

Abstract
Hypoxia is an essential developmental and physiological stimulus that plays a key role in the pathophysiology of cancer, heart attack, stroke, and other major causes of mortality. Hypoxia-inducible factor 1 (HIF-1) is the only known mammalian transcription factor expressed uniquely in response to physiologically relevant levels of hypoxia. We now report that in Hif1a−/− embryonic stem cells that did not express the O2-regulated HIF-1α subunit, levels of mRNAs encoding glucose transporters and glycolytic enzymes were reduced, and cellular proliferation was impaired. Vascular endothelial growth factor mRNA expression was also markedly decreased in hypoxic Hif1a−/− embryonic stem cells and cystic embryoid bodies. Complete deficiency of HIF-1α resulted in developmental arrest and lethality by E11 of Hif1a−/− embryos that manifested neural tube defects, cardiovascular malformations, and marked cell death within the cephalic mesenchyme. In Hif1a+/+ embryos, HIF-1α expression increased between E8.5 and E9.5, coincident with the onset of developmental defects and cell death in Hif1a−/− embryos. These results demonstrate that HIF-1α is a master regulator of cellular and developmental O2 homeostasis.
Keywords: Cardiovascular development, glycolysis, knockout, oxygen, VEGF
Among the most extensively developed physiologic systems are those devoted to O2 homeostasis, which reflects the human body’s constant and absolute requirement for this essential molecule. In particular, mammals have evolved circulatory systems of considerable complexity to ensure that every cell of these large organisms receives sufficient O2 for normal metabolic function. Circulation of blood via the heart and vasculature represents the first physiologic system that is established within the developing embryo. Failure of cardiac, erythroid, or vascular development results in lethality of mouse embryos at mid-gestation, suggesting that O2 delivery may become diffusion limited at this stage, but the degree to which early embryonic development is influenced by tissue hypoxia is unknown. In contrast, there is abundant evidence that in postnatal life hypoxia elicits multiple cellular and systemic physiologic responses, including angiogenesis, erythropoiesis, and glycolysis (for review, see Guillemin and Krasnow 1997). In addition, hypoxia is a pathophysiologic component of many disorders, including heart attack, stroke, and cancer, the major causes of mortality in Western societies. Understanding the molecular mechanisms by which cells sense and respond to hypoxia may provide the basis for novel therapeutic approaches to these disorders.
A growing number of adaptive responses to hypoxia that are understood at the molecular level involve transcriptional activation of gene expression by hypoxia-inducible factor 1 (HIF-1), a heterodimeric basic helix–loop–helix–PAS domain (bHLH–PAS) transcription factor (Wang et al. 1995). mRNAs encoding the HIF-1α and HIF-1β subunits were detected in all human and rodent tissues assayed (Wenger et al. 1996; Wiener et al. 1996). HIF-1 is unique among mammalian transcription factors with respect to the demonstrated specificity and sensitivity of its induction by hypoxia. Expression of the HIF-1α subunit, which is unique to HIF-1, is precisely regulated by cellular O2 concentration such that levels of HIF-1α protein and HIF-1 DNA-binding activity increased exponentially as O2 concentration decreased (Jiang et al. 1996b). HIF-1α transactivation domain function was also hypoxia inducible, an effect that was independent of HIF-1α protein levels (Jiang et al. 1997b; Pugh et al. 1997). In contrast to the specificity of HIF-1α, the HIF-1β subunit, which was identified previously as the aryl hydrocarbon receptor nuclear translocator (ARNT) (Hoffman et al. 1991), also heterodimerizes with several other mammalian bHLH–PAS proteins including the Aryl hydrocarbon receptor (Dolwick et al. 1993), Single-minded-2 (Moffett et al. 1997), and a protein that has been designated EPAS1 (Tian et al. 1997), HLF (Ema et al. 1997), HRF (Flamme et al. 1997), MOP2 (Hogenesch et al. 1997), or HIF-2α (Wenger and Gassmann 1997).
HIF-1 was identified originally by its binding to a hypoxia response element in the human erythropoietin (EPO) gene that was required for transcriptional activation in response to reduced cellular O2 concentration (Semenza and Wang 1992). Subsequently, hypoxia response elements containing functionally essential HIF-1 binding sites with the consensus sequence 5′-RCGTG-3′ (Semenza et al. 1996) were identified in genes encoding transferrin, vascular endothelial growth factor (VEGF), inducible nitric oxide synthase (iNOS), heme oxygenase 1 (HO1), glucose transporter 1 (GLUT1), and the glycolytic enzymes aldolase A, enolase 1, lactate dehydrogenase A, phosphofructokinase L, and phosphoglycerate kinase 1 (for review, see Wenger and Gassmann 1997). Each of these proteins plays an important role in systemic, local, or intracellular O2 homeostasis: EPO increases blood O2-carrying capacity by stimulating erythropoiesis; transferrin delivers iron to the bone marrow for incorporation into hemoglobin; VEGF mediates vascularization; iNOS and HO1 synthesize NO and CO, respectively, which modulate vascular tone; and induction of GLUT1 and glycolytic enzymes allows for increased anaerobic ATP synthesis.
Although the evidence supporting a role for HIF-1α in the transcriptional activation of EPO, VEGF, and other hypoxia-inducible genes in cultured cells is compelling, its involvement in embryonic development has not been determined. The recent identification of the related protein HIF-2α (EPAS/HLF/HRF/MOP2), which can dimerize with HIF-1β and recognize HIF-1 binding sites, has also called into question the unique role of HIF-1α, especially with respect to control of VEGF expression and embryonic vascularization (Ema et al. 1997; Flamme et al. 1997; Hogenesch et al. 1997; Tian et al. 1997). The defects reported in ARNT-deficient mouse embryos (Maltepe et al. 1997) are also difficult to interpret at the transcriptional level because ARNT (HIF-1β) has multiple potential dimerization partners, including HIF-2α, and because the related ARNT2 protein (Hirose et al. 1996) is a potential alternative dimerization partner for HIF-1α. To definitively establish the role of HIF-1 in O2 homeostasis, we have generated Hif1a−/− embryonic stem (ES) cells and mice that lack expression of HIF-1α.
Results
Generation of HIF-1α-deficient ES cells
A targeting vector was designed to disrupt the mouse Hif1a gene by homologous recombination resulting in the replacement of exon 2 by a PGK promoter–neomycin resistance (neo) gene (Fig. 1A). Recombination results in deletion of Hif1a sequences encoding the bHLH domain, which is essential for dimerization of HIF-1α with HIF-1β and binding of HIF-1 to DNA (Jiang et al. 1996a), and introduces termination codons in all three reading frames. Splicing of exon 1 to exon 3 would result in a frameshift and disruption of the coding sequence, thus predicting the generation of a null allele. After electroporation and selection of ES cells in the presence of G418 and gancyclovir, three independent ES cell clones, 7, 19, and H7, were established that were heterozygous for the recombinant allele (Hif1a+/−) as determined by DNA blot hybridization using a 3′ probe not included within the targeting vector (Fig. 1B, lane 2). Hybridization with a neo probe indicated a single homologous recombination event, and karyotype analysis revealed a normal chromosome constitution within clone 7 and 19 cells, whereas H7 cells contained an additional chromosome (data not shown). Clone 7 and 19 cells were utilized for in vivo studies, whereas in tissue culture experiments, clone H7 and 19 cells were analyzed and gave identical results. ES cell clones were subjected to increased G418 selection, and subclones were generated that had converted to homozygosity (Hif1a−/−) for the recombinant allele (Fig. 1B, lane 3).
Figure 1.


Targeted disruption of the Hif1a gene by homologous recombination in ES cells. (A) Structure of HIF-1α protein, wild-type Hif1a locus, targeting vector, and targeted Hif1a locus. Important functional domains in the protein are the bHLH and PAS domains, which are required for dimerization and DNA binding, and the transactivation domains (TAD). Homologous recombination (large crosses) results in replacement of exon 2 with a neomycin resistance gene in the opposite transcriptional orientation (arrow). Targeted clones are G418 and gancyclovir resistant owing to the presence of the neomycin-resistance gene and the absence of the thymidine kinase gene. (B) DNA blot hybridization analysis of ES cell clones. The wild-type and targeted loci are identified by the presence of 14- and 12-kb EcoRV restriction fragments, respectively, that are detected by the 3′ probe shown in A. (C) HIF-1 immunoblot analysis of Hep3B and ES cells. Nuclear extracts were prepared from cells cultured under nonhypoxic (N) (20% O2) or hypoxic (H) (1% O2) conditions for 4 hr. Immunoblot assays were performed using affinity-purified antibodies specific for HIF-1α (top) or HIF-1β (bottom). (D) Immunoblot analysis of HIF-1 expression in Hif1a+/+, Hif1a+/-, and Hif1a−/− ES cells. Nuclear extracts were prepared from cells cultured under nonhypoxic (N) or hypoxic (H) conditions for 4 hr. Immunoblot assays were performed using antibodies specific for HIF-1α (top), HIF-1β (middle), or a control (C) protein, topoisomerase I (bottom). (E) Electrophoretic mobility-shift assay of HIF-1 DNA-binding activity. Nuclear extracts from ES and Hep3B cells were incubated with a double-stranded oligonucleotide probe containing an 18-bp EPO gene sequence. Binding of HIF-1 and a constitutively expressed factor (C) is indicated.
