

Abstract
Fibroblast growth factor-6 (FGF-6) belongs to a family of cytokines that control cell proliferation, cell differentiation, and morphogenetic events. Individual FGFs are either expressed widely or in a restricted pattern during embryonic, fetal, and adult life. FGF-6 exhibits a restricted expression profile predominantly in the myogenic lineage. Important functions in wound healing and tissue regeneration have been proposed for various FGFs in the past, although data from knockout mice have not supported this view. We have inactivated the FGF-6 gene in mice to investigate the role of FGF-6 in skeletal muscle development and regeneration. Wild-type mice up-regulate FGF-6 after skeletal muscle injuries and completely restore experimentally damaged skeletal muscle. In contrast, FGF-6(−/−) mutant mice show a severe regeneration defect with fibrosis and myotube degeneration. The number of MyoD- and Myogenin-expressing activated satellite cells after injury were significantly reduced in mutants. This reduction was not caused by a reduced pool of quiescent satellite cells but presumably by a lack of activation or proliferation. Interbreeding of FGF-6(−/−) mutants with mdx mice leads to striking dystrophic changes in skeletal muscles of double homozygous mice characterized by myotube degeneration, the presence of large amounts of mononuclear cells, and deposition of collagen. RNA analysis revealed an up-regulation of MyoD mRNA in mdx but not in FGF-6(−/−)/mdx double mutant mice. We conclude that FGF-6 is a critical component of the muscle regeneration machinery in mammals, possibly by stimulating or activating satellite cells.
Keywords: FGF-6, muscle regeneration, MyoD, mdx
A number of different growth factors is found in skeletal muscle of mice during development and adult life and are believed to control various aspects of organogenesis such as proliferation of muscle stem cells, migration of myogenic precursor cells, differentiation of myotubes, and regeneration of damaged muscle. The list of growth factors known to affect the behavior of skeletal muscle cells or to be expressed in skeletal muscle tissue is extensive, including members of the fibroblast growth factor (FGF) and transforming growth factor (TGF) families, human growth factor (HGF), leukemia inhibitory factor (LIF), platelet derived growth factor (PDGF), nerve growth factor (NGF), and several others (for review, see Arnold and Braun 1993; Grounds and Yablonka-Reuveni 1993). Most of these growth factors, however, are not only expressed in skeletal muscle but also in several other tissues. Physiological functions in skeletal muscle in vivo have been shown only for relatively few. For example, mice with null mutations in insulin-like growth factor 1 (IGF1) and IGF2 genes as well as in the gene encoding their receptor insulin-like growth factor receptor I (IGFRI) show muscle hypoplasia, although a widespread growth retardation in these mutants suggests a more general role in growth control of various organs (Baker et al. 1993). Mutation of the grb2-binding domain in the c-met receptor unveiled a role for HGF/Met in secondary fiber development during late myogenesis (Maina et al. 1996).
In contrast, inactivation of TGFβ1, TGFβ2, TGFβ3, LIF, PDGF, and NGF which all have proven activities on skeletal muscle tissue in vivo or cultured muscle cells in vitro, had no overt effects on normal muscle cell development or muscle physiology. Likewise, effects of FGFs on myoblast proliferation and differentiation in vitro are widely accepted (for review, see Olson 1992), although data from previous FGF knock-out mice did not reveal crucial roles in development and/or maintenance of muscle cells. The FGF family comprises 10 different members that mediate diverse biological responses such as mitogenesis, angiogenesis, wound healing, and inductive events in a number of different tissues (Baird and Klagsbrun 1991; Yamagushi and Roussant 1995). Several members of the FGF family (i.e., FGF-1, FGF-2, FGF-4, FGF-5, FGF-6, FGF-7, and FGF-8) are expressed in developing skeletal muscle (Mason 1994; Grass et al. 1996). Whereas FGF-4, FGF-7, and FGF-8 are expressed transiently in myotomes and FGF-1, FGF-2, and FGF-5 are found only at low levels or in distinct muscle cells, FGF-6 is the only member of this family that shows relatively high expression restricted to the myogenic lineage (deLapeyrière et al. 1993). A number of different genes encoding FGFs and FGF receptors has been mutated in mice including FGF-3, FGF-4, FGF-5, FGF-7, FGFR-1, and FGFR-3 (Hebert et al. 1994; Feldman et al. 1995; Yamagushi and Rossant 1995; Guo et al. 1996). Mutations in growth factors and receptors expressed in skeletal muscle resulted either in early embryonic lethality (FGF-4 and FGFR-1), thereby preventing analysis of their functions in muscle cells, or in no apparent phenotypic alterations of muscle development and its maintenance (FGF-5 and FGF-7).
Skeletal muscle regeneration after injury is characterized by the proliferation of muscle precursor cells and differentiation of these cells, followed by fusion with each other to form young multinucleated myotubes or with the ends of existing damaged myofibers. Similar to skeletal muscle development during embryogenesis, a tight control of proliferation and differentiation during regeneration is critical for the generation of functional tissue with the correct amount and types of cells. Recently, it has been shown that the myogenic determination factor MyoD is required for myogenic precursor cell (mpc) function in adult skeletal muscle (Megeney et al. 1996). Apparently, MyoD-deficient mice (Rudnicki et al. 1992) accumulate increased numbers of myogenic cells as a consequence of increased muscle stem-cell self-renewal and a reduction of satellite cell differentiation. As a result, MyoD mutant mice develop a severe deficiency in their skeletal muscle regeneration capacity.
Various pathophysiological changes associated with skeletal muscle regeneration have been described. Initially, damaged tissue is infiltrated by fibroblasts, inflammatory cells, and macrophages. Necrotic tissue is removed, revascularization starts, and proliferation of muscle precursor cells is initiated. It is not known which individual molecules are involved in this process, however, or how proliferation of mpcs is controlled (for review, see Grounds and Yablonka-Reuveni 1993). Important roles have been postulated for bFGF and IGF-1, as injections of neutralizing antibodies inhibit muscle regeneration (Lefaucheur and Sebille 1995a). Likewise, supplementation of growth factors like LIF, SF/HGF, and bFGF by bolus injections or pumps leads to increase of muscle fiber density and size after injury (Lefaucheur and Sebille 1995b). Most of these approaches, however, suffer from a number of inherent complications making an accurate assessment of the physiological role of these growth factors difficult. The generation of mutant mice by homologous recombination opens a new avenue. It is now possible to inactivate single candidate genes and analyze the regeneration capacity of muscle tissue in mutants.
In an attempt to elucidate the role of FGF-6 during normal muscle cell development and in regenerative processes, we have inactivated the FGF-6 gene by homologous recombination in mouse embryonic stem (ES) cells. Regeneration of skeletal muscle was studied in FGF-6 mutants after freeze–crush injury and after interbreeding of FGF-6 mutants to mdx mice. On the basis of striking dystrophic changes in regenerating muscle of FGF-6(−/−) mutants and in FGF-6(−/−)/mdx mice and a reduction of MyoD- and Myogenin-expressing activated muscle precursor cells, we propose that FGF-6 is required for efficient muscle regeneration either by activation of satellite cells or stimulation of proliferation of mpcs.
Results
Inactivation of the FGF-6 gene
The vector used to target the FGF-6 gene was designed to replace 3.8 kb of the genomic locus with a neomycin resistance cassette. The deleted region comprised most of exon I, starting at a XhoI restriction site, and the complete exon II (Fig. 1A). Deletion of virtually the complete receptor-binding domain prevents receptor binding of any potential protein product encoded by exon III. Removal of the splice donor site of exon I and disruption of the reading frame would also prevent the translation of a leader sequence necessary for secretion.
