

Abstract
Culture-independent microbiological technologies that interrogate complex microbial populations without prior axenic culture, coupled with high-throughput DNA sequencing, have revolutionized the scale, speed, and economics of microbial ecological studies. Their application to the medical realm has lead to a highly productive merger of clinical, experimental, and environmental microbiology. The functional roles played by members of the human microbiota are being actively explored through experimental manipulation of animal model systems and studies of human populations. In concert, these studies have appreciably expanded our understanding of the composition and dynamics of human-associated microbial communities (microbiota). Of note, several human diseases have been linked to alterations in the composition of resident microbial communities, so-called dysbiosis [1]. However, how changes in microbial communities contribute to disease etiology remains poorly defined. Correlation of microbial composition represents integration of only two datasets (phenotype and microbial composition). This article explores strategies for merging the human microbiome data with multiple additional datasets (e.g. host single nucleotide polymorphisms [SNP] and host gene expression) and for integrating patient-based data with results from experimental animal models to gain deeper understanding of how host-microbe interactions impact disease.
Dysbiosis and disease etiology
Human-associated microbial communities, particularly those of the gastrointestinal (GI) tract, provide myriad beneficial services to ourselves. For instance, gut microbes transform otherwise indigestible plant polysaccharides into absorbable short-chain fatty acids (SCFA) [2] and participate in the development and maintenance of immune homeostasis [3, 4]. Disruption of any of these mutualistic relationships through shifts in microbial community composition (i.e. dysbiosis [1]) could compromise human health and contribute to disease onset, progression, or duration. Indeed, recent applications of culture-independent sequencing technologies to studies of human health and disease have revealed dysbioses associated with diverse conditions, including antibiotic-associated diarrhea [5, 6], bacterial vaginosis [7–9], celiac disease [10], colorectal cancer [11, 12], cystic fibrosis [13, 14], esophageal disease [15], Crohn’s disease and ulcerative colitis (collectively referred to as inflammatory bowel diseases [IBD]) [16–22], irritable bowl syndrome [23–26], necrotizing enterocolitis [27], non-bacterial prostatitis [28, 29], pre-term birth [30], obesity [31–33], pouchitis [34–36], and psoriasis [37].
What has been lacking in studies of human dysbioses is determination of the biological and clinical significance of these community imbalances. Are the changes in enteric microbiota that are observed in IBD simply a consequence of chronic inflammation and its treatment or are they necessary determinants of initiation and/or perpetuation of pathogenesis? Figure 1 presents four possible causal relationships between disease and dysbiosis. First, dysbiosis could be a primary trigger that leads to pathogenesis (Figure 1, Path A). Although the initial factors that disrupt the commensal microbiota are not well defined, their effects are mediated through induction of detrimental immune responses or compromised mucosal barrier function. For example, depletion of commensal bacteria can lead to increased populations of potentially pathogenic organisms, such as occurs with toxin-secreting Clostridium difficile proliferation following antibiotic treatment [6]. Restoring the normal microbiota would, in this scenario, be expected to lessen pathogenesis. Second, dysbiosis could arise in parallel with pathogenesis (Figure 1, Path B), but not serve as a causal factor of disease. In this case, treating the dysbiosis would not ameliorate disease. Similarly (Figure 1, Path C), the pathological condition itself, or its treatment, could cause a secondary shift in microbial community structure. For example, remodeling of the intestinal mucosal milieu following prolonged inflammation could significantly alter both the intestinal environment and the microbial inhabitants of this niche. Finally, even if not a primary cause of disease (Figure 1, Path D), disruption of the commensal microbial community and the mutualistic functions it contributes to the host could contribute to the duration or severity of a disease state. For example, imbalances in the microbiota could also lead to metabolic alterations (e.g. decrease in luminal SCFA) that in turn compromise the protective functions of the epithelial barrier [38]. Alternatively, these abnormal bacteria could secondarily invade a disrupted mucosal barrier or provide antigens and Toll-like receptor ligands that further stimulate adaptive and innate immune responses thereby augmenting and perpetuating an ongoing inflammatory response [39–41]. In these situations, remediating the dysbiosis should provide palliative relief, but likely would not effect a cure, if the primary etiological factor(s) are not addressed. That many of the syndromes associated with dysbiosis are chronic suggests that loss of the normal functions provided by a commensal community, even if not the ultimate causative agent, could significantly prolong the disease state and complicate its resolution.