Immunoblot analysis of ES cell nuclear extracts was performed to determine HIF-1α and HIF-1β protein levels. Wild-type (Hif1a+/+) ES cells differed from Hep3B human hepatoblastoma cells and other cell lines analyzed previously by the presence of abundant HIF-1α protein under nonhypoxic conditions (Fig. 1C, top panel). In response to hypoxia (exposure to 1% O2 for 4 hr), there was only a modest increase in HIF-1α protein levels in ES cells, but a mobility shift suggestive of a post-translational modification such as phosphorylation was apparent. In contrast, HIF-1α protein levels increased dramatically in hypoxic Hep3B cells. HIF-1β protein levels increased modestly in response to hypoxia in both Hep3B and ES cells (Fig. 1C, bottom panel). Changes in HIF-1β levels may reflect greater protein stability in the context of a HIF-1α:HIF-1β heterodimer, as forced expression of HIF-1α is sufficient to increase HIF-1β levels (Jiang et al. 1996a). Compared with Hif1a+/+ ES cells, HIF-1α protein levels were reduced in Hif1a+/− cells, and no HIF-1α protein was detectable in Hif1a−/− cells (Fig. 1D, top panel). HIF-1β levels progressively decreased in Hif1a+/− and Hif1a−/− cells (Fig. 1D, middle panel). Analysis of a control nuclear protein, topoisomerase I, revealed constant levels of expression under all conditions (Fig. 1D, bottom panel).
To analyze the effect of differences in Hif1a genotype on HIF-1 DNA-binding activity, aliquots of the same nuclear extracts were analyzed by electrophoretic mobility-shift assay using a double-stranded oligonucleotide probe containing the HIF-1 binding site from the EPO gene (Semenza and Wang 1992). HIF-1 DNA-binding activity was present in nuclear extracts prepared from nonhypoxic Hif1a+/+ ES cells but not Hep3B cells (Fig. 1E). In response to hypoxia, high levels of HIF-1 activity were detected in Hif1a+/+ ES and Hep3B cells. Hif1a+/− ES cells manifested diminished HIF-1 activity, and no HIF-1 activity was detected in Hif1a−/− cells. In contrast, a constitutively expressed DNA-binding activity was present in all nuclear extracts assayed. Several conclusions follow from these results. First, mouse ES cells are unique among cell lines studied to date based on their high level of HIF-1α expression under nonhypoxic conditions. Second, homologous recombination disrupted Hif1a gene expression leading to partial and complete loss of HIF-1α protein in Hif1a+/− and Hif1a−/− ES cells, respectively. These results indicate that the targeted gene, designated Hif1atm1jhu, is a null allele. Third, HIF-1 DNA-binding activity was not detected in Hif1a−/− ES cells, indicating the absence of any other protein capable of dimerizing with HIF-1β and recognizing a HIF-1 binding site.
Effect of HIF-1α deficiency on O2-regulated gene expression in ES cells
To analyze the involvement of HIF-1α in the transcriptional regulation of hypoxia-inducible genes, Hif1a+/+, Hif1a+/−, and Hif1a−/− ES cells were exposed to 20% or 1% O2 for 16 hr prior to isolation of total RNA for Northern blot hybridization using probes against mRNAs encoding 15 different glucose transporters and glycolytic enzymes spanning the entire pathway from glucose internalization to lactate production. In Hep3B cells, expression of most of these mRNAs was induced by hypoxia, whereas in Hif1a+/+ ES cells, expression under nonhypoxic conditions was high, and hypoxia had little effect on mRNA expression (Fig. 2). The differences in mRNA expression in the two cell lines thus paralleled the differences in HIF-1α expression documented in Figure 1. Four different patterns of mRNA expression were detected in ES cells: (1) GLUT1, GLUT3, ALDA, PGK1, and ENO1 mRNA levels were all modestly increased in hypoxic Hif1a+/+ cells, whereas in Hif1a+/− cells there was no hypoxic induction, and in Hif1a−/− cells mRNA levels were decreased under both hypoxic and nonhypoxic conditions. (2) Hypoxia did not affect the levels of HK1, HK2, and PKM mRNA in Hif1a+/+ cells, but in Hif1a+/− and Hif1a−/− cells mRNA levels decreased in response to hypoxia. (3) PFKL, ALDC, TPI, GAPDH, and LDHA mRNA levels also did not increase in hypoxic Hif1a+/+ cells, but mRNA levels were decreased in Hif1a−/− cells under both hypoxic and nonhypoxic conditions. (4) GPI and PGM mRNA levels were not induced by hypoxia in Hif1a+/+ cells, and HIF-1α deficiency had no reproducible effect on expression.
Figure 2.

Expression of genes encoding glucose transporters and glycolytic enzymes. The glycolytic pathway is shown at left. Symbols for genes encoding the respective enzymes are coded by font according to the mRNA expression pattern (normalized to 18S rRNA) in ES cells cultured under nonhypoxic (N) or hypoxic (H) conditions for 16 hr (lanes 1–6 at right) as follows: (1) (bold) increased expression in hypoxic Hif1a+/+ cells, loss of induction in Hif1a+/− cells, and loss of basal and induced expression in Hif1a−/− cells; (2) (bold and italicized) no effect of hypoxia on expression in Hif1a+/+ cells but decreased expression in hypoxic Hif1a+/− and Hif1a−/− cells; (3) (italicized) no effect of hypoxia on expression in Hif1a+/+ cells but decreased expression in hypoxic and nonhypoxic Hif1a−/− cells; (4) (plain) no effect of hypoxia or HIF-1α deficiency on expression. mRNA expression in Hep3B cells was also assayed (lanes 7,8). The indicated genes encode the following proteins: (GLUT1 and GLUT3) glucose transporter 1 and 3; (HK1 and HK2) hexokinase 1 and 2; (GPI) glucosephosphate isomerase; (PFKL) phosphofructokinase L; (ALDA and ALDC) aldolase A and C; (TPI) triosephosphate isomerase; (GAPDH) glyceraldehyde-3-phosphate dehydrogenase; (PGK1) phosphoglycerate kinase 1; (PGM) phosphoglucomutase; (ENO1) enolase 1; (PKM) pyruvate kinase M; (LDHA) lactate dehydrogenase A. GLUCOSE (EXT) and GLUCOSE (INT) refer to extracellular and intracellular glucose, respectively.
Each filter was subsequently stripped and rehybridized with an 18S rRNA probe to demonstrate the presence of equal amounts of RNA in each lane (data not shown). In addition, autoradiographs were serially hybridized with different probes. For example, the LDHA blot shown was stripped and rehybridized to generate the PKM blot shown. Therefore, the different patterns of expression reflect varying effects of HIF-1α deficiency on the expression of genes encoding glycolytic enzymes. These results indicate that HIF-1 coordinately regulates the expression of at least 13 genes encoding proteins involved in the anaerobic synthesis of ATP by conversion of extracellular glucose to intracellular lactate. Furthermore, this regulation is complex because in some cases (groups 2 and 3), it appears that the level of gene expression represents the net effect of multiple positive and/or negative regulators such that HIF-1 is required to maintain, rather than to induce, gene expression in hypoxic ES cells. As further evidence of the specific effects of HIF-1α deficiency, expression of ODC mRNA (encoding ornithine decarboxylase) was induced to the same degree in hypoxic Hif1a+/+, Hif1a+/−, and Hif1a−/− ES cells (data not shown). Therefore, hypoxia-inducible ODC gene expression is not HIF-1α-dependent. Expression of other hypoxia-inducible genes (encoding adrenomedullin, cyclo-oxygenase-2, 5′-ecto-nucleotidase, endothelin-1, EPO, HO1, iNOS, platelet-derived growth factor-B, transferrin, and transforming growth factor-β1) was demonstrated in Hep3B cells but could not be detected in wild-type or HIF-1α-deficient ES cells (data not shown).