Figure 1.

Targeted disruption of the FGF-6 gene. (A) Schematic representation of the FGF-6 gene (top), the targeting vector (middle), and the mutant allele (bottom). The gene consists of three exons (crossed boxes). The neo-expression cassette, including the phosphoglycerate kinase promoter and a polyadenylation signal, was used to replace most of exons I and II encoding the receptor-binding domain. (B) Wild-type (7.5 kb) and mutant alleles (5.5 kb) are shown on a Southern blot performed with KpnI-digested DNA from a cross of heterozygous FGF-6 parents by use of probe 1 for hybridization. (Lanes 1,3,6,8) DNA from mice heterozygous for the FGF-6 mutation. (Lanes 2,7) DNA from wild-type mice. (Lanes 4,5) DNA from mice homozygous for the FGF-6 mutation. (C) RT–PCR analysis of FGF-6 expression in wild-type (lane 2) and FGF-6 mutant mice (lane 3) at embryonic day 17.5. Primers specific for FGF-6 and GAPDH were used. No FGF-6 RNA was detected in homozygous mutants.
The targeting vector was electroporated into J1–ES cells. After double selection with G418 and FIAU, resistant colonies were picked and expanded. Twenty-three homologous recombinants were identified with a 5′-external probe by Southern blot analysis of DNA from 323 individual clones. This represents a targeting frequency of 1 in 14 double-selected clones (Fig. 1B). Mutated ES-cell clones were tested for integration of a single neo sequence by hybridization to a neo probe. Three independently derived ES clones heterozygous for the FGF-6 mutation were injected into C57/BL6 host blastocysts to generate chimeric founders. Two clones contributed to the germ line as assessed by breeding chimeras to C57/BL6 mice. Founder mice were either backcrossed to C57/BL6 mice or crossed to 129SvJ mice to generate inbred lines. For the present analysis, animals from F1 and F2 outbred generations were used. Heterozygous FGF-6(+/−) mice appeared normal and were bred to produce mice homozygous for the mutant allele (FGF-6 −/−). Southern blot analysis of 155 offspring at weaning (21 days postpartum) revealed no significant deviation from the predicted 1:2:1 Mendelian distribution (37:76:42) of wild-type (+/+), heterozygous (+/−), and homozygous mutant animals (−/−). The gross phenotypical appearance of homozygous FGF-6 mutants was normal, although FGF-6 mutants seemed to have a shorter snout compared with their littermates. Examination of alizarin-stained skeletons of FGF-6 mutants and age-matched controls, however, did not reveal any significant differences of the size or shape of mandibular and maxillar bones. Homozygous FGF-6 mutants, when interbred, were fertile and gave rise to normal litter sizes.
RT–PCR analysis was performed with RNA from tissues of FGF-6 mutant mice to ascertain the lack of transcripts from the mutated locus. As expected, FGF-6 mRNA was absent in E17.5 embryos (Fig. 1C) and in muscle tissue of adult mice (Fig. 5, below).
Figure 5.

Impaired up-regulation of MyoD and Myogenin mRNAs in skeletal muscle of FGF-6 mutant mice after crush injury. RNA was isolated from the musculus tibialis anterior of normal and FGF-6 mutant mice 3 days after crush injury. RT–PCR was performed to detect expression of FGF-6, of the myogenic factors Myogenin and MyoD, and of GAPDH. Two percent of the reaction volume was removed after various amplification cycles as indicated and hybridized with the corresponding probes. Expression of Myogenin and MyoD was severalfold lower in regenerating muscle of FGF-6 mutant mice compared with regenerating muscle of wild-type mice. The number of amplification cycles after which an aliquot was removed is indicated at the top of each lane. As expected, no FGF-6 mRNA was detected in FGF-6 mutant mice, whereas expression levels of GAPDH were similar in wild-type and FGF-6 mutant mice.
Normal skeletal muscle formation in FGF-6 mutant mice
Skeletal muscle of FGF-6 mutant mice was examined at various stages of development by light and electron microscopy. Hematoxylin/eosin (HE)-stained muscle sections from mutant mice were indistinguishable from those of wild-type mice (data not shown). Likewise, immunohistochemical stainings on cryostat and paraffin sections with various antibodies directed against fast and slow MyHC, actin, titin, and desmin revealed no significant differences. Next, we performed transmission electron microscopy to compare skeletal muscle of wild-type and mutant mice at the ultrastructural level and to count the number of quiescent satellite cells. Although no faithful molecular marker exists for quiescent satellite cells, they can be reliably identified by their location external to the sarcolemma and beneath the basal lamina of muscle fibers (Bischoff 1994). No changes in the ultrastructure of myotubes were noted between wild-types and FGF-6 mutants and morphologically normal satellite cells were found in sections from the Musculus tibialis anterior of adult mutant mice (data not shown).
The number of satellite cells was then counted in mutant mice and wild-type littermates relative to the presence of nuclei within myotubes. In FGF-6 mutant mice, 4.5% of counted nuclei were located in satellite cells compared with 2% in wild-type controls. Although only satellite cells within the basal lamina and not outside can be identified by electron microscopy (Grounds and Yablonka-Reuveni 1993), our data clearly indicate that FGF-6 is dispensable for the development of satellite cells. In contrast, the lack of FGF-6 seems to result in a slight increase of the number of satellite cells. At first, this seems to represent a paradox. FGF-6, however, may be involved in the activation of satellite cells by stimulating their entry into the cell cycle. This entry might result in a continuous removal of cells from the quiescent satellite cell pool and promotion of the progression through the myogenic program. The disruption of this process by lack of FGF-6 may block the flow of satellite cells and/or an increased tendency for self-renewal.
Expression of FGF-6 is stimulated after skeletal muscle injury
Given recent reports that various FGFs like FGF-2 (Walgenbach et al. 1995) and FGF-7 (Werner et al. 1992) are elevated during wound healing and tissue regeneration and the potential role of FGF-6 in the control of muscle stem cell proliferation, we investigated FGF-6 expression in regenerating muscle after crush injury compared with normal muscle in wild-type mice. RNAs were isolated from tissues on the third postoperative day and analyzed by semiquantitative RT–PCR followed by Southern blot hybridization. In the same experiment, we examined the expression pattern of the myogenic determination genes MyoD and Myogenin known to be up-regulated during muscle regeneration (Füchtbauer and Westphal 1992; Grounds and Yablonka-Reuvini 1993). Significantly fewer amplification cycles were needed with RNA from regenerating muscle to yield comparable signals for FGF-6, MyoD, and Myogenin than with RNA from normal muscle, whereas no differences were observed when primers specific for GAPDH were used (Fig. 2). These results show that FGF-6 as well as MyoD and Myogenin mRNAs are concomitantly up-regulated with the regeneration of skeletal muscle after crush injury. The increase of FGF-6 expression may even be underestimated, as surrounding normal tissue may dilute the actual mRNA concentration in the regenerating area.
Figure 2.

FGF-6 expression is increased in regenerating muscle tissue of wild-type mice after crush injury. RNA was isolated from the musculus tibialis anterior of control mice or 3 days after crush injury. RT–PCR was performed to detect expression of FGF-6, of myogenic factors Myogenin and MyoD, and of GAPDH. Two percent of the reaction volume was removed after 1, 15, 20, and 24 amplification cycles and hybridized with corresponding probes. Expression of FGF-6, Myogenin, and MyoD increased several fold in regenerating muscle compared with normal muscle (cf. lanes 3 and 7 or lanes 4 and 8).
