

Abstract
The c-ski proto-oncogene has been implicated in the control of cell growth and skeletal muscle differentiation. To determine its normal functions in vivo, we have disrupted the mouse c-ski gene. Our results show a novel role for ski in the morphogenesis of craniofacial structures and the central nervous system, and confirm its proposed function as a player in skeletal muscle development. Homozygous mutant mice show perinatal lethality resulting from exencephaly, a defect caused by failed closure of the cranial neural tube during neurulation. The timing of the neural tube defect in ski −/− embryos coincides with excessive apoptosis in the cranial neuroepithelium, as well as in the cranial mesenchyme. Homozygous ski mutants also exhibit a dramatic reduction in skeletal muscle mass, consistent with a defect in expansion of a myogenic precursor population. Nestin is an intermediate filament expressed in highly proliferative neuroepithelial stem cells and in myogenic precursors. Interestingly, we find decreased nestin expression in both the cranial neural tube and the somites of ski −/− embryos, compared with their normal littermates, but no reduction of nestin in the caudal neural tube. These results are consistent with a model in which ski activities are required for the successful expansion of a subset of precursors in the neuroepithelial or skeletal muscle lineages.
Keywords: ski proto-oncogene, nestin, neural tube
The ski proto-oncogene encodes a nuclear protein that binds to DNA in association with other cellular factors (Nagase et al. 1990) and modulates transcription (Engert et al. 1995). Several lines of evidence suggest that Ski may function to regulate critical decisions leading to a choice between continued proliferation or terminal differentiation. Overexpression of ski has been shown to induce oncogenic transformation, anchorage independence, enhanced cell proliferation and viability, and skeletal muscle differentiation (Stavnezer et al. 1986; Colmenares and Stavnezer 1989; Colmenares et al. 1991a). Induction of myogenesis by ski seems to require both an early determination function leading to the expression of myogenic regulators, as well as a later activity required for fusion and full terminal differentiation (Colmenares et al. 1991b). A role in muscle development is further supported by in vivo studies, which show that overexpression of ski in skeletal muscle of transgenic mice leads to fiber-type specific muscle hypertrophy (Sutrave et al. 1990). Ski can transactivate expression from muscle-specific reporter genes (Engert et al. 1995); however, its endogenous targets remain unknown. Although this body of work strongly suggests that ski plays a role in the normal development of skeletal muscle, the nature of that role is unclear.
Expression of ski is not tissue-restricted, but it is regulated in a distinct spatial and temporal fashion. Low levels of expression of ski mRNA have been detected in many tissues from adults in several vertebrate species (Li et al. 1986; Grimes et al. 1993; Sleeman and Laskey 1993; Lyons et al. 1994; Namciu et al. 1995). In the mouse, there is a low level of expression throughout the embryo, with superimposed upregulation in specific tissues and stages. The earliest changes are found in neural tissue and skeletal muscle. At embryonic days 8.5–9.5 (E8.5–9.5) ski mRNA levels are high in the neural tube and migrating neural crest (Lyons et al. 1994). Subsequently, ski expression is elevated in areas of the central nervous system that retain proliferative potential, such as the cerebellar granule cells, but also in some postmitotic neuronal regions. In skeletal muscle, ski mRNA levels are high only after E12.5, and return to background levels at E15.5, remaining very low thereafter and in adult skeletal muscle (Namciu et al. 1994). This pattern correlates with the proliferation of secondary myoblasts or differentiation of primary myotubes (Ontell and Kozeka 1984), again suggesting that ski may regulate either or both of these events in skeletal muscle development.
To investigate the function of the ski proto-oncogene in vivo, we have disrupted the gene in embryonic stem (ES) cells, and introduced the resulting mutation into the mouse germ line. Our results show that ski is indeed essential for mammalian development, and its expression is required for normal morphogenesis of the face and brain. ski-deficient mice die at birth and suffer primarily from exencephaly, a cranial neural tube defect (NTD). In the cranial neural tube of ski-deficient embryos we find excessive apoptosis and a decreased number of cells expressing nestin, a marker of neuroepithelial precursors. In addition, ski −/− mutants have severe defects in patterning of both vertebral and craniofacial skeletal structures derived from the cranial neural crest, as well as dramatic reductions in skeletal muscle mass. Thus, the timing and location of defects in ski-deficient mice coincide with sites of upregulated ski expression during development.
Results
Generation of ski-deficient mice
All of the sequences required for biological activity are encoded by the first coding exon (exon 1) of the ski gene in both birds and mammals (Zheng et al. 1997). To mutate the mouse ski gene, therefore, we used a neomycin-resistance expression cassette to disrupt exon 1 (Fig. 1A). Homologous recombination with the mouse genome should result in the introduction of two new BamHI sites within exon 1. In Southern analyses of BamHI-digested DNA, probes outside the targeting vector on the 5′ or 3′ sides detect fragments of 4.5 or 2.6 kb, respectively, in the targeted allele, whereas the wild-type allele yields a 7.1-kb band (Fig. 1B). E14.1 ES cells were transfected with the targeting construct, and of 632 clones screened, a single correctly targeted ES cell clone was identified. To verify that the mutation would eliminate Ski protein expression, we generated ES cell lines homozygous for the mutation by selecting for resistance to increased concentrations of G418 (Mortensen et al. 1992). Western analyses showed that Ski protein was easily detected in wild-type or heterozygous ES cells, but absent from the −/− cells (Fig 1C). These results have been confirmed by use of embryo fibroblasts from homozygous mutant mice (not shown).
Figure 1.

Targeted disruption of the ski gene in mice. (A) Diagram of the targeting vector and map of the targeted allele. (neo) pMC1 neo-expression cassette lacking a polyadenylation signal; (HSV-tk) pMC1-tk expression cassette used for negative selection. Positions of critical BamHI sites, location of the probes, and sizes of diagnostic fragments are indicated. (B) Southern analysis of genomic DNA isolated from offspring of heterozygote matings. DNA was digested with BamHI; blots were probed with the 3′ probe shown in Fig. 1A, as well as with labeled λ DNA, and exposed with a Molecular Dynamics PhosphorImager screen. (C) Western blot probed with G8 monoclonal antibody to Ski. The largest band corresponds to full-length ski; smaller forms may be degradation products. (Bottom) The same membrane stained with Sudan black to confirm equal loading and transfer of samples.
Cells from the singly targeted clone were injected into blastocysts and contributed to the germ line of chimeric mice. Chimeric mice were mated to either C57BL/6J or Swiss black females, and heterozygous offspring carrying the ski mutation were found to be healthy and fertile. Heterozygous mice were intercrossed, and their homozygous mutant offspring were found dead at birth due to exencephaly, that is, the absence of a cranial vault (Fig. 2A,B). Mutant pups had additional abnormalities in facial morphology (Fig. 2B) and skeletal muscles (see Figs. 6 and 7, below). After four generations of backcrosses into the random-bred, Swiss black and inbred C57BL/6 backgrounds, all of the phenotypes described continue to segregate with the targeted ski allele, confirming that the mutation we have introduced is the cause of the observed phenotypes.
Figure 2.