Figure 1. Possible etiological significance of dysbiosis.

Imbalances in microbial communities might be a necessary condition for disease (A) or an inconsequential result of etiological agents (B) or disease itself (C). Because of the myriad beneficial services provided by the human-associated microbiota, loss of these functions as a result of disease development could further exacerbate the severity, duration, or frequency of disease (D).
Delineating the causes and etiological consequences of disease-associated dysbioses remains a crucial challenge in studies of the human microbiota. As with other diseases of uncertain microbial etiology, fulfilling Koch’s postulates in the context of dysbiosis is problematic. First, Koch’s postulates are predicated on the establishment of a parasitic relationship between the host and etiologic agent, whereas some dysbiosis-associated pathologies might instead arise from loss of mutualistic interactions. Second, whereas the defining feature of Koch’s postulates is the demand for isolation of a single pathogen in pure culture, dysbiosis is by definition a community phenomenon. The causal significance of dysbiosis could instead be more effectively evaluated through modifications of Koch’s postulates that have been set forth in the era of risk-factor epidemiology (e.g. those of Hill [42] and Evans [43]) and molecular microbiology (e.g. those of Fredricks and Relman [44]). These guidelines recognize that a constellation of necessary, but not sufficient, causal relationships often is the hallmark of diseases that are chronic, multi-factorial, and/or develop with long latencies. Moreover, the Fredricks-Relman guidelines embrace the utility of molecular-based technologies for detecting microorganisms in the absence of culture, a key feature given that syntrophic and symbiotic relationships among microbial community members are likely to limit the reductionist approach of axenic culture. Thus, the pathogenic capacity of a microbial community as a whole could be examined in the context of these revised criteria.
In evaluating causal relationships, both Hill’s criteria and the Fredricks-Relman guidelines consider the strength, consistency, and specificity of association between the occurrence of a putative pathogen and a disease, as well as their dose-response relationship. The degree to which these criteria are satisfied typically provides the primary evidence in support of a proposed association between dysbiosis and disease (e.g. [16]). Hill’s criterion of biological plausibility, though not emphasized by the author [42], is supported in the case of dysbiosis by continuing, intensive work on the biochemical, physiological, immunological, and genetic bases of host-microbiota interactions [40, 41, 45–49]. However critical to establishing correlations, these criteria would not necessarily differentiate among the causal relationships outlined in Figure 1.
Our contention is that three additional Hill’s criteria – namely the coherence, temporality, and experimental support for causality – are the critical factors required to understand the causal role of dysbiosis. Coherence relates to the fit between a hypothesized dysbiosis-disease causal relationship and all other knowledge concerning the disease. This is essentially a hypothesis generating approach to determining the relationship between dysbiosis and disease. The challenge in patient based research is that the number of potential variables (genetic and environmental factors) to consider is extremely large compared with the number of samples that can be analyzed. Addressing temporality could require prospective collection of samples prior to the onset of disease (e.g. longitudinal follow-up of unaffected relatives of patients). The generation of genetic experimental animal models provides extremely useful tools for testing causality. However, differences between the experimental animal model and humans present a potential challenge. The ultimate goal of these strands of research is to determine whether (i) there are interventions that will alter dysbiosis and (ii) whether these interventions alter the course of the disease. The following sections describe these criteria in greater detail, as exemplified by an interdependent approach to delineate the role of dysbiosis in human IBD.
Coherence: integration of the microbiome and human genomics/post-genomic data
Determining whether enteric dysbiosis modifies the incidence and progression of IBD will require integration and correlation of large-scale surveys of microbial communities with rigorously characterized human genotypes and molecular phenotypes (microarray data, proteomic data, and histologic imaging data) in both Crohn’s disease and non-Crohn’s disease cohorts (Figure 2). Moreover, collection of a very large number of well phenotyped clinical specimens is necessary to accurately link potentially subtle patterns of altered enteric microbiota with host response and disease occurrence. Subsequent mechanistic studies in animal models, including selective colonization of gnotobiotic mice with bacterial species identified by the broad human surveys, combined with targeted in vitro experiments can determine causal relationships and further define mechanisms of action.
Figure 2. Metagenomic tools for studying host-microbiota interactions in human disease.