FGF-6 mutant mice show a severe regeneration defect with enhanced fibrosis and myotube degeneration
To determine the functional significance of the increased FGF-6 expression in skeletal muscle after damage and to evaluate the importance of FGF-6 for skeletal muscle regeneration, we performed crush injury-induced regeneration experiments in FGF-6 mutant mice. All regeneration experiments shown were done on 8- to 10-week-old male mice with wild-type littermates as controls to minimize differences in age, sex, or genetic background.
Skeletal muscle of FGF-6 mutant mice displayed a prominent regeneration defect that was already apparent after 4 days but was most striking 10–14 days after injury. Analysis of HE-stained longitudinal sections of wild-type mice 4 days after injury revealed well-advanced regeneration with many new myofibers that almost crossed the lesion in wild-type mice (Fig. 3A). In mutants, the amount and size of newly formed myotubes were reduced and the number of mononuclear cells was much higher. Two weeks after injury, virtually no sign of previous damage was detectable in wild-type mice indicating a complete regeneration (Fig. 3C). In FGF-6 mutants, however, high numbers of mononuclear cells were visible, in addition to hyaline bodies and remnants of degenerated myotubes (Fig. 3D). Next, we investigated the presence of collagen with the van Gieson procedure, which stains collagen brightly red. As shown in Figure 3E, no major collagen deposits were discernible in wild-type mice 2 weeks after injury. In contrast, large amounts of collagen were detected in FGF-6 mutants indicating fibrosis of injured muscle tissue along with large numbers of degenerated myotubes and mononuclear cells (Fig. 3F). Staining of damaged muscle tissue 2 weeks after injury with a monoclonal antibody against fast MyHC (map My32) revealed the persistence of the lesion in mutants that lacked regenerated myotubes expressing MyHC, whereas wild-type mice showed normal regeneration as indicated by the presence of regularly shaped myotubes expressing fast MyHC (Fig. 6C,D, below).
Figure 3.
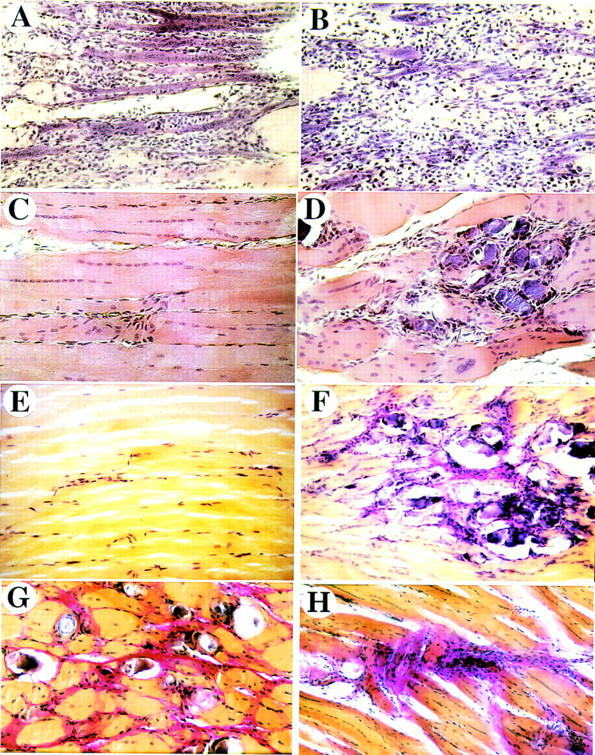
Freeze–crush-induced injuries result in impaired skeletal muscle regeneration of FGF-6 mutant mice. Paraffin sections from wild-type (A,C,E) and FGF-6 mutant mice (B,D,F,G,H) were stained with HE (A–D) and according to the van Gieson procedure (E–H) after 4 days (A,B), 14 days (C–F), and 22 days (G,H) following injury. In wild-type mice well-advanced regeneration is visible after 4 days (A) and regeneration is complete after 14 days (C,E). In FGF-6 mutant mice a regeneration defect is already visible after 4 days B but is most striking at 14 days (D,F) and 22 days (G,H). In mutants, lesions contain large amounts of collagen that stain red in the van Gieson procedure and large amounts of fibroblastoid cells. After 3 weeks, damaged areas in mutants sometimes contain degenerated myotubes (G) or collagen deposits with large amounts of cells (H). In wild-type mice virtually no sign of the previous injury was detectable after 2 weeks (C,E). Magnifications, 200× (A–D) and 100× (E–H).
Figure 6.

Large numbers of PCNA-expressing cells are present in regenerating muscle of FGF-6 mutant mice after crush injury. Cryostat sections from wild-type (A,C) and FGF-6 mutant mice (B,D) were reacted with a monoclonal antibody against PCNA after 3 days (A,B) and with a monoclonal antibody against fast MyHC (mAb MY32) 2 weeks following crush injury (C,D). High amounts of PCNA-positive nuclei indicating proliferating cells were present in regenerating muscle tissues of wild-type and FGF-6 mutant mice. In FGF-6 mutants, PCNA-positive cells most likely represent fibroblastoid cells, because MyoD-positive mpcs are drastically reduced. In contrast to wild-type mice (C) FGF-6 mutants do not display MyHC-expressing differentiated myotubes in damaged muscle areas (D).
Three weeks after injury, the lesion was no longer noticeable in wild-type mice (data not shown). In FGF-6 mutants, however, two types of reactions were found: One group presented a strong fibrosis with large amounts of mononuclear cells; another group presented moderate fibrosis with high numbers of degenerated myotubes and mononuclear cells (Fig. 3G,H).
In total, 38 animals were analyzed after crush injury. In each case of FGF-6 mutant mice, the typical pathological reaction was detected. Wild-type littermates, however, never showed a similar response. They always displayed well-advanced regeneration after 3–8 days and complete reconstitution after 2–3 weeks.
Expression of MyoD and Myogenin is reduced in regenerating muscle tissue of FGF-6 mutants
After damage, injured skeletal muscle tissue is infiltrated by large numbers of mononuclear cells, in particular, eosinophils, neutrophils, macrophages, and fibroblasts. In addition, satellite cells are activated, proliferate, and invade the injured site. Activated satellite cells and mpcs can be identified by the presence of the myogenic determination factor MyoD. In a more advanced state of differentiation, mpcs start to express Myogenin. In adult rodents, MyoD and Myogenin are only found in activated satellite cells and mpcs derived thereof. Nondividing satellite cells cease to express MyoD and Myogenin after birth during young adolescence. Soon after Myogenin expression starts, fusion of myoblasts commences and myotubes begin to mature (Braun et al. 1989; Koishi et al. 1995).
The regeneration defect of FGF-6-deficient mice prompted us to analyze the presence of activated mpcs and the amount of mpcs undergoing differentiation at different times following injury with monoclonal antibodies against MyoD and Myogenin. Three days after injury, substantial numbers of MyoD-positive cells were detected in wild-type mice. Frequently, they were found abundantly in clusters within damaged muscle areas (Fig. 4A). In contrast, only few MyoD-positive cells were observed in regenerating muscle of FGF-6-mutants (Fig. 4B). At the fifth day after injury, the amount of MyoD-expressing cells decreased in wild-type and FGF-6-mutant mice with only occasional MyoD-expressing cells in the mutant (Fig. 4C,D). Nine days after injury, few MyoD-positive cells were still detected in wild-type mice located between regenerated myotubes, whereas in FGF-6 mutants MyoD-positive cells were very rare (Fig. 4E,F).