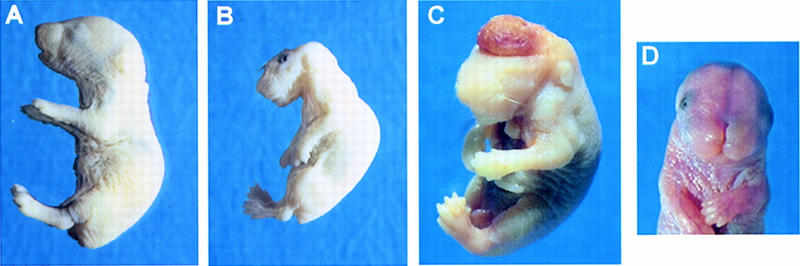
Morphological analysis of wild-type and ski −/− neonates. (A) Lateral view of a wild-type pup. (B) Lateral view of ski −/−, exencephalic pup. Note abnormal curvature of the back, neck, and head; the abnormal square jaw, flaccid limbs, and skinny forelimbs. (C) ski-deficient newborn delivered by C-section, showing the highly vascularized brain mass; this region is normally absent because it is sheared off during birth. Eyes are present and closed but not visible because this mutant is albino; most ski-deficient mice were born with open eyes, but some had normally shut eyelids. An abdominal cut was made to facilitate fixation. (D) ski −/− mouse with a cranial vault and brain, showing frontonasal clefting. Approximately 10%–15% of ski −/− mice had this phenotype (see Tables 1 and 2).
Figure 6.
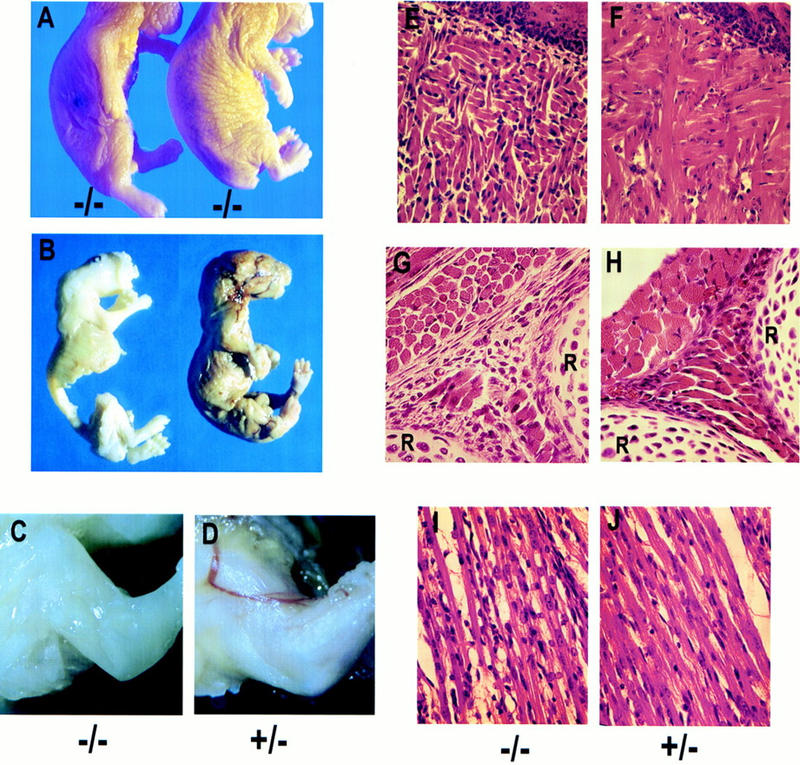
Reduced skeletal muscle mass in ski-deficient newborns. (A) Comparison of two ski −/−, exencephalic newborns with different degrees of muscle deficiency. Note the loose skin along the fore- and hindlimbs of the mouse on the right, which is not emaciated but skinny. (B) Comparison of wild-type and ski-deficient newborns after removal of the skin in preparation for skeletal staining. The ski −/− mouse (left) did not appear emaciated but merely skinny before the skin was removed. (C,D) Close-up comparison of the forelimbs from a ski-deficient pup and its normal heterozygous littermate, with the skin removed after fixation. Mutants appear white because of bleeding from the exposed, angiomatous brain mass; bleeding occurs throughout development but is most severe at birth, when the brain mass is sheared off. (E–J) Histological analysis of muscle groups in the tongue, intercostal region, and forelimb. Sagittal sections; sections from ski −/− newborns are on the left. (E,F) Sections through the tongue, with the epithelial surface on the upper right corner of each section. Note the shorter fibers, and empty space between fibers. (G,H) Sections through the intercostal muscles, with ribs (R) at bottom and right edges; note the reduced diameter of the muscle fibers in cross-section (upper left) and the disorganization and reduced number of fibers in the area between the two ribs (extreme right and bottom). (I,J) Sections through a forelimb muscle; note the reduced diameter of fibers in I, and increased numbers of nuclei not associated with fibers.
Figure 7.

Skeletal abnormalities in ski-deficient mice. In each pair, the ski −/− sample is on the left. (A,B) Skeletal preparations of a ski −/− mouse A and a wild-type littermate B. The mouse in A is missing the frontal, parietal, interparietal, and supraoccipital bones, but the nasal bone is present. Arrowheads indicate the first and second cervical vertebrae (C1 and C2), which are fused in the mutant. Arrow indicates the mandible, which is wider and shorter in the mutant than in the normal littermate. (C,D) Comparison of the basal bones of the skull shows that the supraoccipital bone (s) and the presphenoid bone (ps) are completely absent in the ski-deficient skeleton. bo, basioccipital; bs, basisphenoid. (E) Comparison of the basioccipital bones. Note the anterior arch of the atlas, which is fused to the top of the mutant bone (left). (F) Malformation of the basisphenoid bone.
Exencephaly, facial clefting, and perinatal lethality in ski-deficient mice
ski −/− pups delivered by cesarean section at embryonic day 18.5 (E18.5) were alive, but suffered from exencephaly (absence of the cranial vault) and severe hemorrhage from the exposed, angiomatous brain mass (Fig. 2C), which was normally sheared off at birth. Mutant pups were ∼10% smaller than their normal littermates, and this difference was exaggerated by the abnormal curvature of the spine. Facial morphology was abnormal in all mutants, and included a flat, foreshortened snout and abnormal jaw. This phenotype—exencephaly, abnormal posture, and abnormal facial morphology—was observed in about 85% of ski-deficient mice on either a 129 × Swiss background, or on a 129 × C57BL/6 background (Table 1). The remaining 15% showed facial clefting defects of varying severity, ranging from complete frontonasal clefting (as shown in Fig. 2D), to mild clefting of the lip and nose (Table 1; data not shown). However, even those with mild clefting defects died shortly after birth, presumably from breathing difficulties. Therefore, perinatal lethality of the ski null mutation is completely penetrant regardless of genetic background.
Table 1.