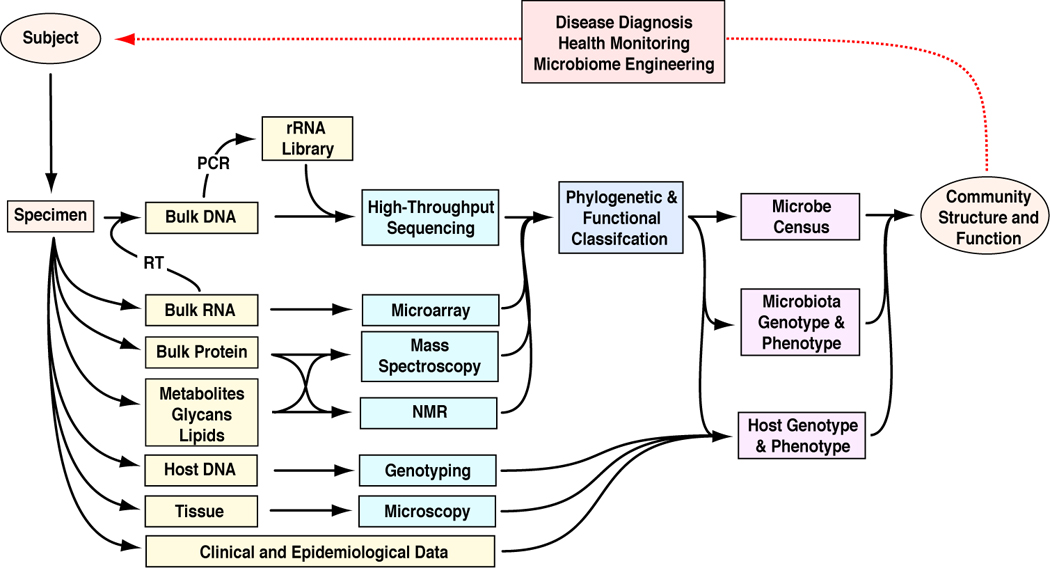
Bacteria are identified through culture independent analysis of 16S rRNA genes amplified from surgical specimens by broad-range PCR. Host genotypes and phenotypes are assessed through analysis of genomic DNA and mRNA of surgical specimens. Together, these data provide insight into how host factors and enteric microbial communities interact during development and/or progression of disease. The ultimate goal of these experiments is to use this knowledge to design, deploy, and monitor novel treatment regimes. Abbreviations: RT, reverse transcription.
The observation that a subset of IBD patients harbor abnormal enteric microbiota, while the microbiota of other IBD patients are apparently ‘normal’ raises several questions [16]. First, do these differences persist within an individual or do all IBD patients experience periodic flares of dysbiosis? Does dysbiosis arise from prolonged treatment with antibiotics or other agents that could alter the microbial habitat of the intestinal tract? Do host genetics, particularly the presence of IBD risk alleles affect the incidence or prevalence of dysbiosis in individuals, regardless of disease state? The correlation of abnormal microbiota with a younger age of surgery [16] was intriguing because alleles of NOD2/CARD15 and ATG16L1 that increase the risk of Crohn’s disease also are associated with younger age of surgery. Host genetic factors that affect innate immunity, such as the NOD2 and ATG16L1 genes, could affect microbial composition, particularly in the ileum. The three prevalent risk alleles of NOD2 (Leu1007fs, R702W, and G908R) are associated with the highest relative risks of ileal Crohn’s disease among the ~100 IBD susceptibility loci identified thus far.
To determine whether human genetic factors are associated with dysbiosis, NOD2 and ATG16L1 genotypic data were integrated with a previously published 16S rRNA sequence dataset [16]. This analysis revealed associations between alterations in intestine-associated microbial composition and disease phenotype, NOD2, and ATG16L1 genotype [22]. Analysis of an independent set of disease unaffected ileum samples collected from patients with three non-overlapping IBD phenotypes undergoing initial surgery: (i) ileal Crohn’s disease, (ii) isolated colitis, and (iii) non-IBD controls. Multivariate analysis of the 16S rRNA sequence dataset selected disease phenotype, C. difficile, and NOD2 genotype as significantly associated with shifts in grossly unaffected ileum-associated microbial composition. In these analyses, potential confounding variables such as obesity and IBD medications were also included. Disease phenotype and NOD2 genotype were also selected as significantly associated with shifts in the relative abundance of the Clostridium coccoides-Eubacterium rectales group measured by PCR.