Figure 4.

Reduction of MyoD-positive muscle precursor cells 3, 5, and 9 days after crush injury in FGF-6 mutant mice. Cryostat sections from wild-type (A,C,E) and FGF-6 mutant mice (B,D,F) were reacted with a monoclonal antibody against MyoD after 3 days (A,B), 5 days (C,D), and 9 days (E,F) following crush injury. Large numbers of MyoD-expressing activated satellite or mpcs are visible in wild-type mice (A), whereas only few MyoD-positive cells are detectable in FGF-6 mutants (B). The amount of MyoD-expressing cells is reduced after 5 (C,D) and 9 days (E,F) following injury. After 9 days, only a few activated mpcs are visible in the lesion of mutants, whereas considerable numbers of MyoD-expressing mpcs are visible between regenerated myotubes in wild-type mice (E). Pictures were taken with Nomarski optics. Magnification, 200×.
Similar results were obtained by use of an antibody against Myogenin even though Myogenin-expressing cells appeared less frequent than MyoD-positive cells and immunoreactive cells seemed not to be clustered. In wild-type mice, the number of cells expressing Myogenin remained stable for several days but significantly decreased in FGF-6 mutants. Nine days after injury, very few, if any, Myogenin-expressing cells were detected in the regenerating muscle of FGF-6 mutants, whereas several mpcs expressing Myogenin were still present in close proximity to regenerating myotubes of wild-type mice (data not shown).
In some regeneration experiments, we also examined RNA levels by semiquantitative RT–PCR. Expression of four different transcripts were analyzed: (1) MyoD; (2) Myogenin; (3) FGF-6; and (4) glycerealdehyde-3phosphate dehydrogenase (GAPDH). Expression of GAPDH was used to assure that similar amounts of cDNA were present in each amplification reaction. The RT–PCR analysis corroborated the data obtained by immunohistochemistry. The amount of MyoD mRNA was much higher in wild-type mice compared with FGF-6 mutant mice 3 days following injury (signal was visible after 21 cycles compared with 27 cycles; Fig. 5). Similarly, the amount of Myogenin expression was clearly reduced in mutant mice (24 cycles in wild-type and 30 cycles in mutant mice) whereas the GAPDH mRNA levels was comparable between wild-type and mutant mice (same number of cycles necessary). As expected, FGF-6 transcripts were absent in mutants, whereas considerable levels of FGF-6 mRNA were present in wild-type animals 3 days after injury. In contrast, by use of primers specific for GAPDH, a plateau level was reached after the same number of amplification cycles in wild-type and mutant mice.
To examine the general proliferation capacity within lesions, we stained with an antibody against proliferating cell nuclear antigen (PCNA) 3 days after injury. PCNA is expressed in replicating cells throughout S phase and thus allows detection of all dividing cells. Counts of PCNA-expressing cells in wild-type and FGF-6 mutant mice revealed a slightly higher number of PCNA-positive cells in mutants (Fig. 6A,B). Note, however, that the number of MyoD- and Myogenin-expressing cells was significantly reduced, and damaged muscle tissue contained fewer or no differentiated myotubes in FGF-6 knockout mice 14 days after injury (Fig. 6C,D). Thus, it seems likely that fibroblasts and other replicating cells are stained by the PCNA antibody in mutants. This conclusion is supported by the presence of many mononuclear cells in regenerating skeletal muscle tissue and large depositions of collagen indicating the activity of fibroblasts in regenerating tissue.
It is known that FGFs have important functions in neovascularization, for example, after ischemia (Yanagisawa-Miwa et al. 1992; Walgenbach et al. 1995). To assess whether angiogenesis in regenerating muscle of FGF-6 mutants was impaired, immunohistochemistry with a vascular endothelial cell marker CD 31 was carried out (Vecchi et al. 1994). We found no significant differences in the number of CD31-stained microvessels between mutant and wild-type mice (data not shown). However, we cannot completely rule out that small differences in the amount or diameter of microvessels exist between mutant and wild-type mice.
Skeletal muscles of FGF-6(−/−)/mdx double mutants are characterized by myotube degeneration, large amounts of mononuclear cells, fibrosis, and attenuated hypertrophy
To further determine the role of FGF-6 in skeletal muscle regeneration, we crossed FGF-6-deficient mice with the mdx mouse strain and with MyoD-deficient mice. mdx mice carry a point mutation in the dystrophin gene that results in continuous damage and subsequent degradation of single myotubes. Unlike the corresponding human disease, Duchenne muscular dystrophy, mdx mice are able to compensate for the loss of myotubes because of their high regeneration capacity, with the exception of the diaphragm, which is the only muscle that undergoes extensive dystrophic alterations in mdx mice (Stedman et al. 1991).
FGF-6(−/−)/mdx mice were found to be viable and fertile and showed no marked clinical signs of severe dystrophy up to 6 months of age except for a dorsal–ventral curvature of the spine observed in many, but not all, double mutants. Histological examination of the diaphragm, limb muscles, and autochtone back muscles of FGF-6(−/−)/mdx mice, however, revealed signs of a severe myopathy (Fig. 7G,H,I) whereas no phenotypical changes were found in skeletal muscles of FGF-6 mutants (Fig. 7A,B,C) and mdx mice (Fig. 7D,E,F). In the diaphragm muscle, a pronounced increase of collagen content was detected as visualized by van Gieson staining (Fig. 7C). Furthermore, mononuclear cells were considerably more frequent in double mutant than in mdx and FGF-6(−/−) mice. Because these cells were mostly embedded in collagen, we conclude that the majority of mononuclear cells were fibroblasts. Interestingly, the diaphragm of FGF-6(−/−)/mdx mice contained only unusually small myotubes in contrast to the diaphragm of mdx mice that contained myotubes of different sizes caused by asynchronous regeneration and hypertrophy. In addition, only a few centrally located nuclei typical for freshly regenerated myotubes were present in the diaphragm of FGF-6(−/−)/mdx mice whereas in mdx mice most of the nuclei were located in the center of the myotubes.
Figure 7.
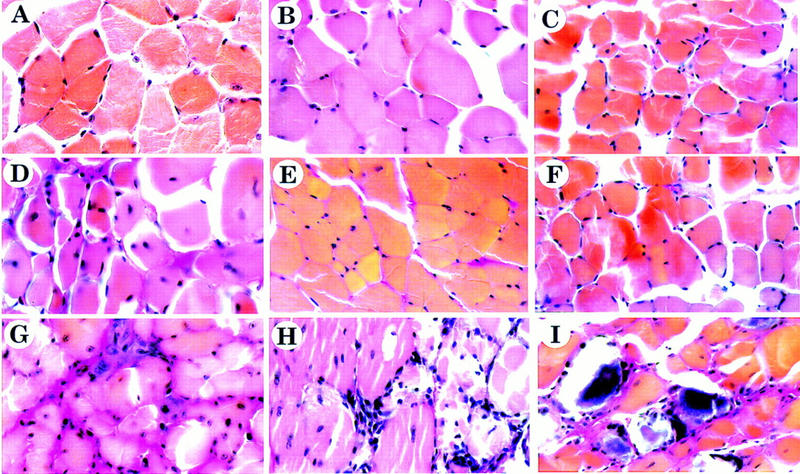
FGF-6(−/−)/mdx double mutant mice show a severe myopathy. Muscle tissue was isolated from diaphragm (A,D,G), musculus tibialis anterior (B,E,H), and musculus erector trunci (C,F,I) of FGF-6(−/−) mice (A–C), mdx mice (D–F), and FGF-6(−/−)/mdx mice (G–I). Sections were prepared and stained according to van Gieson (Bancroft and Stevens 1990). The diaphragm of FGF-6 mutant mice has regularly shaped myotubes. Collagen in larger amount is only found adjacent to a blood vessel (A). Few mononuclear cells are present between myotubes. The diaphragm of mdx mice is characterized by myotubes of different calibers, increased deposition of collagen, and increased numbers of mononuclear cells (D). Note the presence of centrally located nuclei typical for regenerated myotubes. In FGF-6(−/−)/mdx double mutant mice deposition of collagen and mononuclear fibroblastoid cells have increased significantly (G). Muscle sections of FGF-6(−/−) and mdx mice from musculus tibialis anterior (B,E) and musculus erector trunci (C,F) do not reveal any pathological alterations. In sharp contrast, limb muscles of FGF-6(−/−)/mdx mice show large areas with dystrophic changes along with huge amounts of mononuclear cells. G displays the boundary of a dystrophic area containing cellular infiltrations to surrounding healthy muscle tissue. Particularly severe dystrophic changes were found in deep back muscles of FGF-6(−/−)/mdx mice with necrotic myotubes and large amounts of mononuclear cells and collagen (I). It should be noted that dystrophic changes in limb and deep back muscles were not uniformly distributed but occurred only at some locations. Magnifications, 630×.