VEGF mRNA expression in ES cells was detected by blot hybridization and quantitated by densitometric analysis of autoradiographs. The results were corrected for differences in sample preparation or loading by normalizing to results obtained by analysis of 18S rRNA. VEGF mRNA expression was induced by hypoxia in Hif1a+/+ but not in Hif1a−/− ES cells (Fig. 3A). Compared with Hif1a+/+ cells, normalized VEGF mRNA levels were 4.4- and 10.2-fold lower in nonhypoxic and hypoxic Hif1a−/− cells, respectively. In contrast, VEGF mRNA expression increased 2.9- and 4.4-fold in glucose-deprived Hif1a+/+ and Hif1a−/− cells, respectively, indicating that this response is not HIF-1α dependent. These data are consistent with the finding that neither HIF-1α protein nor HIF-1 DNA-binding activity was induced by glucose deprivation of Hif1a+/+ ES cells (data not shown). ES cells were also differentiated into cystic embryoid bodies by suspension culture in methylcellulose for 5 days. In response to hypoxia, VEGF mRNA levels increased in Hif1a+/+ embryoid bodies as described previously (Gassmann et al. 1996) but not in Hif1a−/− embryoid bodies (Fig. 3B). Taken together with previous studies (Semenza et al. 1994, 1996; Forsythe et al. 1996), Figures 2 and 3 demonstrate that HIF-1α is required for O2-regulated transcriptional activation of genes encoding glycolytic enzymes and VEGF.
Figure 3.

Expression of VEGF mRNA in ES cells and cystic embryoid bodies. (A) ES cells. Hif1a+/+ and Hif1a−/− cells were incubated in complete medium at 20% O2 (N), complete medium at 1% O2 (H), or glucose-deficient medium at 20% O2 (−G) for 16 hr. Total RNA was isolated and analyzed by blot hybridization with 32P-labeled VEGF cDNA (top) and 18S rRNA oligonucleotide (bottom) probes. (B) Embryoid bodies. ES cells were cultured for 4 days in methylcellulose to induce differentiation and then exposed to 20% (N) or 0% (H) O2 for 16 hr prior to RNA isolation and blot hybridization as described above.
Effect of HIF-1α deficiency on ES cell proliferation
To analyze the general involvement of HIF-1α in cellular physiology, the growth rates of Hif1a+/+ and Hif1a−/− ES cells were determined under nonhypoxic (20% O2) and hypoxic (1% O2) conditions. Equal numbers of cells of each genotype were plated in multiple paired sets. After 24 hr at 20% O2, one set of plates was removed, and cells were counted (“0 hr” time point). Triplicate sets of Hif1a+/+ and Hif1a−/− cells were then cultured either at 20% or 1% O2 for an additional 24 or 48 hr prior to enumeration by trypan blue staining of live and dead cells present both in the medium and attached to the dishes. The culture medium contained 25 mm HEPES and 25 mm glucose and was changed every 24 hr to minimize changes in pH and glucose concentration. The experiment was repeated three times, and mean cell numbers (relative to 0 hr) were determined. When cultured in the presence of 1% O2, the number of Hif1a−/− cells was significantly reduced relative to Hif1a+/+ cells at both 24 and 48 hr (Fig. 4). In addition, there was a significant, but less marked, difference in the number of cells after 48 hr in the presence of 20% O2. This is consistent with the expression of HIF-1 in Hif1a+/+ cells at 20% O2 (Fig. 1) and the effect of HIF-1α deficiency on glycolytic gene expression under nonhypoxic conditions (Fig. 2). There was no significant difference in the degree of cell death between genotypes (data not shown), suggesting that the differences in cell number reflect differences in rates of cell division.
Figure 4.

Growth of ES cells under nonhypoxic and hypoxic culture conditions. Hif1a+/+ (+) or Hif1a−/− (−) cells (6 × 105) were plated per 10-cm dish. After 24 hr, one plate of each genotype was removed for cell counting (0 hr). The remaining plates were cultured under nonhypoxic (N; 20% O2) or hypoxic (H; 1% O2) conditions. The medium was changed after 24 hr and, for each condition, three plates each were removed after 24 or 48 hr for cell counts, which were normalized to the 0-hr cell count. Four fields were counted from each plate, and the experiment was performed three times. The mean (n = 36) and s.d. (bar) for each experimental condition were calculated, and results for Hif1a−/− cells that were significantly different from Hif1a+/+ cells are indicated: (*) P < 0.05; (**) P < 0.01 (Student’s t-test).
Generation and analysis of HIF-1α-deficient mice
To analyze the effect of HIF-1α deficiency on embryonic development, clone 7 and clone 19 Hif1a+/− ES cells were each injected into blastocysts from C57BL/6 mice to produce chimeras, and for each ES cell clone, germ-line transmission was obtained when chimeric (F0) males were mated to C57BL/6 females. Hif1a+/− F1 mice were identified by DNA blot hybridization analysis, and heterozygous littermates were mated. Among the 113 F2 offspring of Hif1a+/− × Hif1a+/− matings that were genotyped at 4 weeks of age, 34.4% were Hif1a+/+, 65.5% were Hif1a+/−, and none were Hif1a−/−, a clear departure from the expected 1:2:1 Mendelian ratio. Among 54 offspring of Hif1a+/+ × Hif1a+/− matings, 51.8% were Hif1a+/+ and 48.1% were Hif1a+/−, which was not significantly different from the expected 1:1 ratio. These data indicate that whereas the viability of Hif1a+/− mice was not reduced, Hif1a−/− mice were not viable.
Analysis of timed matings between heterozygotes revealed the presence of morphologically abnormal embryos as early as E8.5 (Table 1A). Between E8.75 and E10.0, approximately one quarter of the live embryos were abnormal. At E10.0, dead (asystolic) embryos were first identified. At E10.5, there was a dramatic decrease in the number of abnormal live embryos and a corresponding increase in the number of dead and resorbed embryos. Genotype analysis of live embryos by PCR revealed that all of the abnormal embryos were Hif1a−/− (Table 1B). Between E8.5 and E8.75, the majority of the Hif1a−/− embryos appeared morphologically normal, whereas at later stages all of the Hif1a−/− embryos were abnormal. In contrast, all of the Hif1a+/+ and Hif1a+/− embryos were morphologically normal at all developmental stages analyzed.
Table 1.
Analysis of pregnancies resulting from timed Hifla+/− × Hifla+/− matings
| A. Morphologic analysis of uterine implanations | |||||
|---|---|---|---|---|---|
| Gestational age
|
Number of embryos (no. of litters)
|
Live embryos (%)
|
Dead
|
Resorbed
|
|
| normal
|
abnormal
|
||||
| E8.5 | 90 (12) | 87.8 | 6.7 | 0 | 5.6 |
| E8.75 | 77 (10) | 66.2 | 27.3 | 0 | 6.5 |
| E9.25–E9.75 | 179 (22) | 67.6 | 19.6 | 0 | 12.8 |
| E10.0 | 75 (9) | 61.3 | 26.6 | 1.3 | 10.6 |
| E10.5 | 52 (6) | 61.5 | 5.7 | 13.4 | 19.2 |
| ⩾E11.0 | 44 (5) | 68.2 | 0 | 0 | 31.8 |
| B. Genetic analysis of live embryos | |||||||
|---|---|---|---|---|---|---|---|
| Gestational age
|
Number of embryos (no. of litters)
|
Normal (%)
|
Abormal (%)
|
||||
| +/+ | +/− | −/− | +/+ | +/− | −/+ | ||
| E8.5 | 34 (5) | 17.6 | 52.9 | 29.4 | 0 | 0 | 0 |
| E8.75 | 39 (5) | 25.6 | 43.5 | 17.9 | 0 | 0 | 12.8 |
| E9.25–E9.75 | 49 (9) | 30.6 | 42.8 | 0 | 0 | 0 | 26.5 |
| E10.0–E10.5 | 34 (4) | 19.8 | 56.3 | 0 | 0 | 0 | 23.8 |
Hif1a+/+ and Hif1a−/− embryos each had a mean of 11 somites at E8.5–E8.75. At E9.75–E10.0, Hif1a+/+ embryos had a mean of 29 somites, whereas Hif1a−/− embryos had a mean of 12 somites, indicating that developmental arrest of Hif1a−/− embryos had occurred late on embryonic day 8. By E10, the retarded development of viable Hif1a−/− compared with Hif1a+/+ embryos was striking (Fig. 5A). In addition to developmental arrest, gross morphological abnormalities included pericardial effusion, failure of neural tube closure with cystic degeneration and prolapse of the neural folds, and cystic enlargement of the hindbrain (Fig. 5B,C). Hypoplasia of the branchial arches was also demonstrated by scanning electron microscopy (data not shown). Thus, all Hif1a−/− embryos were morphologically abnormal by E9.0, and all died by E11.0.
Figure 5.