Genotypes and phenotypes of litters from heterozygous intercrosses
| Embryonic day
|
Numbera
|
Percent +/+b (% NTDc)
|
Percent +/−b (% NTDc)
|
Percent −/−b (% NTDc)
|
|---|---|---|---|---|
| 129 × Swiss | ||||
| 9.5 | 125 | 22 (0) | 56 (4) | 23 (86d) |
| 14.5 | 92 | 33 (0) | 52 (0) | 15 (93) |
| 18.5 | 102 | 33 (0) | 49 (2) | 18 (83) |
| Newborn | 147 | 31 (0) | 51 (3) | 18 (88) |
| 129 × C57BL/6 | ||||
| 14.5 | 46 | 28 (0) | 48 (5) | 24 (73) |
| 18.5 | 44 | 27 (0) | 57 (0) | 16 (86) |
| Newborn | 55 | 40 (0) | 58 (3) | 2e (100) |
Total number of embryos included.
Percent of total embryos with a given genotype.
Percent of embryos with a given genotype that exhibited an NTD.
Homozygous embryos without NTDs always had facial clefting.
C57BL/6 mothers presumably cannibalized abnormal pups.
Homozygous −/− pups totaled only 18% of the newborn offspring from heterozygous intercrosses on a mixed Swiss × 129 background, instead of the expected 25%, indicating some embryonic lethality (Table 1). At E14.5, the proportion of −/− embryos was similarly reduced, but at E9.5 they made up 23% of the total. Therefore, ∼25% of ski-deficient embryos died between E9.5 and E14.5. On a C57BL/6 × 129 mixed background, a similar rate of embryonic lethality was observed but occurred somewhat later; homozygous mutant embryos comprised 24% of the total at E14.5 but only 16% by E18.5.
We have observed no neural tube defects in wild-type mice, but a small proportion of ski +/− heterozygotes on both genetic backgrounds have exencephaly or facial clefting (Table 1), suggesting that loss of a single ski allele can lead to predisposition to neural tube defects. We have found no evidence of spina bifida, kinky tails, or other caudal neural tube defects in the ski −/− embryos or newborns.
Overexpression of ski has been shown to transform hematopoietic cells (Larsen et al. 1993), and ski expression is modulated during differentiation of megakaryocytes (Namciu et al. 1994). To determine whether the blood loss in ski −/− embryos reflected an underlying defect in hematopoiesis, we have performed differential counts on peripheral blood smears, and have examined histological preparations of livers and spleens. We found no differences between normal and mutant embryos. Differentiation of myeloid and erythroid precursors appeared normal in ski −/− embryos, megakaryocytes and platelets were present, and clotting times were comparable to those of normal ski +/+ and +/− littermates.
Neural tube defects in ski-deficient embryos
Exencephaly is a cranial neural tube defect, resulting from failed closure of the neural folds during neurulation. In the mouse, the neural tube initiates closure at E8.5, beginning at the cervical/hindbrain boundary (Morriss-Kay et al. 1994). Two additional de novo closure sites occur at the caudal and rostral limits of the forebrain (Juriloff et al. 1991). Closure then spreads along the neural folds in the rostral and caudal directions. By E9.5, closure is normally complete. To determine whether exencephaly in ski −/− mice was caused by a neural tube defect, we examined embryos at the time of neurulation. At E8.5, ski-deficient embryos could not be distinguished from their normal littermates. By E9.5, however, wild-type and heterozygous embryos usually had completely closed cranial neural tubes, while ski −/− embryos showed open and everted cranial neural folds (Fig. 3). In all cases, the closure defect involved the second de novo closure point at the midbrain/forebrain boundary, and extended rostrally to the anterior neuropore, or caudally to the cervical/hindbrain boundary, or both (see Figs. 2 and 3).
Figure 3.

Morphological analysis of E9.5 embryos. Left (A,C,E,G) show a ski −/− embryo, right (B,D,F) a normal +/− littermate. (A) Lateral view showing open neural folds starting at the midbrain and extending rostrally and impaired development of the forebrain. Except for the head, size of the mutant and normal embryos is comparable. (B) Normal heterozygous littermate of embryo shown in A. (C,D) Dorsal views showing the open and everted neural folds of the ski −/− embryo. The neural tube of the normal embryo in D is completely closed; the discontinuity is a photographic artifact. (E,F) Frontal sections through the plane of the paper of embryos shown in C and D, stained with hematoxylin and eosin. The arrowhead shows the ventricular surface of the neuroepithelium, which in the ski −/− embryo becomes exposed to the exterior. Arrows indicate the cranial mesenchyme. (G) Frontal section, at higher magnification, showing the caudal neural tube of the ski −/− embryo in E. (nt) Neural tube (caudal); (n) notochord; (s) somite. Tears in the neuroepithelium are sectioning artifacts.
Histological examination revealed no differences in the neuroepithelium of ski −/− embryos at E9.5 in comparison to normal littermates, other than the eversion that exposed the inner ventricular surface in the mutants (Fig. 3E,F, arrowheads). In the mesenchyme adjacent to the everted neuroepithelium, there was some disorganization and cells appeared sparser than in control embryos (Fig. 3E,F). This effect was likely secondary to the increased volume in the cranial region caused by the everted tissue, however, because facial mesenchyme (Fig. 3E,F, bottom) and somitic mesenchyme (Fig. 3G) appeared well-organized and normal. Closure of the caudal neural tube appeared normal in all embryos (Fig. 3G shows a ski −/− mutant), consistent with the absence of spina bifida or kinky tails in the newborn ski −/− mice.
Excessive apoptosis during neurulation in ski-deficient embryos
Enforced overexpression of ski in cultured cells has been shown to enhance both cell growth rates and viability (Stavnezer et al. 1986; Colmenares and Stavnezer 1989), and elevated ski expression has been documented in the neural tube during neurulation (Lyons et al. 1994). To determine whether the neural tube defect might be related to a decrease in cell proliferation or viability, we examined embryos at E9.5, the earliest time at which ski-deficient embryos developed abnormalities. BrdU labeling at E9.5 showed similar labeling indices of 44% and 39% in the rostral neuroepithelium of wild-type and ski −/− embryos, respectively (data not shown). Labeling indices in the cranial mesenchyme and in the caudal neural tube, which closed normally in all embryos, were also comparable between wild-type and ski −/− embryos.
In contrast, we observed a significant excess of programmed cell death in both the neural tube and craniofacial mesenchyme of E9.5 ski-deficient embryos compared with their normal littermates. During neurulation in normal embryos, apoptosis has been observed along the closing edges of the neural folds. In wild-type and normal ski +/− embryos, labeling of DNA ends with the TUNEL assay revealed a small number of apoptotic cells along the closing neural folds, as expected. However, ski −/− embryos showed moderately increased cell death along the unfused neural folds (not shown), and dramatically increased apoptosis in both the cranial neural tube (Fig. 4A,B), and in the craniofacial mesenchyme (Fig. 4B, arrow), particularly in the frontonasal masses. In the neuroepithelium, programmed cell death was most abundant in the region of differentiating cells moving away from the ventricular zone, towards the future mantle layer.
Figure 4.