In limb muscles of FGF-6(−/−)/mdx mice, extensive areas with dystrophic changes were observed along with large amounts of mononuclear cells. In contrast to the diaphragm, deposition of collagen was moderate. Figure 7G shows the boundary of a dystrophic area containing cellular infiltrations to surrounding muscle tissue that appeared relatively healthy. Dystrophic changes were particularly severe in deep back muscles of FGF-6(−/−)/mdx mice with necrotic myotubes and large amounts of mononuclear cells and collagen (Fig. 7I). Interestingly, the histological picture strongly resembled limb muscle tissue of FGF-6 mutants after freeze–crush injury (cf. with Fig. 3D). In body wall muscles of double mutants, large patches containing huge numbers of cells embedded in collagen were found (data not shown). It should be noted that in contrast to the diaphragm, dystrophic changes in limb, deep back, and body wall muscles were not uniformly distributed within skeletal muscles but occurred at several yet restricted locations. The presence of degenerated myotubes, large amounts of mononuclear cells, and the pronounced fibrosis indicate that the regeneration capacity of skeletal muscles is severely compromised in FGF-6(−/−)/mdx mice.
To analyze whether a combined knockout of MyoD and FGF-6 affects muscle development, we interbred MyoD(−/−) mice (Rudnicki et al. 1992) with FGF-6(−/−) mice. Double homozygous mice were detected after weaning with no significant deviation from the predicted Mendelian distribution. MyoD(−/−)/FGF-6(−/−) mice were fertile but showed a significant reduction of litter sizes. Preliminary analysis of MyoD(−/−)/FGF-6(−/−) mice revealed no obvious malformations (data not shown).
MyoD expression is up-regulated in skeletal muscles of mdx mice but not in FGF-6(−/−) and FGF-6(−/−)/mdx mice
We next wanted to characterize the regeneration potential of FGF-6, mdx, and FGF-6(−/−)/mdx mice on the basis of the presence of activated mpcs expressing MyoD in different muscles. Radioactive in situ hybridization was performed with paraffin sections from diaphragm muscle of FGF-6, mdx, and FGF-6(−/−)/mdx mice with a MyoD cRNA probe (Fig. 8A,B,C). Whereas only background signals were detected on sections from FGF-6 mutant mice, strong hybridization signals were observed on sections of mdx mice. Interestingly, hybridization signals dropped to background levels when diaphragm muscle from FGF-6(−/−)/mdx double mutant mice was used. Because quantification of RNA expression is difficult to achieve by in situ hybridization, a Northern blot analysis was performed with RNA isolated from back muscles of FGF-6, mdx, FGF-6(−/−)/mdx mice, and wild-type E17.5 embryos (Fig. 8D). Again, very low MyoD expression was observed in FGF-6 mutant mice similar to MyoD expression in back muscles of adult, wild-type mice (data not shown). In mdx mice, a strong up-regulation of MyoD mRNA was detected whereas FGF-6(−/−)/mdx mice remained at a low basal level and did not show a significant up-regulation of MyoD expression. Thus, failure of MyoD up-regulation in FGF-6(−/−)/mdx mice, despite a stimulus for regeneration supplied by degenerating myotubes, appears to be caused by the absence of a signal in FGF-6 mutant mice necessary to stimulate MyoD-expressing cells.
Figure 8.

Up-regulation of MyoD in mdx mice is inhibited in FGF-6(−/−)/mdx double mutants. (A–C) In situ hybridization of the diaphragm muscle of adult FGF-6 mutant (A), mdx mutant (B), and FGF-6(−/−)/mdx double mutant mice (C). The level of MyoD mRNA corresponds to the amount of black silver grains in bright field pictures. FGF-6(−/−) mice show only background signals (A). mdx mutants are characterized by an up-regulation of MyoD in skeletal muscles caused by continuous regeneration of myotubes (B). Lack of FGF-6 leads to reduction of MyoD mRNA in mdx mice and to an impairment of muscle regeneration (C). MyoD RNA expression in back muscles of adult FGF-6, mdx, and FGF-6(−/−)/mdx mice, and wild-type E17.5 embryos was quantitated by Northern blot analysis (D). MyoD expression was very low in FGF-6 mutant (lane 1). In mdx mice, a strong up-regulation of MyoD mRNA was detected (lane 2) whereas FGF-6(−/−)/mdx mice did not show a significant up-regulation of MyoD expression (lane 3). As a positive control, MyoD expression at E17.5 is shown. No Myogenin expression was detected in back muscles of adult mice. Hybridization with a GAPDH probe and ethidium bromide-staining of RNA served as loading controls and demonstrated the integrity of RNA samples, respectively.
Discussion
In the present study we describe the inactivation and overexpression of FGF-6 in mice to analyze its function during development and in adult animals. Mice lacking FGF-6 show a pronounced regeneration defect after experimentally induced crush injury of muscle and a significantly reduced appearance of MyoD-expressing mpcs. Furthermore, interbreeding of FGF-6 mutants with mdx mice resulted in a severe myopathy in double-homozygous FGF-6(−/−)/mdx mice.
Our studies indicate that FGF-6 is required in certain pathological situations such as skeletal muscle damage for normal regeneration, whereas physiological functions of FGF-6 during development might be overlapping with other growth factors. Although the exact role of FGF-6 in the regeneration process is not clear, our data imply that FGF-6 may be involved in the activation of satellite cells and the expression of MyoD (Johnson and Allen 1995). Because FGFs induce proliferation in a wide variety of cells, it appears likely that FGF-6 acts by stimulating the entry of satellite cells into the cell cycle rather than directly promoting MyoD expression. Generally, treatment of myogenic cell lines with recombinant FGF in culture down-regulates MyoD expression. It should be mentioned, however that at certain concentrations, FGF-6 stimulates expression of myogenic factors in vitro (Pizette et al. 1996).
