.5940. [DOI] [PubMed] [Google Scholar]
- 47.Popa-Nita O, Proulx S, Pare G, et al. Crystal-induced neutrophil activation: XI. Implication and novel roles of classical protein kinase C. J Immunol. 2009;183:2104–2114. doi: 10.4049/jimmunol.0900906. [DOI] [PubMed] [Google Scholar]
- 48.Querfeld C, Rizvi MA, Kuzel TM, et al. The selective protein kinase C beta inhibitor enzastaurin induces apoptosis in cutaneous T-cell lymphoma cell lines through the AKT pathway. J Invest Dermatol. 2006;126:1641–1647. doi: 10.1038/sj.jid.5700322. [DOI] [PubMed] [Google Scholar]
- 49.Mattioli B, Giordani L, Quaranta MG, et al. Leptin exerts an anti-apoptotic effect on human dendritic cells via the PI3K-Akt signaling pathway. FEBS Lett. 2009;583:1102–1106. doi: 10.1016/j.febslet.2009.02.029. [DOI] [PubMed] [Google Scholar]
- 50.Maya-Monteiro CM, Almeida PE, D'Avila H, et al. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem. 2008;283:2203–2210. doi: 10.1074/jbc.M706706200. [DOI] [PubMed] [Google Scholar]
- 51.Lam QL, Liu S, Cao X, et al. Involvement of leptin signaling in the survival and maturation of bone marrow-derived dendritic cells. Eur J Immunol. 2006;36:3118–3130. doi: 10.1002/eji.200636602. [DOI] [PubMed] [Google Scholar]
- 52.Cohen G, Raupachova J, Wimmer T, et al. The uraemic retention solute para-hydroxy-hippuric acid attenuates apoptosis of polymorphonuclear leukocytes from healthy subjects but not from haemodialysis patients. Nephrol Dial Transplant. 2008;23:2512–2519. doi: 10.1093/ndt/gfn098. [DOI] [PubMed] [Google Scholar]
- 53.Fernandez-Riejos P, Najib S, Santos-Alvarez J, et al. Role of leptin in the activation of immune cells. Mediators Inflamm. 2010 doi: 10.1155/2010/568343. doi:10.1155/2010/568343. [DOI] [PMC free article] [PubMed] [Google Scholar]