Thus, NOD2 and ATG16L1 alleles have determining effects on the microbiota that are independent of the occurrence of overt disease. An important implication of this result is that the dysbiosis that has been widely reported in connection with Crohn’s disease [17] is not solely the result of environmental effects such as treatment history or diet. Interestingly, the abundance of an individual enteric genus is rarely significantly correlated with both NOD2 and ATG16L1 genotype, suggesting complex interactions between host genotype and microbial community composition [22]. Moreover, several genera are significantly associated with disease phenotype, but not with NOD2 or ATG16L1 genotype, suggesting either that disease itself influenced the relative frequencies of these genera or that other genetic determinants were involved. Therefore, further associations between other Crohn’s disease polymorphisms and environmental factors likely will emerge as study populations expand.
An association between enteric microbial profiles and genetic predisposition suggests that dysbiosis arises either as a consequence of direct genetic effects on microbial composition, perhaps through altered Paneth cell function [50–52] or as a direct result of the pathogenic process. Thus, the question of whether dysbiosis contributes to Crohn’s disease pathogenesis or is an innocuous byproduct remains to be settled. Furthermore, how mucosal barrier dysfunction or inflammation per se could lead to dysbiosis also is not clear. The plethora of luminal commensal bacteria are largely tolerated by the mucosal immune system and ignored by the systemic immune systems of normal hosts [49, 53, 54], but are essential drivers of pathogenic mucosal and systemic inflammatory responses in genetically predisposed hosts [39–41, 55]. Enteric microbial communities play a critical role in this process by stimulating the development of GI lymphoid tissues [56, 57], fortifying the physical epithelial barrier [58], and regulating the quality and magnitude of the mucosal immune response [3, 4] to commensal bacteria and potential pathogens. Shifts in microbial community composition that arise from dysfunctional innate mucosal immunity suggest a selective disadvantage to those microbial groups that serve to nurture gut barrier integrity. To fully understand the mechanism(s) by which host genetic factors influence microbial composition and the functional consequences of the changes in microbial composition it is critical that the microbiome data be linked also with host and bacterial transcriptomics [59, 60], proteomics [61–63], and metabolomics [64–66].
Experimental support: human interventions and animal models
The strongest evidence that dysbiosis contributes to human disease could be obtained through double-blind, randomized controlled experiments with agents that normalize dysbiotic profiles in individuals with disease or create dysbiosis in normal individuals. Under these conditions, the causal relations between exposure to dysbiosis and subsequent development of pathology then can be defined. However, ethical and practical considerations (how to artificially create a dysbiosis that precisely matches that of a particular disease, how to sustain microbial alterations with antibiotics, probiotics and prebiotics and how to reproduce environmental triggers that likely initiate inflammatory processes [40, 52]) limit this approach. Consequently, quasi-experimental studies in which dysbiosis is induced as a consequence of medical (e.g. C. difficile-associated diarrhea resulting from antibiotic exposure[6]) or surgical procedures (e.g. pouchitis following ileal-pouch anal anastomosis [34]) likely will be the dominant means of manipulating human microbiota prior to disease occurrence.
In parallel with quasi-experimental studies of humans, well-designed direct experiments with animal models of disease can contribute important insights into the timing and mechanistic basis of disease pathophysiology in relation to altered microbiota [67]. Multiple models of experimental colitis exhibit dysbiosis similar to that seen in human IBD, with contraction of several Clostridium subsets and expansion of Enterobacteriaceae, including Escherichia coli [68, 69]. Studies in gnotobiotic mice and rats implicate commensal enteric bacteria in the pathogenesis of immune-mediated chronic intestinal inflammation [67, 70, 71] and demonstrate that individual bacterial species relevant to the dysbiosis of human IBD have differential abilities to induce or prevent experimental enterocolitis [70, 72]. Of note however, not all species that are altered in abundance in experimental colitis can elicit pathogenesis, whereas those that are sufficient to cause colitis may not be affected by disease state [67]. Therefore, dysbiosis might best be viewed as a starting point for careful experimentation, rather than an endpoint.
Host genetic background is a determinant both of enteric bacterial profiles and inflammatory sequellae to individual bacterial species. For example, NOD2-deficient mice display altered mucosally-associated bacteria and Bacteroides spp. composition in this model [73]. These differences were not, however, detected in a comparison of fecal microbiota, suggesting that the effect of NOD2 deficiency on microbial composition was restricted to the ileum, and perhaps reflect changes in mucosa-adherent communities [73]. Similarly, germ-free (sterile) IL-10 deficient mice develop proximal colitis when mono-colonized with either several adherent/invasive E. coli strains, including one human ileal Crohn’s disease isolate, and aggressive distal colitis and duodenal inflammation when selectively colonized with Enterococcus faecalis, but no disease with Bacteroides vulgatus [72].