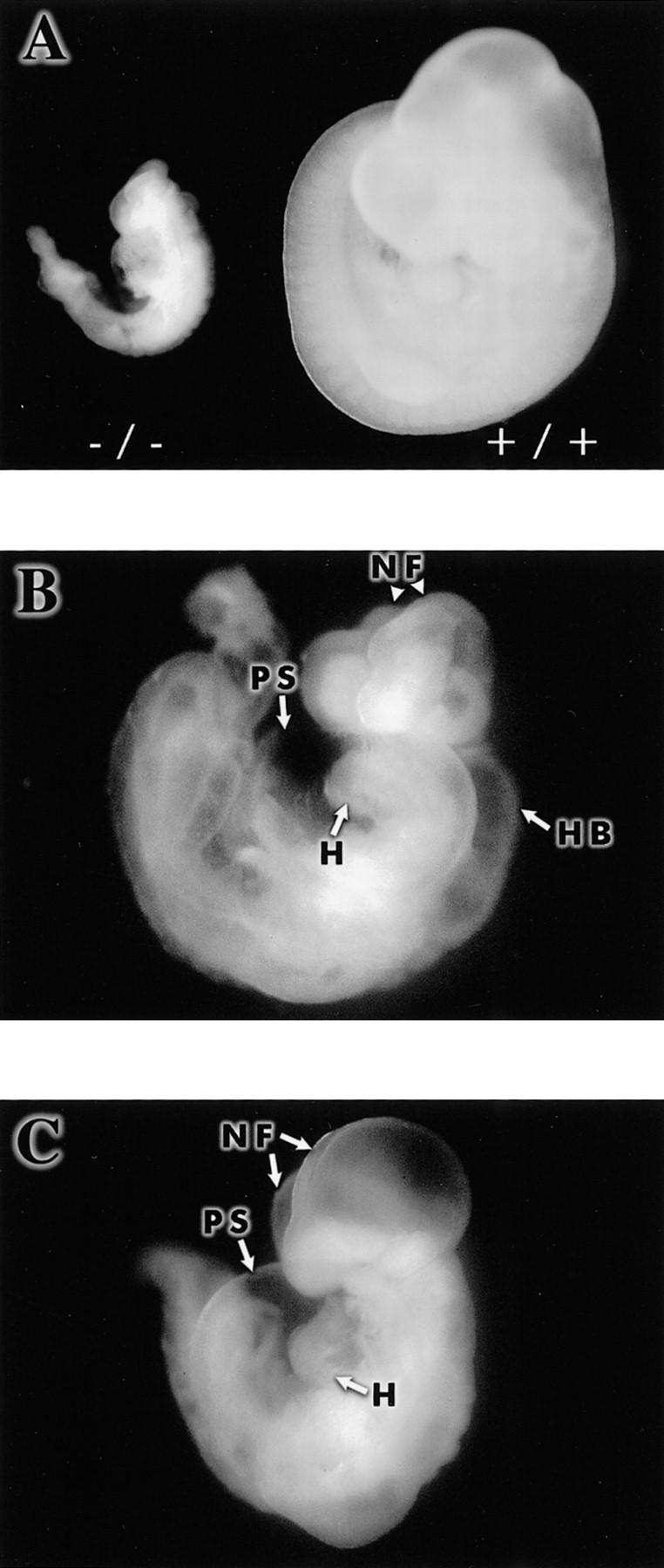
Abnormal development of Hif1a−/− embryos. (A) Comparison of viable Hif1a−/− and Hif1a+/+ embryos at E10.0. (B) Prolapsed neural folds (NF), cystic enlargement of the hindbrain (HB), and pericardial effusion in an E9.75 Hif1a−/− embryo. (H) Heart; (PS) pericardial sac. (C) Cystic appearance of cephalic region and pericardial effusion in an E9.75 Hif1a−/− embryo.
Histologic analysis of E9.75–E10.0 Hif1a−/− and stage-matched Hif1a+/+ embryos
Histologic analysis demonstrated specific defects in Hif1a−/− embryos involving the cephalic mesenchyme, blood vessels, and presumptive myocardium. Among the most striking abnormalities was a marked reduction of viable mesenchymal cells in the cranial region (Fig. 6C,E) relative to stage-matched controls (Fig. 6A). Prolapse of the lateral neural folds was associated with buckling of the somatic ectoderm ventral to the neurosomatic junction (box in Fig. 6E). The presence of extensive mesenchymal cell death was confirmed by supravital staining with Nile blue sulfate (Fig. 7; data not shown). These results indicate that mesenchymal cell death was responsible for the failure of neural tube closure that was uniformly observed in Hif1a−/− embryos. A second striking histologic abnormality identified in the cephalic region of Hif1a−/− embryos was the presence of large endothelial-lined vascular structures that in some cases occupied virtually the entire mesenchymal compartment, which was almost completely devoid of viable cells (Fig. 6C,E). These anomalous vascular structures were also present in other regions of the embryo (especially along the path of the dorsal aortae), but they were most prevalent and prominent in the head region.
Figure 6.

Histologic analysis. Sections through the cranial region of stage-matched Hif1a+/+ (A) and E9.75–E10.0 Hif1a−/− (C,E) embryos are shown. A normal blood vessel (arrowhead in A) can be compared with the anomalous vascular structures contained within the cephalic mesenchyme (arrowheads in C and E) of Hif1a−/− embryos, which also manifest prolapsed neural folds (NF) and a deficiency of cranial mesenchyme (CM) relative to the Hif1a+/+ embryo. Sections through the cardiac region of stage-matched Hif1a+/+ (B) and E9.75–E10.0 Hif1a−/− (D,F) embryos are shown. Hyperplasia of presumptive myocardium (M) and anomalous tissue (arrows in F) are evident within the enlarged pericardial cavity (PC) and along the pericardium (P). (AV) Atrioventricular canal; (BA1) first branchial arch; (NT) neural tube; (OV) otic vesicle; (OT) ventricular outflow tract.
Figure 7.

Supravital dye staining. Ten-somite Hif1a+/+ (left) and Hif1a−/− (right) embryos have been treated with Nile blue sulfate. Punctate staining indicative of cell death is apparent within the mesenchymal compartment of the fore- and midbrain region in the left neural fold of the Hif1a−/− embryo and is absent in the corresponding region of the Hif1a+/+ embryo. Nonspecific trapping of dye in the heart lumen also allowed analysis of cardiac morphogenesis. Arrows over the embryos have been placed the same distance apart to demonstrate the constriction between ventricle and outflow tract of the Hif1a−/− embryo. The Hif1a−/− heart is not stained, suggesting absence of a patent lumen.
A third malformation observed in E9.75 Hif1a−/− embryos was hyperplasia of the presumptive myocardium. Unlike Hif1a+/+ embryos (Fig. 6B), in which the cardiac mesoderm was consistently organized into 2–3 concentric cell layers, the presumptive myocardium of Hif1a−/− embryos showed anomalous organization and excessive cell accumulation, with as many as 12 cell layers observed in some regions of the developing heart (Fig. 6D,F). Abnormal collections of cells were also observed within and along the pericardial cavity (Fig. 6F). The lumen of the heart tube in all Hif1a−/− embryos examined was not obviously patent, in contrast to the patent lumen observed in all Hif1a+/+ embryos. Whereas minimal cell death was observed in cardiac tissues, the nonspecific, extracellular trapping of Nile blue sulfate revealed a marked constriction at the junction of the ventricle and outflow tract (Fig. 7), which was present in 21% of E9.25 and 40% of E9.75 Hif1a−/− embryos. These observations indicate that in Hif1a−/− embryos, abnormal cellular proliferation resulted in a marked reduction in size of the ventricular cavity and outflow tract.
Analysis of vascular development by whole-mount PECAM immunohistochemistry
Whole-mount immunoperoxidase staining using a monoclonal antibody against the endothelial cell marker PECAM (platelet endothelial cell adhesion molecule) was performed to investigate the vascular anomalies in Hif1a−/− embryos. At the earliest developmental stage examined (E8.5–E8.75), no differences between Hif1a+/+ and Hif1a−/− embryos were observed (Fig. 8A,B). By E9.25, however, significant deficiencies in vascularization were noted, especially within the cephalic region of Hif1a−/− embryos. The heads of Hif1a−/− embryos contained only a sparse network of small vessels (Fig. 8D), whereas a profusion of vessels was detected throughout the mesenchymal compartment of Hif1a+/+ embryos (Fig. 8C). Instead of the normal vascular network, large anomalous endothelial-lined vascular structures were present within the cephalic mesenchyme of Hif1a−/− embryos, confirming the results of histologic analysis. In addition to the cranial anomalies, the branchial arch vessels in all mutant embryos were either severely hypoplastic or absent (Fig. 8D). The dorsal aortae were present but abnormal in morphology, and localized regions of dilatation were observed. The cranial vascular deficiencies observed in E9.25 embryos were more pronounced in E9.75 embryos, and all Hif1a−/− embryos examined demonstrated irregularities in the diameter and/or position of the dorsal aortae and intersomitic vessels (data not shown). These data indicate that in Hif1a−/− embryos vascularization is initiated properly but fails progressively throughout the embryo between E8.75 and E9.75, leading to embryonic lethality between E10.0 and E11.0.
Figure 8.