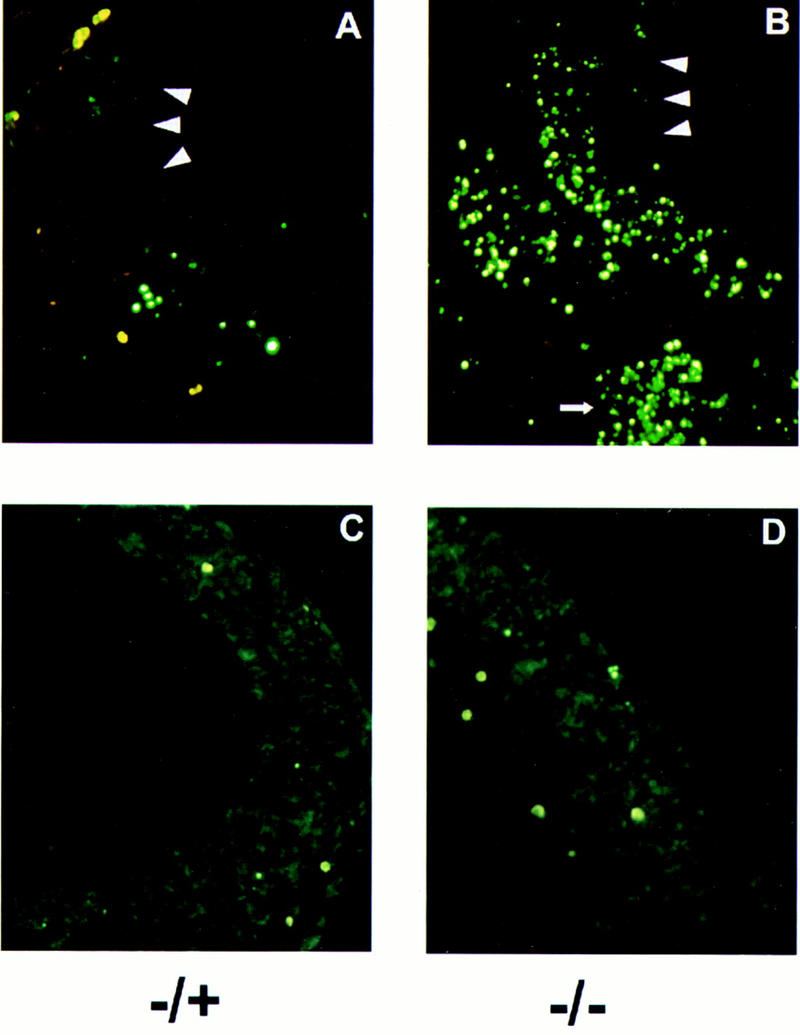
Excessive apoptosis in ski-deficient embryos at neurulation. Frontal frozen sections were prepared from embryos at E9.5, and analyzed with the TUNEL assay. (A,B) Ventricular edges of the neuroepithelium are indicated by arrowheads. Red blood cells are stained bright yellow; apoptotic cells that have incorporated labeled dUTP fluoresce green. DAPI staining (not shown) revealed that cells labeled with dUTP also showed condensed chromatin characteristic of apoptosis. The small arrow at the bottom of B shows abundant apoptosis in the craniofacial mesenchyme. In the neuroepithelium, apoptosis is concentrated along the mantle layer. (C,D) Frontal sections of embryos at E10.5, processed as described above. The amount of apoptosis is comparable in the −/− mutant and its heterozygous littermate at this stage.
The increase in apoptosis suggested two possibilities: excessive cell death could result directly or indirectly from the absence of Ski protein, leading to the neural tube defect; or apoptosis could be secondary to the NTD, and caused by exposure of the neuroepithelium to the amniotic environment. To distinguish between these possibilities, we examined embryos at E10.5, because in the latter case prolonged exposure might lead to increased cell death in the developing neuroepithelium. TUNEL staining of embryos at E10.5 revealed no significant difference in the number of apoptotic cells between ski −/− and normal littermates (Fig.4C,D), suggesting that excessive apoptosis is both temporally and causally related to the failed closure of the cranial neural folds.
Gene expression in the neural tube of ski-deficient embryos
Because excessive apoptosis was most abundant in the region associated with differentiating cells, we wished to determine whether cell death in this region might cause depletion of differentiated cells in the neuroepithelium of ski-deficient embryos. Therefore, we examined the expression of two markers of neural differentiation, nestin and tubulin. Nestin is an intermediate filament expressed at high levels in proliferating neuroepithelial stem cells and down-regulated during subsequent differentiation of these precursors into the neuronal or glial lineages (Lendahl et al. 1990; Zimmerman et al. 1994). Class III β-tubulin is one of the earliest markers of terminal neuronal differentiation (Geisert and Frankfurter 1989). We used antibodies specific for these markers to perform immunostaining of E9.5 embryos.
In heterozygous ski +/− embryos, nestin was detected in the mantle zone of the neuroepithelium in both the cranial (Fig. 5B) and caudal (Fig. 5D) portions of the neural tube. In contrast, ski −/− mutants showed a marked decrease in nestin-positive cells in the cranial neural tube (Fig. 5A). No such decrease was evident in the caudal neural tube (Fig. 5, cf. C and D). The decrease in nestin-positive cells indicates a reduction of neuroepithelial stem cells. Interestingly, the levels and limits of expression of β-III tubulin, a late marker of neuronal differentiation, were comparable in heterozygous and homozygous mutant embryos (Fig. 5E,F). These results are most consistent with a defect involving premature differentiation of nestin-positive stem cells, which may then undergo programmed cell death.
Figure 5.

Expression of nestin and β-III tubulin in heterozygous and ski-deficient E9.5 embryos. Frontal sections correspond to those shown in Fig. 3 (E–G), except that the caudal neural tube (C,D) is rotated sideways to show a larger area; large arrowheads in A, B, E, and F point to the ventricular surface of the neuroepithelium. Frozen sections were prepared and labeled with monoclonal antibodies to nestin or β-III tubulin, followed by FITC-labeled second antibody. In each pair, the ski −/− sample is on the left. (A,B) Expression of nestin in the cranial neural tube is evident at the outer edge of the mantle zone, where differentiated cells accumulate. Nestin-positive cells along the future mantle zone (away from the ventricular zone) are less abundant in the mutant A than in the +/− embryo shown in B. In addition, the −/− sample shows some staining of cells in the intermediate and exposed ventricular zone, which is not seen in the normal heterozygote and may indicate premature differentiation. (C,D) Expression of nestin in the somitic myotome and caudal neural tube. Small arrowheads point to the somitic myotomes; note nestin expression in myotome of the ski −/− embryo C is drastically reduced compared with the heterozygous embryo D, although expression in the caudal and closed neural tube is comparable in both sections. Note, however, the reduced size of the spinal cord in C. (E,F) Expression of β-III tubulin is comparable in ski-deficient E and heterozygous F embryos at E9.5.
We have used moncolonal antibodies and immunostaining to examine the levels of expression of additional gene products whose levels are regulated in the neuroepithelium or cranial mesenchyme during neurulation. These include neurofilaments, neural cell adhesion molecule (N-CAM), and chondroitin sulfate proteoglycan. As was the case with β-III tubulin, we have found no differences between ski −/− embryos and their normal littermates in the expression of any of these markers at E9.5 (data not shown).