Recently, a defect in the differentiation of satellite cells of MyoD mutant mice has been described (Megeney et al. 1996). MyoD-deficient mice (Rudnicki et al. 1992) appear to accumulate increased numbers of myogenic cells as a result of increased muscle stem-cell self-renewal and a reduction in satellite cell differentiation. As a consequence, the skeletal muscle regeneration capacity of MyoD-deficient mice is compromised. Interestingly, the phenotypes of FGF-6 mutant and MyoD mutant mice appear to overlap partially. In both knockout mice the amount of satellite cells in undamaged muscle is normal or elevated, both mutants suffer from a regeneration defect and show a deficit in mpc activation. Differences become apparent after interbreeding with mdx mice that show persisting degeneration of myotubes. Whereas MyoD(−/−)/mdx mice show an abnormal waddling gait by 3–5 months of age, we have not observed a similar phenotype in FGF-6(−/−)/mdx mice, but another feature of MyoD(−/−)/mdx mice, a profound lordosis and kyphosis, is also often seen in FGF-6(−/−) and FGF-6(−/−)/mdx mice. In addition, FGF-6(−/−)/mdx mice show a highly abnormal diaphragm muscle without the marked reduction of muscle thickness as described for MyoD(−/−)/mdx mice (Megeney et al. 1996). Side-by-side comparison of MyoD(−/−), FGF-6(−/−), MyoD(−/−)/mdx, and FGF-6(−/−)/mdx mice on the same genetic background will show whether the reaction to muscle cell damage is identical or different.
In this context, a further analysis of double mutants for FGF-6 and MyoD will be informative. So far, we have not observed any apparent signs of muscular dystrophy or a massive exacerbation of the individual phenotypes in FGF-6(−/−)/MyoD(−/−) double mutants. If the phenotype of double mutants and the regeneration defect is not augmented compared with each single mutation, then it is likely that both genes act in the same pathway. If the severity of the phenotype increases after muscle injury, MyoD and FGF-6 might be involved in parallel pathways of satellite cell activation.
Electron microscopy of skeletal muscle from FGF-6 mutant mice revealed the presence of normal or slightly elevated amounts of satellite cells. This finding implies that FGF-6 is not necessary for the development of satellite cells or for proliferation of the satellite cell pool during embryonic development. Because relatively large amounts of satellite cells are present in undamaged muscle of adult mutant mice, it also appears that self-renewal of satellite cells is directed by different mechanisms than mpc activation and proliferation induced by injury. Interestingly, this feature of FGF-6 mutants is also shared by MyoD mutants supporting the view that FGF-6 and MyoD may act in the same pathway required for satellite cell activation in adults but not for the generation or self-renewal of satellite cells.
Histological analysis of skeletal muscle of FGF-6 mutants after injury and immunohistochemical staining with a PCNA antibody revealed the presence of large numbers of proliferating cells that did not express satellite cell markers. The presence of these fibroblastoid cells might be explained by compensatory, supranormal activities of other growth factors that may lead to enhanced growth of fibroblasts and relatively retarded growth of mpcs. Our analysis of FGF-6(−/−)/mdx mice seems to support this view: Large numbers of mononuclear cells were found in skeletal muscles of double mutants that are likely to result from compensatory growth factor circuits built up in response to continuous damage of skeletal muscle in mdx mice (Stedman et al. 1991). In addition, a compromised regeneration program together with the presence of degenerated myotubes may represent a permanent stimulus for tissue infiltration by inflammatory cells.
In agreement with these findings, hybridization with a MyoD probe by in situ hybridization on diaphragm sections and by Northern blot analysis with RNA isolated from back muscle showed an up-regulation of MyoD-expression in mdx mice but failure of an up-regulation in FGF-6(−/−)/mdx double mutant mice. After completion of muscle formation in adults, MyoD expression is low. Normal turnover processes do not seem to require high MyoD concentrations. In contrast, the necessity for continous new muscle cell formation in mdx mice increases the amount of MyoD-expressing cells, a process that is severely compromised by the lack of FGF-6. This interpretation is further supported by the presence of degenerated myotubes found at numerous locations in limb, deep back, and body wall muscles of FGF-6(−/−)/mdx double-mutant mice. Because mdx mice lack Dystrophin, which is thought to be required for the stabilization of the sarcolemma differences in histological appearance and differences in the severity of myopathy in various muscle groups might be the result of distinct mechanical challenges encountered by individual muscles. On the basis of our experimental data, we propose that FGF-6 stimulates the entry of satellite cells into the cell cycle. This step appears to be necessary for activation of MyoD expression and for progression through the myogenic differentiation pathway. Failure of adequate myoblast proliferation and compensatory counter regulation by other growth factors might lead to the proliferation of different cell types like fibroblasts. According to this model, the difference in severity of the phenotypic appearance of MyoD and FGF-6 mutant mice might be explained by the establishment of different compensatory mechanisms. The presence of FGF-2 and other growth factors in skeletal muscle of FGF-6-deficient mice may also lead to the activation of satellite cells, albeit with lower efficiency. In contrast, MyoD mutant mice have a more restricted choice for compensation because myoblasts depend critically on either MyoD or Myf-5 (Rudnicki et al. 1993; Braun and Arnold 1996).
The strong angiogenic properties of FGFs have been documented in numerous reports (Yanagisawa-Miwa et al. 1992; Nabel et al. 1993). Most studies have concentrated on FGF-1 and FGF-2 formerly known as acidic and basic FGFs. It has also been suggested that FGF-6 may possess angiogenic activities because injection of FGF-6 transformed NIH-3T3 fibroblasts into nude mice gives rise to vascularized tumors (Iida et al. 1992). In addition, overexpression of FGF-6 from the muscle specific human myogenin promotor in chimeric mice resulted in increased vascularization, formation of hemangiomas, and embryonic lethality (T. Floss and T. Brown, unpubl.).
Along with their suggested role in angiogenesis during development, FGFs may also have important functions for neovascularization (Walgenbach et al. 1996). In principle, it is possible that defects in neovascularization may be responsible for impaired tissue regeneration, for example, after skeletal muscle injury. As a consequence, proliferation of myoblasts might be hampered and expression of MyoD might be reduced. Such an alternative model, however, appears less likely to explain the regeneration defect of FGF-6 mutants for several reasons: (1) No apparent signs for blood vessel malformation or ischemia were detected in FGF-6 mutants making a critical or unique function of FGF-6 for the formation of blood vessels unlikely; and (2) high amounts of proliferating cells were detected in regenerating muscle tissue of FGF-6 mutants after crush injury; and (3) staining of regenerating skeletal muscle with antibodies against CD31 that is present on endothelial cells revealed no significant difference between mutant and wild-type animals. It is difficult, however, to exactly estimate the extent and rate of angiogenesis and the resulting cumulative diameter of capillary tips, capillary sprouts, and small blood vessels to be effective in tissue perfusion (Pettet 1996). Therefore, we cannot completely rule out the possibility that defects in neovascularization following crush injury contribute to the regeneration defect observed in FGF-6 mutants.
The inactivation of FGF-6 has revealed a role of this growth factor in the regeneration of skeletal muscle. Rescue experiments by injection of recombinant growth factor or retroviruses will show whether exogenously supplied FGF-6 can compensate for the mutation. If this is the case, FGF-6 might prove to be useful for the treatment of muscle injuries or muscle dystrophies, particularly if FGF-6 evokes the response preferentially on myoblasts. In either case, FGF-6 mutant mice and FGF-6(−/−)/mdx double-mutant mice should be useful systems to study various aspects of muscle regeneration and might open the way to new or improved therapeutical regimens.