Both traditional probiotic bacterial species (Lactobacillus spp. and Bifidobacterium spp.) as well as a commensal species that is decreased in human Crohn’s disease (Faecalibacterium prausnitzii) can attenuate experimental colitis [17]. Reports of transmission of colitis [74, 75], obesity [76], and metabolic syndrome [77] by fecal transfer to normal recipients strongly implicate dysbiosis as a primary etiologic factor in disease pathogenesis, although secondary changes in luminal microbiota following nonspecific intestinal inflammation or injury have been documented. An exciting observation that a common enteric viral pathogen, norovirus, can induce functional and phenotypic alterations in mice with decreased ATG16L1 expression offers proof of principle that environmental triggers can modulate genetically-defined disease susceptibility [52]. Recent evidence that human fecal microbial composition can be conserved in gnotobiotic mouse recipients provides an elegant opportunity to explore the pathogenicity of dysbiotic bacteria in human IBD patients in murine models [78]. These selective constitution studies will help define mechanisms of the pathogenicity of human bacterial profiles and microbial subsets in recipients by modeling interactions of human genes, environmental stimuli and human microbiota.
Temporality: modeling causal relationships in a systems biological framework
In addition to experimental animal and human microbial manipulation approaches, longitudinal observational studies represent a critical means of assessing the causal relationship between dysbiosis and disease occurrence. In principle, these studies would determine whether a particular exposure (e.g. alterations in microbiota) precedes development of overt disease. Despite their clear utility, few if any such trials have been conducted to date, although technological innovations in studying human microbiota now make large-scale cohort and/or case-control studies feasible. However, the chronic nature and the protracted preclinical phase of many dysbiosis-associated diseases could complicate interpretation of results, because timing of disease onset is imprecise in these cases, especially when human genetic loci help determine disease susceptibility.
Although the technology of culture-independent metagenomics is well established, development of statistical tools for analyzing microbial community organization has lagged behind the evolution of DNA sequencing technologies. An important component of this work will be continued development of computational and statistical methods for analysis of host-microbe dynamics. Moreover, robust methods are needed to integrate and mine data generated through myriad ‘omics technologies. As Sauer et al. have noted in regards to genetic analysis "The (traditional) reductionist approach has successfully identified most of the components and many of the interactions but … the pluralism of causes and effects in biological networks is better addressed by observing, through quantitative measures, multiple components simultaneously and by rigorous data integration with mathematical models" [79]. A potentially promising approach could be the development of statistical methods for analyzing microbiome data from a biological causal pathway perspective [80, 81]. This analytical methodology is based on structural equations modeling (SEM), a set of interrelated regression equations with random independent as well as dependent variables that allow formulation and testing of directional causal pathway hypotheses [82]. Although widely used in social, economic, and behavioral sciences (e.g. [80, 83–90]), SEM techniques have been applied only sparingly to ecological data (e.g. [91–93]), despite their original proposal by the American geneticist Dr. Sewall Wright [94]. Given the increasing quantitative nature of biological studies and the availability of multimodality genetic, genomic, proteomic, and other relevant morphological and physiological data, SEM is one of the most suitable statistical modeling and analysis tools for the study of the biological networks from a systems biology point of view.
In the example shown in Figure 3, the aim is to study the interactions among host genotype, gene/protein expression, physiology, morphology, gastro-intestinal microbiome and host phenotype using structural equation modeling. The composition of the microbiome is treated as an unknown (latent) variable that can be modeled through multiple observed variables using a combination of experimental methods (e.g. different sequencing platforms, quantitative PCR, and microarrays). Each node with an incoming arrow can be analyzed through a regression equation with the given node as the response variable and the originating nodes of the incoming arrow(s) as the independent variables (regressors). If the node is categorical, such as phenotype, the resulting equation will be a logistic regression. A numerical node such as microbiome, will lead to a linear regression equation. The path coefficients (α, β, and λ; Figure 3) are simply the usual regression coefficients. These regression equations can be analyzed individually, as commonly done, in a sectional approach. However, a unified simultaneous analysis of this entire equation system is the optimal solution in terms of the power and accuracy of the resulting statistical inference (K. Sharpe, PhD thesis, State University of New York at Stony Brook, 2010).