Skeletal muscle abnormalities in ski-deficient mice
Gain-of-function studies in vitro and in vivo have suggested that ski might play a role in the development of skeletal muscle (Colmenares and Stavnezer 1989; Sutrave et al. 1990). In vivo, overexpression of ski in skeletal muscles of transgenic mice leads to skeletal muscle hypertrophy (Sutrave et al. 1990). Interestingly, ski −/− mutants showed a reduction in skeletal muscle mass, with variable penetrance and expressivity depending on genetic background. In offspring from 129 × Swiss intercrosses, ∼15% of ski −/− mutant mice were extremely emaciated at birth (Fig. 6A, left) while another 20% appeared somewhat skinny (Fig. 6A, right). The reduction of skeletal muscle mass was very severe in emaciated mice, and became obvious in the skinny mice after removal of the skin, and in comparison with normal littermates (Fig. 6B). Between 50% and 60% of ski −/− mutants on a 129 × C57BL6 background were obviously skinny, and tended to have flaccid limbs (see Fig. 2A,B). Reduced muscle mass was never observed among ski +/− heterozygous mice, even among those few that suffered from exencephaly.
Histological examination of skeletal muscles from skinny mutants showed reductions in the diameter of muscle fibers (Fig. 6, cf. G and H, and I and J), and increased space between fibers (Fig. 6E–J). In addition, as is evident in the tongue, intercostal muscles, and forelimb muscles, many fibers appeared shorter and disorganized, and their number was reduced and replaced by nonmuscle tissue (Fig. 6E–J, cf. nuclei not in myofibers).
Nestin expression has also been documented in myogenic cells, particularly in muscle precursors in the myotome and dermatome (Hockfield and McKay 1985; Sejersen and Lendahl 1993; Kachinsky et al. 1994). We found a significant reduction in the expression of nestin in the developing myotomes of ski −/− mutants in comparison with heterozygous littermates (Fig. 5C,D, arrowheads), although, as discussed above, there was no such reduction in the adjacent caudal neural tube. We have detected no differences in somitic programmed cell death between ski-deficient and normal heterozygous embryos.
To determine whether the reduction in skeletal muscle mass was accompanied by changes in expression of the myogenic regulatory factors, we prepared RNA from skeletal muscles of normal and ski-deficient embryos at E13.5–16.5, the stages when ski mRNA levels are elevated in skeletal muscle. Northern analyses by use of probes to the myogenic regulatory genes myoD, myogenin, MRF-4, and myf-5, and to p21Cip, revealed no changes in expression of these mRNAs in ski −/− mutants compared with their normal littermates (data not shown). Similarly, we have found no difference between normal and mutant E15.5 fetuses in the expression of desmin, MyoD, and myosin heavy chain as detected by immunostaining (not shown). Thus, although fibers formed are smaller and less organized, terminal differentiation appears qualitatively normal.
Skeletal abnormalities and partial homeotic transformations in ski −/− embryos
To examine more closely the defects in the cranial vault, and to determine the reason for the altered facial morphology of ski −/− mutants, we stained skeleton preparations of normal or mutant neonates with alcian blue and alizarin red. In exencephalic newborns, the frontal, parietal, and interparietal bones were obviously absent, as shown in Figure 7 (A,B). Additional defects were found in the basal cranial bones derived in part from the neural crest. The basioccipital and basisphenoid bones were malformed and undersized, whereas the presphenoid bone was absent in most of the exencephalic mutants (Fig. 7C–F). The mandibular bone was shorter and considerably thicker in ski-deficient mice (Fig. 7A, arrow).
Skeletal abnormalities extended caudally from the basioccipital bone to the first three cervical vertebrae (C1, C2, and C3) in ski −/− mutants (Fig. 7A,B, arrowheads). Defects in cervical vertebrae ranged from mild (broadening of C2) to very severe, as in the fusion of C1 and C2 (Fig. 7A,B). These changes can be characterized as partial anterior homeotic transformations because the identity of the posterior vertebrae resembled, but usually did not completely adopt, the appearance of the more anterior segment. ski −/− pups born with facial clefting had no vertebral homeotic transformations, but did show deformities of the basioccipital, basisphenoid, and presphenoid bones (data not shown). The penetrance of these skeletal abnormalities, shown in Table 2, was independent of genetic background, and very high; 100% of ski −/− neonates examined had some form of abnormality involving the atlas and axis, and 78% lacked the presphenoid bone, whereas the remaining 22% had deformed presphenoids.
Table 2.
Bone malformations in ski-deficient mice
|
|
+/+ (%) (n = 26)
|
+/− (%) (n = 46)
|
−/− (%) (n = 32)
|
|---|---|---|---|
| Vertebral abnormalities | |||
| broad C2 | 0 | 0 | 28 |
| C1 and C2 fusion | 0 | 0 | 34 |
| C1, C2, and C3 fusion | 0 | 0 | 9 |
| C2 to C1 transformation | 0 | 0 | 3 |
| fusion of aaaa to basioccipital | 0 | 3 | 37 |
| Presphenoid defects | |||
| absence of presphenoid | 0 | 0 | 78 |
| presphenoid malformations | 0 | 0 | 9 |
| cleft faceb | 0 | 0 | 13 |
| basisphenoid malformations | 0 | 0 | 50 |
(aaa) Anterior arch of the atlas.
Cleft face mutants had a presphenoid bone, but it was malformed in all cases.
Expression of the AP-2 transcription factor is normally concentrated in the cranial neural crest, and the neural tube defect in AP-2 knockout mice involves altered expression of N-CAM in the neuroepithelium and neural crest (Schorle et al. 1996; Zhang et al. 1996). We were particularly interested in examining their expression, because AP-2 is the only other gene whose knockout produces both exencephaly and facial clefting. By use of antibodies to both N-CAM and AP-2, we have detected no major differences in expression of these markers between normal and ski-deficient embryos. Therefore, the defects observed in neural crest-derived structures are not related to a global defect in the generation or migration of cranial neural crest cells.
Discussion
In this study we have shown that the ski proto-oncogene has important pleiotropic functions during mammalian development. We provide the first direct evidence that ski is required for development of the central nervous system, and specifically for closure of the cranial neural tube. We also show that ski is indeed required for the normal development of skeletal muscle in vivo. In addition, we have found that ski plays a role in specifying the identity of certain neural-crest-derived craniofacial structures. Our results reveal a striking correlation between the sites and timing of upregulated ski expression—in neural tube, neural crest, and skeletal muscle—and the developmental defects observed in its absence.
ski is required for cranial neural tube closure
Neurulation is a complex developmental process involving elevation of the neural plate to form lateral folds, followed by apposition and fusion of the neural folds (Copp 1993; Morriss-Kay et al. 1994). This process requires the concerted participation of multiple tissues in the embryo, including the neuroepithelium, neural crest, surface ectoderm, and cranial as well as ventral mesenchyme (Copp and Bernfield 1994). Both congenital and environmental conditions (such as temperature or maternal nutrition) can result in NTDs. Thus, it is not surprising that NTDs represent some of the most common human abnormalities found in ∼1 per 1000 births (Copp 1993; Copp and Bernfield 1994). As many as 12 separate genes have been estimated to direct neurulation, many of these with modifiers.
ski joins a varied group of genes required for neural tube closure during mouse development (Chen and Behringer 1995; Ishibashi et al. 1995; Sah et al. 1995; Schorle et al. 1996; Stumpo et al. 1995; Zhang et al. 1996; Zhao et al. 1996). On the basis of our results, we propose that ski is a particularly attractive candidate for involvement in human NTDs. First, the penetrance of exencephaly in ski −/− mutants is very high, even on a variable genetic background such as 129 × Swiss. Second, ski +/− heterozygotes show an increased risk for exencephaly, suggesting that it may be involved even in sporadic cases of NTDs. Finally, the appearance of NTDs in association with facial clefting syndromes in humans (Sedano and Gorlin 1988), may define a subset of birth defects whose etiology may involve mutations in ski.