PECAM immunohistochemistry. Comparison of E8.5–E8.75 Hif1a+/+ (A) and Hif1a−/− (B) embryos reveals no apparent differences in vascularization. In contrast, the vascularization of the E9.25 Hif1a−/− embryo (D) is markedly abnormal when compared with a stage-matched Hif1a+/+ embryo (C). Anomalous endothelial-lined vascular structures are present in the cranial region (arrow in D), no branchial arch vessels have formed, the lumen of the heart tube is not apparent, and the diameter of the dorsal aorta (DA) is irregular. (BA1) First branchial arch; (V) Ventricle.
Analysis of HIF-1α expression in Hif1a+/+ and Hif1a−/− embryos
To globally assess HIF-1α expression during embryonic development, protein lysates were prepared from Hif1a+/+ conceptuses, including both embryo and extraembryonic membranes, at various stages between E8.5 and E18. Lysate aliquots were assayed for expression of HIF-1α (Fig. 9, top panel) and HIF-1β protein (Fig. 9, middle panel). In addition, the lysates were assayed for expression of topoisomerase I as a control nuclear protein (Fig. 9, bottom panel). Proteins were detected by chemiluminescence and quantitated by laser densitometry. For each sample, HIF-1α and HIF-1β protein levels were normalized to the expression of topoisomerase I. In contrast to the high level of expression in Hif1a+/+ ES cells, expression of HIF-1α was considerably lower in E8.5 Hif1a+/+ embryos. Compared with expression at E8.5, HIF-1α levels increased 7- to 8-fold at E9.5–E10 and 12- to 18-fold at E11–E12. At E18, HIF-1α expression was 10-fold higher than at E8.5. Between E8.5 and E18, HIF-1β levels varied by less than twofold when normalized to topoisomerase I. HIF-1α protein was not detected in lysates of E10 Hif1a−/− embryos, and HIF-1β levels were decreased by 75% relative to E10 Hif1a+/+ embryos. In contrast, topoisomerase I was expressed at normal levels in Hif1a−/− embryos, demonstrating that loss of HIF-1α and HIF-1β expression was not an artifact of sample preparation or loading. These studies demonstrate developmental regulation of HIF-1α expression in Hif1a+/+ embryos and complete absence of HIF-1α expression in Hif1a−/− embryos. Decreased HIF-1β levels in HIF-1α-null ES cells and embryos complement previous findings of increased HIF-1β levels in HIF-1α-overexpressing cells (Jiang et al. 1996a) and decreased HIF-1α levels in HIF-1β-null cells (Forsythe et al. 1996), suggesting that these proteins may have decreased stability in the absence of their dimerization partner.
Figure 9.

Immunoblot analysis of HIF-1 expression in Hif1a+/+ and Hif1a−/− embryos. Aliquots (15 μg) of nuclear extracts from nonhypoxic (N) and hypoxic (H) Hif1a+/+ ES cells (lanes 1,2) and aliquots (120 μg) of lysates prepared from Hif1a+/+ (WT; lanes 3–8) and Hif1a−/− (M; lane 9) embryos of the indicated gestational age were analyzed using antibodies specific for HIF-1α (top), HIF-1β (middle), or as a control (C), topoisomerase I (bottom).
Discussion
HIF-1α expression in ES cells
Previous studies have demonstrated the specificity and sensitivity of HIF-1α expression as a function of O2 concentration. It was thus a considerable surprise to identify high levels of HIF-1α in nonhypoxic ES cells, which indicates the existence of a novel, cell type-specific pathway leading to HIF-1α expression. We have demonstrated recently HIF-1α expression in nonhypoxic rat fibroblasts transformed by the V-SRC oncoprotein (Jiang et al. 1997a). ES cells are derived from inner cell mass/embryonic ectoderm of the blastocyst, which represents a population of actively dividing cells. HIF-1α expression may be activated by one or more signal transduction pathways involved in cellular proliferation. The decreased proliferation of Hif1a−/− ES cells suggests that HIF-1α may be involved in the regulation of cell division, either directly or indirectly via effects on intermediary metabolism. However, HIF-1α deficiency had no apparent effect on preimplantation development. The significance of constitutive HIF-1α expression in ES cells will thus require further investigation.
As a consequence of constitutive HIF-1α expression, nonhypoxic ES cells demonstrated high-level expression of mRNAs encoding glycolytic enzymes. The Warburg effect refers to the high rate of aerobic glycolysis that is characteristic of tumor cells (Warburg 1956). Rous sarcoma virus infection of rat cells was associated with aerobic glycolysis, and this phenotype was correlated with V-SRC-mediated transformation (Carroll et al. 1978). Increased glycolytic gene expression mediated by HIF-1α expression may be required for maximal rates of cellular proliferation by both ES and V-SRC-transformed cells under hypoxic conditions. In ES cells, HIF-1α was required for normal expression of almost all genes involved in the metabolism of extracellular glucose to intracellular lactate, a remarkable example of coordinate transcriptional regulation. Furthermore, HIF-1α was required to maintain expression of several genes under hypoxic conditions such that in its absence expression of these genes was hypoxia repressible. These results may reflect the constitutive expression of HIF-1α in ES cells, as the expression patterns clearly differed from those observed in Hep3B cells. However, these results suggest that HIF-1α may be required for expression of other genes that do not show an obvious pattern of hypoxic induction.
Regulation of Vegf gene expression by HIF-1
Experimental data indicate that VEGF is essential for vasculogenesis and angiogenesis in the embryo and fetus as well as for neovascularization of ischemic tissues and tumors in the adult organism (for review, see Ferrara and Davis-Smyth 1997). An understanding of the molecular basis for hypoxia-induced VEGF expression is of critical importance because of the potential therapeutic utility of stimulating or inhibiting VEGF expression in ischemic and neoplastic tissues, respectively. Previous studies have provided evidence supporting a role for HIF-1 in mediating VEGF transcriptional activation in hypoxic cells. First, an essential HIF-1 binding site was identified 1 kb 5′ to the human, mouse, and rat VEGF genes (Levy et al. 1995; Liu et al. 1995; Forsythe et al. 1996; Shima et al. 1996). Second, forced expression of HIF-1α and HIF-1β activated transcription from the human VEGF promoter in nonhypoxic cells and resulted in a superactivation in hypoxic cells (Forsythe et al. 1996). Third, loss of HIF-1β expression and HIF-1 DNA-binding activity was associated with a marked decrease in VEGF mRNA levels under hypoxic conditions (Forsythe et al. 1996; Salceda et al. 1996; Wood et al. 1996; Maltepe et al. 1997). Fourth, expression of V-SRC was associated with increased HIF-1α protein, HIF-1 DNA-binding activity, VEGF mRNA, and transcriptional activity of a reporter gene containing the VEGF hypoxia-response element that was dependent on an intact HIF-1 binding site (Jiang et al. 1997a). Fifth, VEGF and HIF-1α expression was coinduced in association with increased myocardial vascularization in anemic fetal sheep (Martin et al. 1997). The demonstration that HIF-1α deficiency resulted in a complete loss of induced expression provides definitive proof that HIF-1 mediates Vegf gene transcriptional activation in hypoxic ES cells. Induction of VEGF expression via increased mRNA stability under hypoxic conditions was demonstrated previously (Ikeda et al. 1995; Stein et al. 1995; Levy et al. 1996). The absence of any increase in VEGF mRNA expression in HIF-1α-deficient ES cells indicates either that post-transcriptional mechanisms are not operative in ES cells, that post-transcriptional mechanisms are of limited effect in the absence of transcriptional activation, or that the post-transcriptional mechanisms are also dependent on HIF-1α, perhaps for transcription of a gene encoding a hypoxia-induced mRNA binding protein.
HIF-1α is required for proper cardiovascular development
Analysis of Hif1a−/− embryos revealed multiple defects in cardiovascular development, including pericardial effusion; disorganized cardiac morphogenesis with myocardial hyperplasia and ventricular obstruction; formation of dilated vascular structures, especially in the cephalic region; and marked reduction in the overall number of cephalic blood vessels including hypoplasia of the arch vessels. Pericardial effusion may represent a secondary defect, as it has been reported in knockout mice without intracardiac defects (Warren et al. 1994). In contrast, hyperplasia of the presumptive myocardium, which has not been reported previously to our knowledge, almost certainly represents a primary defect rather than a response to ventricular outflow obstruction. VEGF-deficient embryos also manifested ventricular outflow obstruction but had a patent ventricular cavity and decreased, rather than increased, ventricular wall thickness (Carmeliet et al. 1996; Ferrara et al. 1996). The hyperplastic and highly disorganized appearance of the presumptive myocardium suggests strongly that this defect represents a cause, rather than a consequence, of ventricular outflow obstruction in Hif1a−/− embryos and that HIF-1α may play a direct role in cardiac morphogenesis.