Materials and methods
Construction of the targeting vector, electroporation of ES cells, and generation of mice
The FGF-6 gene was isolated from a genomic phage library from J1-ES cell DNA (a kind gift of R. Jaenisch, Whitehead Institute, Cambridge, MA) by screening with a mouse FGF-6 cDNA probe (Grass et al. 1997). Several overlapping phage clones were isolated that covered about 40 kb of the locus. A 5-kb EcoRI fragment was identified that contained part of exon I and 5′-upstream sequences. From this fragment, a 1.8-kb XbaI–XhoI fragment was released, modified with XhoI linkers, and inserted into the transfer vector pPNT. To generate a 3′-homology fragment, a 6-kb EcoRI fragment covering parts of intron 2 and exon 3 was identified and inserted into the corresponding restriction site of pPNT.
ES cell line J1 was grown on embryonic feeder cells as described (Braun et al. 1992). Electroporation and selection of ES cells were all done as described previously (Braun et al. 1992). A 5.5-kb mutant KpnI fragment was detected in 23 of 323 clones compared with the 7.5-kb wild-type fragment by use of a 5′-flanking probe (a 1.0-kb EcoRI–XbaI fragment). Three randomly chosen recombined ES cell clones were injected into C57/BL6 mouse blastocysts. Two clones contributed to the germ line and were independently used to establish inbred and C57/BL6 hybrid mouse strains. mdx mice were purchased from Harlan-Winkelmann (Paderborn, Germany). The generation of MyoD-deficient mice has been described before (Rudnicki et al. 1992).
Induction of muscle regeneration, histological analysis, and immunohistochemical staining
Regeneration of skeletal muscle was induced by a single freeze–crush injury of the musculus tibialis anterior by use of standard procedures as described by McGeachie and Grounds (1987). For all regeneration experiments 10–12 week-old male mice were used. For histological analysis, tissues were fixed in 4% formalin for several days before embedding in paraffin wax (Bancroft and Stevens 1990). Embryos were fixed in 4% paraformaldehyde, dehydrated through a graded alcohol series, and infiltrated with paraffin. Serial sections were taken at 7 μm. HE stainings and van Gieson stainings to visualize collagen were performed as described (Bancroft and Stevens 1990). For immunohistochemistry, tissues were dissected at different time points after crush injury, processed as described (Braun and Arnold 1995), and sectioned on a cryostat. Sections of regenerating muscles were stained with a mouse monoclonal antibody against MyoD (Medac), a monoclonal antibody against Myogenin (kindly supplied by W.E. Wright, Southwestern Medical Center, Dallas, TX), a monoclonal antibody (MY32) against fast MyHC (purchased from Sigma), and a monoclonal antibody against PCNA (Dakopads). Bound antibodies were visualized with a Vectastain elite kit by use of diaminobenzidine as substrate. Pictures from sections stained with Myogenin and MyoD antibodies were taken with Nomarski optics to visualize cellular structures. Transmission electron microscopy was performed on musculus tibialis anterior isolated from 12 week-old male mutant mice and control littermates. Muscles were dissected and fixed in freshly prepared 5% paraformaldehyde/3% glutaraldehyde in HEPES-buffered saline for 1 hr on ice and processed with standard procedures (Bancroft and Stevens 1990). Approximately 500 nuclei were counted from mutant and wild-type animals.
RNA analysis
Isolation of total RNA from embryos and muscle tissues were done according to standard procedures (Sambrook et al. 1989). mRNA was isolated with the Micro-Fast track system as recommended by the manufacturer (Invitrogen). RT–PCR was essentially done as described by Heymann et al. (1996) in a 50-μl volume. Amplification parameters were: denaturation 95°C for 45 sec, annealing 2 min at 55°C (FGF-6), 52°C (Myogenin, MyoD), 57°C (GAPDH), extension 72°C for 2 min. Aliquots of 2% of the reaction volume were removed from the assay after different cycles as indicated, extracted with chloroform, and analyzed on a 2% agarose gel. Gels were blotted and hybridized with the corresponding probes to verify the identity of the PCR products. Primers for detection of reverse transcriptase products were derived from different exons to distinguish RT–PCR products from genomic DNA contaminations. Primer sequences were: FGF-6, AACACACGAGGAGAACCC and CGTAGGCGTTGTAGTTGTTTGG; MyoD, AGGCTCTGCTGCGCGACC and TGCAGTCGATCTCTCAAAGCACC; Myogenin, GAGCGCGATCTCCGCTACAGAGG and CTGGCTTGTGGCAGGCCCAGG; GAPDH, ACTCCCACTCTTCCACCTTC and TCTTGCTCAGTGTCCTTGC. The expected product sizes were as follows: FGF-6, 292 bp; MyoD, 491; Myogenin, 229; GAPDH, 185 bp. In situ hybridizations were performed with 35S-UTP-labeled antisense RNA probes as described (Grass et al. 1996). Northern blot analysis was done as outlined previously (Braun et al. 1989). For quantification, autoradiographs were densitometrically scanned with a Herolab Enhanced Analysis System.
Acknowledgments
The excellent technical assistance of S. Heymann and P. Gatzlaff is gratefully acknowledged. We are indebted to Dr. M. Rohde (GBF Braunschweig) for expert electron microscopy. We further thank E. Bober for critically reading the manuscript, Eli Lilly for their gift of FIAU, R. Jaenisch for the genomic phage library, W. E. Wright for the Myogenin monoclonal antibody, and A. Mantovani, Milano for his kind gift of mAb MEC 7.46 and MEC 13.3 against CD31. This work was supported by the SFB 271: “Molekulare Mechanismen Morphoregulatorischer Prozesse” Teilprojekt B2 and by DFG grant Br 1413 to T. B.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL t.braun@tu-bs.de; FAX 00 49 531 3 91 81 78.