Apoptosis in ski-deficient mice
Previous studies have reported excessive apoptosis only in the cranial mesenchyme of mice with exencephaly (Chen and Behringer 1995; Zhao et al. 1996). Our data show that excessive cell death in ski −/− embryos occurs in both cranial mesenchyme and cranial neural tube, and its timing coincides with the failure of neural tube closure. Thus, excessive apoptosis may cause or contribute to the neural tube defect in ski-deficient mutants. The fact that we observe no excessive apoptosis in the caudal neural tube, which closes normally to form the spinal cord, is consistent with this hypothesis. The elevated expression of ski mRNA in the neural tube at this time strongly argues that excessive apoptosis results from the absence of intrinsic Ski functions in a subset of neuroepithelial cells.
An in vivo role for ski in skeletal muscle development
Previous work has suggested a variety of functions for ski in the process of muscle development. Overexpression of ski in skeletal muscles of transgenic mice resulted in fiber-type specific hypertrophy, an effect occurring well after terminal differentiation (Sutrave et al. 1990; Leferovich et al. 1995). On the other hand, ski-induced myogenesis in quail fibroblasts suggested a role in both proliferation and myogenic determination (Colmenares and Stavnezer 1989). Analysis of ski expression during mouse development showed that upregulation of ski mRNA in muscle coincided with secondary myoblast proliferation (Namciu et al. 1995). Our work now shows a reduction in skeletal muscle mass in ski-deficient mice, resulting from a decrease in fiber size and density. This phenotype is most consistent with a defect in expansion or survival of secondary myoblasts, which could, in turn, result from either a proliferative deficiency or from premature differentiation. This type of defect would not affect the differentiation of primary myofibers, but could explain the observed subsequent reduction in fiber size and organization. In addition, the observed decrease in nestin expression in the myotome at E9.5 suggests the possibility of an additional, earlier defect in myogenic progenitors.
The defects caused in skeletal muscle by the absence of ski do not phenocopy any of the phenotypes observed in mice lacking the myogenic regulatory factors. Either myoD or myf5 are required for specification of the skeletal muscle lineage (Rudnicki et al. 1993), whereas myogenin is essential for terminal differentiation and fiber formation in vivo (Hasty et al. 1993; Nabeshima et al. 1993), and MRF4 appears to be dispensable during development for the formation of skeletal muscle (Zhang et al. 1995; Olson et al. 1996). The defects in ski-deficient mice seem, perhaps, closest to the myogenin mutants, in that it is principally secondary myogenesis that is affected (Venuti et al. 1995). The myogenin mutation has a more global effect on the terminal differentiation of secondary myotubes, however, whereas the effect of the ski mutation seems to be more quantitative. Thus, it is clear that ski is not essential for myogenic determination, or for primary myoblast differentiation. Instead, it may play a role in regulating the balance among proliferation, terminal differentiation, and survival in both the neural and skeletal muscle lineages.
ski is required for normal facial morphogenesis
The skeletal abnormalities that lead to craniofacial dysmorphology in ski −/− mice arise in bony structures derived from the cranial neural crest. However, mutant mice do not show widespread defects in all neural crest derivatives. Multiple studies suggest that patterning of craniofacial structures may require combinatorial expression of multiple transcription factors to specify the identity of any given segment. In this regard, disrupted expression of several homeodomain genes, such as Msx-1 (Satokata and Maas 1994; Mina et al. 1995), Hoxb7 (McLain et al. 1992), Dlx-2 (Qiu et al. 1995), and Cart1 (Zhao et al. 1996), has been reported to cause phenotypes that overlap with some of the defects present in ski-deficient mice. Our work suggests that some of these regulatory pathways require the activity of the ski proto-oncogene, and identifies a specific set of homeobox-containing genes as potential targets and interacting partners.
Vertebral defects phenocopy mutations in Hox4 paralogs
The defects in cervical vertebrae of ski-deficient mice closely resemble those observed in mice lacking one or more of the Hox4 paralogs (Ramirez-Solis et al. 1993; Horan et al. 1994; Kostic and Capecchi 1994; Horan et al. 1995a,b). In fact, they represent a phenocopy of a hypomorphic mutation in Hoxb4 (Ramirez-Solis et al. 1993). These data suggest that ski activity may contribute to modulating the levels or specifying the rostral domains of expression of the Hox4 paralogous genes. It will be extremely interesting to determine whether Hox4 expression is regulated by ski.
Regulatory pathways that require Ski activity
The activities of the ski proto-oncogene have offered an intriguing opportunity to study the regulated balance between cell growth and differentiation. Expression of ski is not tissue-restricted, and its biological activity likely depends on its expression levels and on its required association with other transcription factors. Given these features, the participation of ski in regulatory pathways involving sets of basic helix–loop–helix (bHLH) and homeobox-containing genes is novel and intriguing. Our findings have identified a set of transcription factors that may represent targets or partners of ski, and have also provided the basis for a genetic approach to test these interactions.
Materials and methods
Disruption of the ski gene in mouse ES cells
A λ clone isolated from a 129-mouse genomic library and containing the first coding exon of c-ski (Namciu et al. 1995) was used to generate the targeting vector shown in Figure 1A. A pMC1neo-expression cassette lacking a polyadenylation site was inserted into a SacI site in exon 1 with BamHI linkers; the cloning generated a small deletion in the ski coding sequence. This vector contained 6.7-kb of homology and included 5.9-kb upstream, and 0.8-kb downstream from the insertion site. An MC1tkpA cassette encoding the herpes virus thymidine kinase gene (HSV-tk) was added to the 3′ homology arm to enrich for homologous recombinants by use of negative selection. The vector was linearized and electroporated into E14.1 ES cells, which were then selected in G418 plus gancyclovir. 632 clones were screened with a probe outside the targeting vector within the 3′ intron, and two homologous recombinants were identified; however, only one of these was found to be correctly targeted after digestion with several restriction enzymes and probing with a fragment on the 5′ side of the insertion.