One of the most striking histologic findings was the presence of massively enlarged vascular structures in the cephalic region of Hif1a−/− embryos. These structures were remarkably similar to those described for Vegf−/− embryos (Carmeliet et al. 1996). The marked reduction in vascularization of Hif1a−/− embryos is also consistent with a deficiency of VEGF production. The demonstration that Hif1a−/− ES cells and embryoid bodies are deficient in expression of VEGF mRNA provides a potential molecular basis for the observed defects in embryonic vascularization, although analysis of Vegf expression in situ will be required to confirm this hypothesis. Vascularization of Hif1a−/− embryos appeared normal at E8.5, but by E9.25 only abnormally dilated vascular structures were present in the cephalic region. In contrast, the dorsal aortae were poorly developed or absent in E8.5 Vegf+/− and Vegf−/− embryos, respectively, and dilated vascular structures were observed in E9.5 Vegf−/− embryos (Carmeliet et al. 1996). Recent studies have demonstrated that VEGF is a survival factor for newly formed blood vessels such that VEGF withdrawal results in capillary regression (Benjamin and Keshet 1997). It is possible that Hif1a−/− embryos produced sufficient VEGF to initiate vasculogenesis but that VEGF production became limiting owing to absence of HIF-1-mediated transcriptional activation or loss of VEGF-producing mesenchymal cells or both. The increased expression of HIF-1α in E9.5, compared with E8.5, Hif1a+/+ embryos is of interest in this regard.
Although decreased vascularization and decreased VEGF mRNA expression were reported for Arnt−/− embryos lacking HIF-1β (ARNT), the enlarged vascular structures and absence of normal cephalic vascularization seen in Vegf−/− and Hif1a−/− embryos by E9.25 were not observed in Arnt−/− embryos (Maltepe et al. 1997). One explanation for these results is that in Arnt−/− embryos, heterodimerization of HIF-1α with the protein product of the Arnt2 gene, which is homologous to Arnt but is expressed primarily within the nervous system (Hirose et al. 1996), may allow for adequate VEGF synthesis in the cephalic region, whereas in Hif1a−/− embryos, the absence of both HIF-1α:HIF-1β (ARNT) and HIF-1α:ARNT2 heterodimers has a more significant effect on Vegf transcription.
HIF-1α is required for embryonic cell survival
A defining feature of Hif1a−/− embryos is the overwhelming degree of cell death that occurred in a progressive manner beginning around E8.5 and leading to embryonic death around E10.5. Hif1a−/− embryos manifested severely prolapsed neural folds that, if the embryos developed to term, would result in exencephaly. In addition, the branchial arches, which are the anlage of the developing craniofacial skeleton, were hypoplastic and discontinuous (L.E. Kotch, N.V. Iyer, and G.L. Semenza, unpubl.). The basis for these malformations was the massive death of cells within the cephalic mesenchyme, which normally supports the neural folds during the process of neural tube closure and contributes to the formation of the branchial arches. By E9.5, the deficiency of cells was so profound that the cephalic region of the embryo appeared translucent (Fig. 5C). In addition to the death of mesenchymal cells, abnormally dilated vascular structures were also present. It is not clear whether these structures represent a cause or a consequence of the mesenchymal cell death. Tissue cannot remain viable without adequate perfusion, and the vascular defects may have been responsible for overwhelming death of the mesenchymal population. However, excessive cell death was detected in E8.5 Hif1a−/− embryos prior to the appearance of any vascular defects (L.E. Kotch, N.V. Iyer, and G.L. Semenza, unpubl.), suggesting that an intact mesenchyme may be required to provide essential cellular or humoral support of vascular integrity, as demonstrated in mice deficient for platelet-derived growth factor-B (Lindahl et al. 1997).
Defective embryonic angiogenesis or erythropoiesis has been associated with cell death previously (Warren et al. 1994; Henkemeyer et al. 1995; Carmeliet et al. 1996; Maltepe et al. 1997; Wang et al. 1997). Although it is very difficult to make comparisons between studies, the extent of cell death, both within the cephalic mesenchyme and throughout the embryo, appears to be most severe within Hif1a−/− embryos. Furthermore, scanning electron microscopy and Nile blue sulfate staining revealed the presence of increased cell death prior to the appearance of vascular defects (L.E. Kotch, N.V. Iyer, and G.L. Semenza, unpubl.). These results suggest a complex interrelationship between mesenchymal cell survival and embryonic vascularization. HIF-1α deficiency, in addition to interfering with the development of a circulatory system required for adequate tissue oxygenation, may also render many cell populations within the embryo unable to adapt to O2 deprivation. A major intracellular adaptation involves increased expression of genes encoding glucose transporters and glycolytic enzymes, a process that requires HIF-1α. We hypothesize that the cephalic mesenchyme of Hif1a−/− embryos may be particularly susceptible to hypoxia-mediated cell death and that a deficiency of mesenchyme in concert with a deficiency of VEGF may result in major vascular defects, leading to more severe hypoxia and extensive cell death, ultimately resulting in embryonic lethality.
HIF-1α regulates essential developmental and physiological aspects of O2 homeostasis
The results presented here demonstrate that HIF-1α is essential for cellular and developmental aspects of O2 homeostasis. In addition, preliminary studies indicate that Hif1a+/− mice have impaired cardiovascular responses to chronic hypoxia (A.Y. Yu, N.V. Iyer, and G.L. Semenza, unpubl.). Thus, HIF-1α is required for the formation of key physiological systems during embryonic development and for their subsequent utilization during postnatal life. It remains to be determined whether the expression of HIF-1α within the developing cardiovascular system occurs in response to a hypoxic stimulus or whether it is part of a hard-wired developmental program. In either case, these results indicate that HIF-1α is a master regulator of O2 homeostasis. Other master regulatory factors have been identified that play key roles in the formation of specific tissues, such as the involvement of GATA1 and MYOD in erythroid and muscle development, respectively (Orkin 1992; Weintraub 1993). Several master regulators have been shown to play roles both in development and postnatal physiology, including PIT1, which is required for the development of the somatomammotrophs of the anterior pituitary as well as their subsequent production of growth hormone and prolactin (Andersen and Rosenfeld 1994). HIF-1α, rather than specifying a cell type, plays a more global role in development and physiology that is based on the fundamental requirement for O2 that is shared by all cells.
The evolution of multicellular organisms presented the challenge of delivering O2 to cells too far from the external environment to receive adequate oxygenation by diffusion. Whereas there does not appear to be a HIF-1α homolog in Saccharomyces cerevisiae, bHLH–PAS proteins with sequence similarity to HIF-1α have been identified in both invertebrate and vertebrate species. In Drosophila melanogaster, O2 is not blood borne through a circulatory system but instead is delivered directly to all cells via tracheal tubes, formation of which requires TRH, a bHLH–PAS protein with sequence similarity to HIF-1α (for review, see Guillemin and Krasnow 1997). Fruit flies and humans have thus evolved different methods of O2 delivery since diverging >500 million years ago but nevertheless express proteins that are structurally and functionally related, suggesting that the challenge of tissue oxygenation in multicellular organisms was solved in part by the evolution of HIF-1α-like regulatory factors.
Materials and methods
Construction of the targeting vector
DNA spanning Hif1a exons 2 and 3 (Wenger et al. 1997) was isolated from a 129/Sv mouse genomic library (partial Sau3A digest of AB-1 ES cell DNA in λGEM11) and used to construct targeting vector TVTK8. The 5′ arm of TVTK8 was constructed by subcloning a 3-kb EcoRI fragment from intron 1 into the EcoRI site of pBluescript SK(−) (Stratagene). The 5′ EcoRI site originated from the λGEM11 polylinker. The 3′ arm of TVTK8 consisted of Hif1a sequences from intron 2 and exon 3 and was subcloned as a HindIII fragment. A dose-sensitive neomycin resistance gene driven by a PGK promoter from pNTK (Mortensen et al. 1992; Ausubel et al. 1994) was inserted into the EcoRV site between the two arms of TVTK8. A PGK promoter–herpes simplex virus thymidine kinase coding sequence cassette was inserted as an XhoI–SalI fragment into the XhoI site located 3′ to the Hif1a sequences in TVTK8. TVTK8 was linearized with XhoI for electroporation.
ES cell culture and transfection
J1 ES cells (Li et al. 1992) were cultured in DMEM medium containing 25 mm glucose and 110 mg/liter of sodium pyruvate, supplemented with 15% ES cell-qualified fetal bovine serum (FBS), 875 nm insulin, 1% penicillin–streptomycin, 1% nonessential amino acids, 40 μl/liter of monothioglycerol, and 1000 U/ml of LIF (GIBCO) on neomycin-resistant, irradiated mouse embryonic fibroblasts (Bradley 1987; Robertson 1987). Cells (5 × 107) in 0.7 ml were electroporated with 25 μg of linearized TVTK8 at 240 V and 540 μF in a Gene Pulser (Bio-Rad). After 48 hr, transfected cells were cultured in medium containing 300 μg/ml of G418 (Geneticin, GIBCO) and 2 μm gancyclovir (Mansour et al. 1988). Individual clones were picked and expanded in a 96-well plate with feeder layer. DNA extracted from individual clones (Ramirez-Solis et al. 1992) was analyzed by blot hybridization.