References
- Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nature Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- Arnold HH, Braun T. Myogenic control genes in vertebrates. In: Wassarman PM, editor. Advances in developmental biology. Greenwich, UK: JAI Press; 1993. pp. 111–158. [Google Scholar]
- Baird A, Klagsbrun M. The fibroblast growth factor family. NY Acad Sci. 1991;638:1–513. [Google Scholar]
- Baker J, Liu J-P, Robertson EJ, Efstradiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Bancroft JD, Stevens A. Theory and practice of histological techniques. Edinburgh, UK: Churchill Livingston; 1990. [Google Scholar]
- Bischoff R. The satellite cell and muscle regeneration. In: Engel AG, Franzini-Armstrongl C, editors. Myology. New York, NY: McGraw-Hill; 1994. pp. 97–133. [Google Scholar]
- Braun T, Arnold HH. Inactivation of Myf-6 and Myf-5 genes in mice leads to alterations in skeletal muscle development. EMBO J. 1995;14:1176–1186. doi: 10.1002/j.1460-2075.1995.tb07101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Myf-5 and MyoD genes are activated in distinct mesenchymal stem cells and determine different skeletal muscle cell lineages. EMBO J. 1996;15:310–318. [PMC free article] [PubMed] [Google Scholar]
- Braun T, Bober E, Buschhausen-Denker G, Kohtz S, Grzeschik KH, Arnold HH. Differential expression of myogenic determination genes in muscle cells: Possible autoactivation by the myf gene products. EMBO J. 1989;8:3617–3636. doi: 10.1002/j.1460-2075.1989.tb08535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T, Rudnicki MA, Amold HH, Jaenisch R. Targeted inactivation of the mouse regulatory gene Myf5 results in abnormal distal rib development and early postnatal death in homozygous mouse mutants. Cell. 1992;71:369–382. doi: 10.1016/0092-8674(92)90507-9. [DOI] [PubMed] [Google Scholar]
- deLapeỳriere O, Ollendorff V, Planche J, Ott MO, Pizette S, Coulier F, Birnbaum D. Expression of the FGF6 gene is restricted to developing skeletal muscle in the mouse embryo. Development. 1993;118:601–611. doi: 10.1242/dev.118.2.601. [DOI] [PubMed] [Google Scholar]
- Feldman B, Poueymirou W, Papaioannou V, Chiara T, Goldfarb M. Requirement of FGF-4 for postimplantation mouse development. Science. 1995;267:246–249. doi: 10.1126/science.7809630. [DOI] [PubMed] [Google Scholar]
- Füchtbauer E-M, Westphal H. MyoD and Myogenin are coexpressed in regenerating skeletal muscle of the mouse. Dev Dynam. 1992;193:34–39. doi: 10.1002/aja.1001930106. [DOI] [PubMed] [Google Scholar]
- Grass S, Arnold HH, Braun T. Alterations in somite patterning of myf-5-deficient mice: A possible role for FGF-4 and FGF-6. Development. 1996;12:141–150. doi: 10.1242/dev.122.1.141. [DOI] [PubMed] [Google Scholar]
- Grounds MD, Yablonka-Reuvini Z. Molecular and cellular biology of muscle regeneration. In: Partridge T, editor. Molecular and cellular biology of muscular dystrophy. London, UK: Chapman and Hall; 1993. pp. 210–256. [Google Scholar]
- Guo L, Degenstein L, Fuchs E. Keratinocyte growth factor is required for hair development but not for wound healing. Genes & Dev. 1996;10:165–175. doi: 10.1101/gad.10.2.165. [DOI] [PubMed] [Google Scholar]
- Hebert J, Rosenquist T, Gotz J, Martin G. FGF5 as a regulator of the hair growth cycle: Evidence from targeted and spontaneous mutations. Cell. 1994;78:1017–1025. doi: 10.1016/0092-8674(94)90276-3. [DOI] [PubMed] [Google Scholar]
- Heymann S, Koudrava M, Köster M, Braun T. Regulation and function of SF/HGF during migration of limb muscle precursor cells in chicken. Dev Biol. 1996;180:566–578. doi: 10.1006/dbio.1996.0329. [DOI] [PubMed] [Google Scholar]
- Iida S, Yoshida T, Naito K, Sakamoto H, Katoh O, Hirohashi S, Sato T, Onda M, Sugimura T, Terada M. Human hst-2 (FGF-6) oncogene: cDNA cloning and characterization. Oncogene. 1992;7:303–309. [PubMed] [Google Scholar]
- Johnson S, Allen RE. Activation of skeletal muscle satellite cells and the role of fibroblast growth factor receptors. Exp Cell Res. 1995;2:449–453. doi: 10.1006/excr.1995.1251. [DOI] [PubMed] [Google Scholar]
- Koishi K, Zhang M, McLennan IS, Harris JA. MyoD protein accumulates in satellite cells and is neurally regulated in regenerating myotubes and skeletal muscle fibers. Dev Dynam. 1995;202:244–254. doi: 10.1002/aja.1002020304. [DOI] [PubMed] [Google Scholar]
- Lefaucheur JP, Sebille A. Basic fibroblastic growth factor promotes in vivo muscle regeneration in murine muscular dystrophy. Neurosci Lett. 1995a;202:121–124. doi: 10.1016/0304-3940(95)12223-0. [DOI] [PubMed] [Google Scholar]
- ————— Muscle regeneration following injury can be modified in vivo by immune neutralization of basic fibroblastic growth factor, transforming growth factor beta-1 or insulin-like growth factor I. J Neuroimmunol. 1995b;5:85–91. doi: 10.1016/0165-5728(94)00166-l. [DOI] [PubMed] [Google Scholar]
- Maina F, Casagranda F, Audero E, Simeone A, Comoglio PM, Klein R, Ponzetto C. Uncoupling of grb2 from the met receptor in vivo reveals complex roles in muscle development. Cell. 1996;87:531–542. doi: 10.1016/s0092-8674(00)81372-0. [DOI] [PubMed] [Google Scholar]
- Mason I. The ins and outs of fibroblast growth factors. Cell. 1994;78:547–552. doi: 10.1016/0092-8674(94)90520-7. [DOI] [PubMed] [Google Scholar]
- McGeachie JK, Grounds MD. Initiation and duration of muscle precursor replication after mild and severe injury to skeletal muscle of mice. An autoradiographic study. Cell Tissue Res. 1987;248:125–130. doi: 10.1007/BF01239972. [DOI] [PubMed] [Google Scholar]
- Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes & Dev. 1996;10:1173–1183. doi: 10.1101/gad.10.10.1173. [DOI] [PubMed] [Google Scholar]
- Nabel EG, Yang Z, Plautz G, Forough R, Zhan Y, Haudenschild CC, Maciag T, Nabel GJ. Recombinant fibroblast growth factor-1 promotes intimal hyperplasia and angiogenesis in arteries in vivo. Nature. 1993;362:844–846. doi: 10.1038/362844a0. [DOI] [PubMed] [Google Scholar]
- Olson EN. Interplay between proliferation and differentiation within the myogenic lineage. Dev Biol. 1992;154:261–272. doi: 10.1016/0012-1606(92)90066-p. [DOI] [PubMed] [Google Scholar]
- Pettet G, Chaplain MAJ, McElwain DLS, Byrne HM. On the role of angiogenesis in wound healing. Proc R Soc Lond B. 1996;263:1487–1493. doi: 10.1098/rspb.1996.0217. [DOI] [PubMed] [Google Scholar]
- Pizette S, Coulier F, Birnbaum D, deLapeyrière O. FGF-6 modulates the expression of fibroblast growth factor receptors and myogenic genes in muscle cells. Exp Cell Res. 1996;224:143–151. doi: 10.1006/excr.1996.0122. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to upregulation of the myogenic HLH gene Myf5 and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettien RA, Petrof B, Narusawa M, Leferovitch JM, Sladky JT, Kelly AM. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- Vecchi A, Garlanda C, Lampugnani MG, Resnati M, Matteucci C, Stoppacciaro A, Schnurch H, Risau W, Ruco L, Matovani A, Dejana E. Monoclonal antibodies specific for endothelial cells of mouse blood vessels. Their application in the identification of adult and embryonic endothelium. Eur J Cell Biol. 1994;63:247–254. [PubMed] [Google Scholar]
- Walgenbach KJ, Gratas C, Shestak K, Becker D. Ischaemia-induced expression of bFGF in normal skeletal muscle: A potential paracrine mechanism for mediating angiogenesis in ischaemic skeletal muscle. Nature Med. 1995;1:453–459. doi: 10.1038/nm0595-453. [DOI] [PubMed] [Google Scholar]
- Werner S, Peters KG, Longaker MT, Fuller-Pace F, Banda MJ, Williams LT. Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc Natl Acad Sci. 1992;89:6896–6900. doi: 10.1073/pnas.89.15.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagushi T P, Rossant J. Fibroblast growth factors in mammalian development. Curr Opin Genet Dev. 1995;5:485–491. doi: 10.1016/0959-437x(95)90053-j. [DOI] [PubMed] [Google Scholar]
- Yanagisawa-Miwa A, Uchida Y, Nakamura F, Tomaru T, Kido H, Kamijo T, Sugimoto T, Kaji K, Utsuyama M, Kurashima C, Ito H. Salvage of infarcted myocardium by angiogenic action of basic fibroblast growth factor. Science. 1992;257:1401–1403. doi: 10.1126/science.1382313. [DOI] [PubMed] [Google Scholar]