Generation of ski-deficient mice
The single correctly targeted clone was used for injection into C57BL6/J blastocysts. Chimeric mice were bred to either Swiss black or C57BL6/J mates. Germline transmission was detected by the agouti color of the offspring. Tail DNA was isolated from agouti pups and used for Southern blotting. Embryos were genotyped by Southern blotting of yolk sac DNA. Chimeric mice were also bred with 129/J mates to establish the ski mutation on an inbred background. The phenotype of ski −/− mice in this strain was identical to that observed previously, but genetic analysis in this strain was abandoned because of poor breeding.
Histological analysis and immunostaining
Embryos and newborns were processed for histological analysis as described (Ramirez-Solis et al. 1993). Briefly, embryos or mice were fixed in fresh 4% paraformaldehyde, dehydrated through ethanol, and embedded in paraffin. Four to 5 μm sections were cut and stained with hematoxylin and eosin. For immunostaining, fixed embryos were transferred to 30% sucrose in phosphate-buffered saline (PBS), embedded in OCT, frozen, and sectioned at 10 μm. Sections were preincubated in PBS plus 5% horse serum, then stained as described (Colmenares and Stavnezer 1989). After staining, sections were mounted in Mowiol plus DABCO. The following monoclonal antibodies were used: antibody to nestin (RAT401) and antibody to MyoD were obtained from the Developmental Studies Hybridoma Bank; antibody to the 160-kD neurofilament, HNK-1 antibody to N-CAM, antibody to desmin, and antibody to β-III tubulin were from Sigma.
TUNEL assay
Frozen sections were thawed, permeabilized in PBS with 0.1% Triton X-100, and incubated in TUNEL assay mix (Boehringer Mannheim) prepared according to the manufacturer’s instructions. Samples were then stained with DAPI and mounted in Mowiol plus DABCO.
Skeletal preparations
Neonates or fetuses at E18.5 were sacrificed, skinned, eviscerated, and fixed in 95% ethanol. Skeletons were then stained with alizarin red S and alcian blue, essentially as described (Hogan et al. 1994). However, a milder alkaline solution was used for digestion and clearing to prevent the skeletons of ski −/− mice from falling apart; this effect is presumably caused by the small amount of muscle in homozygous mutant mice.
Western analysis
Lysates isolated from ski −/− ES cell clones were run on a 7.5% SDS-PAGE gel and transferred to a PVDF membrane. After blocking, the membrane was probed with G8 monoclonal anti-Ski antibody, which was then detected by chemiluminescence using Western Light (Tropix).
Acknowledgments
We thank Ed Stavnezer for encouragement, critical comments, and for sharing unpublished data. We thank Tom Doetschman and John Duffy for their help with ES cells and with knockout technology; Trevor Williams for the AP-2 antibody; Bruce Trapp and Akiko Nishiyama, for sharing their expertise and reagents; Donna Driscoll, for critical reading of the manuscript, and Jim Lang for expert help with photography. We are also grateful to Ros Smith and Lakshmi Amaravadi for sharing their data prior to publication, and for stimulating discussions. This work was supported by a grant from the National Institutes of Health (HD30728 to C.C).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL colmenc@cesmtp.ccf.org; FAX (216) 445-6269.
References
- Chen ZF, Behringer RR. Twist is required in head mesenchyme cranial neural tube morphogenesis. Genes & Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Colmenares C, Stavnezer E. The ski oncogene induces muscle differentiation in quail embryo cells. Cell. 1989;59:293–303. doi: 10.1016/0092-8674(89)90291-2. [DOI] [PubMed] [Google Scholar]
- Colmenares C, Sutrave P, Hughes SH, Stavnezer E. Activation of the c-ski oncogene by overexpression. J Virol. 1991a;65:4929–4935. doi: 10.1128/jvi.65.9.4929-4935.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmenares C, Teumer JK, Stavnezer E. Transformation-defective v-ski induces MyoD and myogenin expression but not myotube formation. Mol Cell Biol. 1991b;11:1167–1170. doi: 10.1128/mcb.11.2.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ. Neural tube defects. Trends Neurosci. 1993;16:381–383. doi: 10.1016/0166-2236(93)90001-3. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Bernfield M. Etiology and pathogenesis of human neural tube defects: insights from mouse models. Curr Opin Pediatr. 1994;6:624–631. doi: 10.1097/00008480-199412000-00002. [DOI] [PubMed] [Google Scholar]
- Engert JC, Servaes S, Sutrave P, Hughes SH, Rosenthal N. Activation of a muscle-specific enhancer by the Ski proto-oncogene. Nucleic Acids Res. 1995;23:2988–2994. doi: 10.1093/nar/23.15.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisert EE, Jr, Franfurter A. The neuronal response to injury as visualized by immunostaining of class III beta-tubulin in the rat. Neurosci Lett. 1989;102:137–141. doi: 10.1016/0304-3940(89)90068-2. [DOI] [PubMed] [Google Scholar]
- Grimes HL, Ambrose MR, Goodenow MM. C-ski transcripts with and without exon 2 are expressed in skeletal muscle and throughout chick embryogenesis. Oncogene. 1993;8:2863–2868. [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- Hockfield S, McKay RD. Identification of major cell classes in the developing mammalian nervous system. J Neurosci. 1985;5:3310–3328. doi: 10.1523/JNEUROSCI.05-12-03310.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Constantini F, Lacy E. Manipulating the mouse embryo. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Horan GS, Wu K, Wolgemuth DJ, Behringer RR. Honeotic transformation of cervical vertebrae in Hoxa-4 mutant mice. Proc Natl Acad Sci. 1994;91:12644–12648. doi: 10.1073/pnas.91.26.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan GS, Kovacs EN, Behringer RR, Featherstone MS. Mutations in paralogous Hox genes result in overlapping homeotic transformations of the axial skeleton: Evidence for unique and redundant function. Dev Biol. 1995a;169:359–372. doi: 10.1006/dbio.1995.1150. [DOI] [PubMed] [Google Scholar]
- Horan GS, Ramirez-Solis R, Featherstone MS, Wolgemuth DJ, Bradley A, Behringer RR. Compound mutants for the paralogous hoxa-4, hoxb-4, and hoxd-4 genes show more complete homeotic transformations and a dose-dependent increase in the number of vertabrae transformed. Genes & Dev. 1995b;9:1667–1677. doi: 10.1101/gad.9.13.1667. [DOI] [PubMed] [Google Scholar]
- Ishibashi M, Ang SL, Shiota K, Nakanishi S, Kageyama R, Guillemot F. Targeted disruption of mammalian hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix-loop-helix factors, premature neurogenesis, and severe neural tube defects. Genes & Dev. 1995;9:3136–3148. doi: 10.1101/gad.9.24.3136. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ, Tom C, MacDonald KB. Normal mouse stains differ in the site of initiation of closure of the cranial neural tube. Teratology. 1991;44:225–233. doi: 10.1002/tera.1420440211. [DOI] [PubMed] [Google Scholar]