Generation and analysis of Hif1a−/− ES cells and embryoid bodies
Hif1a+/− ES cells were cultured in medium containing 2 mg/ml of G418 for 14 days (Mortensen et al. 1992; Ausubel et al. 1994). Resistant colonies were picked and analyzed by DNA blot hybridization. Hif1a+/− cells used in subsequent experiments were picked from the same plates as the Hif1a−/− clones. For analysis of growth rates, the medium was supplemented with 25 mm HEPES, and 6 × 105 cells were plated on gelatin-coated 10-cm Petri dishes (Falcon). In cells cultured for 48 hr at 1% O2, the medium was changed at 24 hr in a hypoxic chamber (Plas Labs) to ensure a constant O2 concentration. At the end of the experiment, cells were trypsinized and stained with trypan blue. For generating embryoid bodies, ES cells were trypsinized, and fibroblasts were removed by adherence to tissue culture dishes. ES cells (2.5 × 105) were cultured in 12 ml of Iscove’s MDM–methylcellulose (MethoCult, Stem Cell Technologies) for 4 days and exposed to 20% O2 or 0% O2 (5% CO2/balance N2) for 16 hr, and total RNA was isolated.
RNA blot hybridization
ES cells or embryoid bodies were lysed in guanidine isothiocyanate, and total RNA was isolated by phenol–chloroform extraction (Chomczynski and Sacchi 1987). Fifteen-microgram aliquots of RNA were fractionated by 2.2 m formaldehyde, 1.4% agarose gel electrophoresis and transferred to nitrocellulose filters. IMAGE Consortium cDNA clones (Lennon et al. 1996) were obtained (Research Genetics, Inc.), and the inserts were isolated and 32P-labeled by random-primer synthesis using a commercial kit (Life Technologies, Inc.). Hybridization was performed in Quik-Hyb (Stratagene) at 67°C. Blots were washed in 0.1× SSC, 0.1% SDS at room temperature for 1 hr and then at 55°C for 1 hr, and exposed to film for autoradiography. Blots were stripped of radioactivity and hybridized with a 32P-labeled oligonucleotide that was complementary to 18S rRNA (Forsythe et al. 1996).
Generation and analysis of mutant mice
Clone 7 or clone 19 J1 ES cells were injected into E3.5 C57BL/6 mouse embryos using standard methods (Bradley 1987; Hogan et al. 1994). Injected blastocysts were surgically implanted into the uterus of pseudopregnant CD1 foster mothers. Chimeric males, identified by coat color and DNA blot hybridization analysis, were mated to C57BL/6 females, and offspring were analyzed by DNA blot hybridization. Genomic DNA isolated from mouse tails or cultured cells was digested with EcoRV, fractionated by 0.7% agarose gel electrophoresis, transferred to a nylon membrane, and hybridized to a probe containing Hif1a or neo sequences. Probe labeling and hybridization were performed as described above. For timed matings, Hif1a+/− or Hif1a+/+ mice were mated, and female mice were examined for the presence of vaginal plugs twice daily. Depending on whether vaginal plugs were detected at 10:30 a.m. or 6:30 p.m., the gestational age of the embryos at 12:00 noon was considered to be E0.5 or E0.0, respectively.
Immunoblot and electrophoretic mobility-shift assays
Confluent ES or Hep3B cells were incubated at 1% or 20% O2 for 4 hr and nuclear extracts were prepared (Wang and Semenza 1995). For immunoblot assays, 15-μg aliquots of nuclear extracts were fractionated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. The blot was probed with affinity-purified anti-HIF-1α or anti-HIF-1β antibodies (Jiang et al. 1996b) followed by a 1:2000 dilution of goat anti-rabbit immunoglobulin for detection with ECL reagents (Amersham). Antitopoisomerase I (TopoGEN, Columbus, OH) was used at 1:1000 dilution followed by goat anti-human immunoglobulin antibodies at 1:2000 dilution. For electrophoretic mobility-shift assays, 5-μg aliquots of nuclear extracts were incubated with 32Plabeled oligonucleotide probe W18, containing 18 bp from the EPO hypoxia-response element (Semenza and Wang 1992).
Genotype analysis of mouse embryos by PCR
Genomic DNA isolated from embryos or yolk sacs was used to amplify neo and Hif1a exon 2 sequences. Both neo (463-bp) and Hif1a (317-bp) sequences were amplified from DNA of Hif1a+/− embryos, whereas only Hif1a or only neo sequences were amplified from DNA of Hif1a+/+ and Hif1a−/− embryos, respectively. The sequences of Hif1a exon 2 primers were 5′-ACTGGCTGCTATTGGGCGAAGTG-3′ and 5′-GTAAAGCACGAGGAAGCGGTCAG-3′. Conditions for PCR were 94°C for 30 sec, 52°C for 20 sec, and 72°C for 22 sec, for 40 cycles. The sequences of neo primers were 5′-TGTAGTCTCCTGCTAAAAG-3′ and 5′-TTATTCGAGTTAAGACAAAC-3′. Conditions for PCR were 94°C for 30 sec, 67°C for 15 sec, and 72°C for 15 sec, for 30 cycles.
Histology
Eight stage-matched Hif1a−/− and Hif1a+/+ embryos, each from a different pregnancy, were examined histologically. Embryos were fixed in Bouin’s solution, rinsed in 70% alcohol, dehydrated through a graded alcohol series, and embedded using a Polysciences JB4 kit. Embedded tissue was sectioned at 4 μm on a Sorvall microtome, and sections were stained in methylene blue-acid fuchsin for 15 min at pH 4.0 and examined using a Nikon photomicroscope.
Nile blue sulfate staining
Viable embryos were harvested, extraembryonic membranes were removed, and the embryos were staged by somite number. The yolk sacs were collected for genotype determination. Embryos were incubated in a 1:50,000 solution of Nile blue sulfate in lactated Ringer’s solution at 37°C for 30 min and then rinsed in Ringer’s solution (Kotch et al. 1995).
PECAM immunoperoxidase staining
Three Hif1a−/− and Hif1a+/+ embryos each were collected on E8.5, E9.25, and E9.75 for whole-mount immunoperoxidase staining using the anti-mouse PECAM monoclonal antibody MEC13.3 (PharMingen). Embryos were submersed into Dent fixative (5:1 methanol/DMSO) for at least 24 hr and bleached using a solution of 10% H2O2 in Dent fixative. Specimens were washed in buffer, incubated overnight with MEC13.3 at a concentration of 10 μg/ml, rinsed, incubated overnight in horseradish peroxidase-coupled anti-rat immunoglobulin G, and rinsed again (Schlaeger et al. 1995). A color reaction was initiated by submersion of specimens into a H2O2-containing DAB solution and was terminated by dehydration with methanol.
Immunoblot assays
Four or five embryos were pooled for all time points except E18 (one embryo) and homogenized in 20 mm HEPES (pH 7.5), 1.5 mm MgCl2, 0.2 mm EDTA, 100 mm NaCl, 2 mm dithiothreitol (DTT), 0.4 mm phenylmethylsulfonyl fluoride (PMSF), and 1 mm Na3VO4. NaCl (4 m) was added to a final concentration of 0.45 m. The homogenate was centrifuged 30 min at 10,000g, and the supernatant was mixed with an equal volume of 20 mm HEPES (pH 7.5), 1.5 mm MgCl2, 0.2 mm EDTA, 0.45 m NaCl, 40% (vol/vol) glycerol, 2 mm DTT, 0.4 mm PMSF, and 1 mm Na3VO4. Protein concentrations were determined using a commercial kit (Bio-Rad). Protein (120 μg) was fractionated by 7% SDS-PAGE. Immunoblot analysis was performed as described (Jiang et al. 1996b) except that a 1:500 dilution of anti-HIF-1α and a 1:2000 dilution of anti-HIF-1β antibodies were used. Anti-human topoisomerase I (TopoGEN, Columbus, OH) was used at a 1:1000 dilution followed by goat anti-human immunoglobulin antibodies at 1:2000 dilution.
Acknowledgments
We thank A. Cuenca, S. Desai, J. Lutz, T. Menard, R. Roe, and D. Wu for technical assistance. We are grateful to R. Jaenisch for providing J1 cells, J.-F. Lu for providing gancyclovir, C. Moore and G. Stetten for performing karyotype analysis, R. Mortensen for providing pNTK, K. Sulik for use of histology facilities at the University of North Carolina at Chapel Hill, H. Zhong and J. Simons for providing topoisomerase antibodies, and J. Chatham, C. Dang, S. Farmer, C. Hayes, S. Lanzkron, S. Sharkis, T. Sato, J. Strandberg, and T. Townes for helpful discussions. G.L.S. is an Established Investigator of the American Heart Association, and this work was supported in part by grants from the American Heart Association National Center, the National Institutes of Health (R01-DK39869 and R01-HL55338 to G.L.S.), and the Swiss National Science Foundation (to M.G.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
Analysis of other ES cell lines did not reveal high-level expression of HIF-1α at 20% O2, suggesting that this property may be restricted to the J1 line and is not a general characteristic of ES cells.