- Kachinsky AM, Dominov JA, Miller JB. Myogenesis and the intermediate filament protein, nestin. Dev Biol. 1994;165:216–228. doi: 10.1006/dbio.1994.1248. [DOI] [PubMed] [Google Scholar]
- Kostic D, Capecchi MR. Targeted disruptions of the murine Hoxa-4 and Hoxa-6 genes result in homeotic transformations of components of the vertebral column. Mech Dev. 1994;46:231–247. doi: 10.1016/0925-4773(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Larsen J, Meyer S, Steinlein P, Beug H, Hayman MJ. Transformation of chicken bone marrow cells by the v-ski oncogene. Oncogene. 1993;8:3221–3228. [PubMed] [Google Scholar]
- Leferovich JM, Lana DP, Sutrave P, Hughes SH, Kelly AM. Regulation of c-ski transgene expression in developing and mature mice. J Neurosci. 1995;15:596–603. doi: 10.1523/JNEUROSCI.15-01-00596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- Li Y, Turck CM, Teumer JK, Stavnezer E. Unique sequence, ski, in Sloan-Kettering avian retroviruses with properties of a new cell-derived oncogene. J Virol. 1986;57:1065–1072. doi: 10.1128/jvi.57.3.1065-1072.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons GE, Micales BK, Herr MJ, Horrigan S, Namciu S, Shardy D, Stavnezer E. Proto-oncogene c-ski is expressed in both proliferating and post-mitotic neuronal populations. Dev Dynam. 1994;201:354–365. doi: 10.1002/aja.1002010407. [DOI] [PubMed] [Google Scholar]
- McLain K, Schreiner C, Yager KL, Stock JL, Potter SS. Ectopic expression of Hox-2.3 induces craniofacial and skeletal malformations in transgenic mice. Mech Dev. 1992;39:3–16. doi: 10.1016/0925-4773(92)90021-b. [DOI] [PubMed] [Google Scholar]
- Mina M, Gluhak J, Upholt WB, Kollar EJ, Rogers B. Experimental analysis of Msx-1 and Msx-2 gene expression during chick mandibular morphogenesis. Dev Dynam. 1995;202:195–214. doi: 10.1002/aja.1002020211. [DOI] [PubMed] [Google Scholar]
- Morriss-Kay G, Wood H, Chen WH. Normal neurulation in mammals. Ciba Found Symp. 1994;181:51–63. doi: 10.1002/9780470514559.ch4. [DOI] [PubMed] [Google Scholar]
- Mortensen RM, Conner DA, Chao S, Geisterfer-Lowrance AA, Seidman JG. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima Y, Hanaoka K, Hayasaka M, Esumi E, Li S, Nonaka I. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature. 1993;364:532–535. doi: 10.1038/364532a0. [DOI] [PubMed] [Google Scholar]
- Nagase T, Mizuguchi G, Nomura N, Ishizaki R, Ueno Y, Ishii S. Requirement of protein co-factor for the DNA-binding function of the human ski proto-oncogene product. Nucleic Acids Res. 1990;18:337–343. doi: 10.1093/nar/18.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namciu S, Lieberman MA, Stavnezer E. Induction of the c-ski proto-oncogene by phorbol ester correlates with induction of megakaryocyte differentiation. Oncogene. 1994;9:1407–1416. [PubMed] [Google Scholar]
- Namciu S, Lyons G, Heyman HC, Colmenares C, Stavnezer E. Enhanced expression of mouse c-ski accompanies terminal skeletal muscle differentiation in vivo and in vitro. Dev Dynam. 1995;204:291–300. doi: 10.1002/aja.1002040307. [DOI] [PubMed] [Google Scholar]
- Olson EN, Arnold HH, Rigby PW, Wold BJ. Know your neighbors: Three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell. 1996;85:1–4. doi: 10.1016/s0092-8674(00)81073-9. [DOI] [PubMed] [Google Scholar]
- Ontell M, Kozeka K. The organogenesis of murine striated muscle: a cytoarchitectural study. Am J Anat. 1984;171:133–148. doi: 10.1002/aja.1001710202. [DOI] [PubMed] [Google Scholar]
- Qiu M, Bulfone A, Martinez S, Meneses JJ, Shimamura K, Pedersen RA, Rubenstein JL. Null mutation of Dlx-2 results in abnormal morphogenesis of proximal first and second branchial arch derivatives and abnormal differentiation in the forebrain. Genes & Dev. 1995;9:2523–2538. doi: 10.1101/gad.9.20.2523. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis R, Zheng H, Whiting J, Krumlauf R, Bradley A. Hoxb-4 (Hox-2.6) mutant mice show homeotic transformation of a cervical vertebra and defects in the closure of the sternal rudiments. Cell. 1993;73:279–294. doi: 10.1016/0092-8674(93)90229-j. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nature Genet. 1995;10:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nature Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–238. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- Sedano HO, Gorlin RJ. Frontonasal malformation as a field defect and in syndromic associations. Oral Surg Oral Med Oral Pathol. 1988;65:704–710. doi: 10.1016/0030-4220(88)90014-x. [DOI] [PubMed] [Google Scholar]
- Sejersen T, Lendahl U. Transient expression of the intermediate filament nestin during skeletal muscle development. J Cell Sci. 1993;106:1291–1300. doi: 10.1242/jcs.106.4.1291. [DOI] [PubMed] [Google Scholar]
- Sleeman JP, Laskey RA. Xenopus c-ski contains a novel coiled-coil protein domain, and is maternally expressed during development. Oncogene. 1993;8:67–77. [PubMed] [Google Scholar]
- Stavnezer E, Barkas AE, Brennan LA, Brodeur D, Li Y. Tranforming Sloan-Kettering viruses generated from the cloned v-ski oncogene by in vitro and in vivo recombinations. J Virol. 1986;57:1073–1083. doi: 10.1128/jvi.57.3.1073-1083.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpo DJ, Bock CB, Tuttle JS, Blackshear PJ. MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc Natl Acad Sci. 1995;92:944–948. doi: 10.1073/pnas.92.4.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutrave P, Kelly AM, Hughes SH. Ski can cause selective growth of skeletal muscle in transgenic mice. Genes & Dev. 1990;4:1462–1472. doi: 10.1101/gad.4.9.1462. [DOI] [PubMed] [Google Scholar]
- Venuti JM, Morris JH, Vivian JL, Olson EN, Klein WH. Myogenin is required for late but not early aspects of myogenesis during mouse development. J Cell Biol. 1995;128:563–576. doi: 10.1083/jcb.128.4.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Behringer RR, Olson EN. Inactivation of the myogenic bHLH gene MRF4 results in up-regulation of myogenin and rib anomalies. Genes & Dev. 1995;9:1388–1399. doi: 10.1101/gad.9.11.1388. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hagopian-Donaldson S, Serbedzija G, Elsemore J, Plehn-Dujowich D, McMahon AP, Flavell RA, Williams T. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 1996;381:238–241. doi: 10.1038/381238a0. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Behringer RR, de Crombrugghe B. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nature Genet. 1996;13:275–283. doi: 10.1038/ng0796-275. [DOI] [PubMed] [Google Scholar]
- Zheng, G.Z., J. Teumer, C. Colmenares, C. Richmond, and E. Stavnezer. 1997. Identification of a core functional and structural domain of the v-Ski oncoprotein responsible for both transformation and myogenesis. Oncogene (in press). [DOI] [PubMed]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]