Footnotes
E-MAIL gsemenza@gwgate1.jhmi.jhu.edu; FAX (410) 955-0484.
References
- Andersen B, Rosenfeld MG. Pit-1 determines cell types during development of the anterior pituitary gland: A model for transcriptional regulation of cell phenotypes in mammalian organogenesis. J Biol Chem. 1994;269:29335–29338. [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, Albright LM, Coen D, Varki MA, Janssen K. Current protocols in molecular biology. New York, NY: John Wiley and Sons; 1994. [Google Scholar]
- Benjamin LE, Keshet E. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: Induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc Natl Acad Sci. 1997;94:8761–8766. doi: 10.1073/pnas.94.16.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley A. Production and analysis of chimaeric mice. In: Robertson EJ, editor. Teratocarcinomas and embryonic stem cells: A practical approach. 1st ed. Oxford, UK: IRL Press; 1987. pp. 113–152. [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertesenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Ash JF, Vogt PK, Singer SJ. Reversion of transformed glycolysis to normal by inhibition of protein synthesis in rat kidney cells infected with temperature-sensitive mutant of Rous sarcoma virus. Proc Natl Acad Sci. 1978;75:5015–5019. doi: 10.1073/pnas.75.10.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dolwick KM, Swanson HI, Bradfield CA. In vitro analysis of Ah receptor domains involved in ligand-activated transcription. Proc Natl Acad Sci. 1993;90:8566–8570. doi: 10.1073/pnas.90.18.8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH–PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci. 1997;94:4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Flamme I, Frohlich T, von Reutern M, Kappel A, Damert A, Risau W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1α and developmentally expressed in blood vessels. Mech Dev. 1997;63:51–60. doi: 10.1016/s0925-4773(97)00674-6. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang B-H, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann M, Fandrey J, Bichet S, Wartenberg M, Marti HH, Bauer C, Wenger RH, Acker H. Oxygen supply and oxygen-dependent gene expression in differentiating embryonic stem cells. Proc Natl Acad Sci. 1996;93:2867–2872. doi: 10.1073/pnas.93.7.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin K, Krasnow MA. The hypoxic response: Huffing and HIFing. Cell. 1997;89:9–12. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, Pawson T. Vascular system defects and neuronal apoptosis in mice lacking Ras GTPase-activating protein. Nature. 1995;377:695–701. doi: 10.1038/377695a0. [DOI] [PubMed] [Google Scholar]
- Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H, Saijo Y, Gotoh O, Sogawa K, Fujii-Kuriyama Y. cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt) Mol Cell Biol. 1996;16:1706–1713. doi: 10.1128/mcb.16.4.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu F-F, Snader F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu Y-Z, Perdew GH, Bradfield CA. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signalling pathway. J Biol Chem. 1997;272:8581–8593. doi: 10.1074/jbc.272.13.8581. [DOI] [PubMed] [Google Scholar]
- Ikeda E, Achen MG, Breier G, Risau W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J Biol Chem. 1995;270:19761–19766. doi: 10.1074/jbc.270.34.19761. [DOI] [PubMed] [Google Scholar]
- Jiang B-H, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996a;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- Jiang B-H, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996b;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- Jiang B-H, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 and transcription of genes encoding VEGF and enolase 1: Involvement of HIF-1 in tumor progression. Cancer Res. 1997a;57:5328–5335. [PubMed] [Google Scholar]
- Jiang B-H, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1α: Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997b;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- Kotch LE, Chen S-Y, Sulik KK. Ethanol-induced teratogenesis: Free radical damage as a possible mechanism. Teratology. 1995;52:128–136. doi: 10.1002/tera.1420520304. [DOI] [PubMed] [Google Scholar]
- Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. consortium: An integrated molecular analysis of genomes and their expression. Genomics. 1996;33:151–152. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270:13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- Levy AP, Levy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem. 1996;271:2746–2753. doi: 10.1074/jbc.271.5.2746. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-Bdeficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Circ Res. 1995;77:638–643. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int- 2 in mouse embryo-derived stem cells: A general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- Martin, C., A.Y. Yu, B.-H. Jiang, L. Davis, D. Kimberly, A.R. Hohimer, and G.L. Semenza. 1997. Cardiac hypertrophy in chronically anemic sheep: Increased vascularization is associated with increased myocardial expression of vascular endothelial growth factor and hypoxia-inducible factor 1. Am. J. Obstet. Gynecol. (in press). [DOI] [PubMed]
- Moffett P, Reece M, Pelletier J. The murine Sim-2 gene product inhibits transcription by active repression and functional interference. Mol Cell Biol. 1997;17:4933–4947. doi: 10.1128/mcb.17.9.4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen RM, Conner DA, Chao S, Geisterfer-Lowrance AAT, Seidman JG. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH. GATA-binding transcription factors in hematopoietic cells. Blood. 1992;80:575–581. [PubMed] [Google Scholar]
- Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia-inducible factor 1: Definition of regulatory domains within the α subunit. J Biol Chem. 1997;272:11205–11214. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis R, Rivera-Perez J, Wallace JD, Wims M, Zheng H, Bradley A. Genomic DNA microextraction: A method to screen numerous samples. Anal Biochem. 1992;201:331–335. doi: 10.1016/0003-2697(92)90347-a. [DOI] [PubMed] [Google Scholar]
- Robertson EJ. Embryo-derived stem cell lines. In: Robertson EJ, editor. Teratocarcinomas and embryonic stem cells: A practical approach. 1st ed. Oxford, UK: IRL Press; 1987. pp. 71–112. [Google Scholar]
- Salceda S, Beck I, Caro J. Absolute requirement of aryl hydrocarbon receptor nuclear translocator protein for gene activation by hypoxia. Arch Biochem Biophys. 1996;334:389–394. doi: 10.1006/abbi.1996.0469. [DOI] [PubMed] [Google Scholar]
- Schlaeger TM, Qin Y, Fujiwara Y, Magram J, Sato TN. Vascular endothelial cell lineage-specific promoter. Development. 1995;121:1089–1098. doi: 10.1242/dev.121.4.1089. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Roth PH, Fang H-M, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- Semenza GL, Jiang B-H, Leung SW, Passantino R, Concordet J-P, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- Shima DT, Kuroki M, Deutsch U, Ng Y-S, Adamis AP, D’Amore PA. The mouse gene for vascular endothelial growth factor: Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J Biol Chem. 1996;271:3877–3883. doi: 10.1074/jbc.271.7.3877. [DOI] [PubMed] [Google Scholar]
- Stein I, Neeman M, Shweiki D, Itin A, Keshet E. Stabilization of vascular endothelial growth factor mRNA by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Mol Cell Biol. 1995;15:5363–5368. doi: 10.1128/mcb.15.10.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes & Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang B-H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J. 1997;16:4374–4383. doi: 10.1093/emboj/16.14.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- Warren AJ, Colledge WH, Carlton MBL, Evans MJ, Smith AJH, Rabbits TH. The oncogenic cysteine-rich LIM domain protein Rbtn2 is essential for erythroid development. Cell. 1994;78:45–57. doi: 10.1016/0092-8674(94)90571-1. [DOI] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesis: Redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Wenger RH, Gassmann M. Oxygen(es) and the hypoxia-inducible factor 1. Biol Chem. 1997;378:609–616. [PubMed] [Google Scholar]
- Wenger RH, Rolfs A, Marti HH, Guenet J-L, Gassmann M. Nucleotide sequence, chromosomal assignment, and mRNA expression of mouse hypoxia-inducible factor 1α. Biochem Biophys Res Commun. 1996;223:54–59. doi: 10.1006/bbrc.1996.0845. [DOI] [PubMed] [Google Scholar]
- Wenger RH, Rolfs A, Kvietikova I, Spielmann P, Zimmermann DR, Gassmann M. The mouse gene for hypoxia-inducible factor-la: Genomic organization, expression, and characterization of an alternative first exon and 5′ flanking sequence. Eur J Biochem. 1997;246:155–165. doi: 10.1111/j.1432-1033.1997.t01-1-00155.x. [DOI] [PubMed] [Google Scholar]
- Wiener CM, Booth G, Semenza GL. In vivo expression of mRNAs encoding hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1996;225:485–488. doi: 10.1006/bbrc.1996.1199. [DOI] [PubMed] [Google Scholar]
- Wood SM, Gleadle JM, Pugh CW, Hankinson O, Ratcliffe PJ. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression: Studies in ARNT-deficient cells. J Biol Chem. 1996;271:15117–15123. doi: 10.1074/jbc.271.25.15117. [DOI] [PubMed] [Google Scholar]