Figure 3. Incorporating multimodality microbiome measurements.
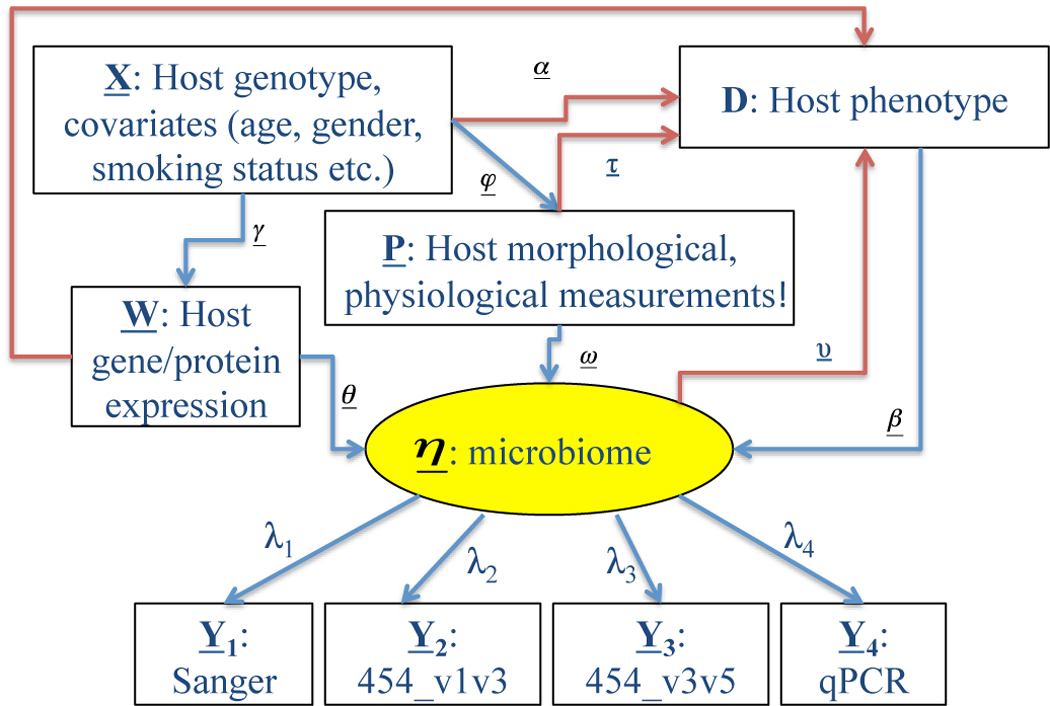
Schematic diagram of potential determinants of human microbiome and host phenotype (e.g. disease). The central oval (microbiome) depicts an unmeasured latent variable representing the kinds and quantities of microbes that constitute the microbiome. These are inferred by various experimental techniques, including Sanger sequencing, 454 pyrosequencing (using different 16S rRNA primer sets), and quantitative PCR (qPCR). Red arrows denote influences of host and microbiome on host phenotype whereas blue arrows denote causal relationships between host factors and microbiome. The relative contributions of these factors can be modeled using structural equation modeling, as described in the text.
SEMs can be readily extended to handle hierarchical (i.e. longitudinal) datasets using methods developed for magnetic resonance imaging (MRI) time series data [80]. Moreover, future work potentially can produce a powerful, unified SEM method that enables the simultaneous analysis of a pathway network with both continuous (possibly non-normal) and categorical dependent variables by extending the newly emerging generalized linear latent and mixed models (GLLAMM; [95]). These methods assess statistical relationships among complex datasets with mixed data types (i.e. categorical and continuous measures), for instance allowing for simultaneous correlation of human gene expression and microbiome data with disease outcomes, such as presence of a particular disease. Although these methods can not prove assertions about causality, they provide a robust framework for assessing and potentially rejecting such hypotheses. In the clinical context, the ability to model the interactions between complex datasets and multi-factorial disease states holds great promise to both elucidate fundamental features of etiopathogenesis and discover novel biomarkers of disease onset, severity, and progression.
Concluding remarks and future directions
Rather than discovering exotic new pathogens, the application of microbial metagenomics to human health has instead provided compelling, though not conclusive, evidence that disruption of host-microbe mutualism might be central to a variety of pathologies. In these cases microbial communities, not individual parasitic microorganisms, could play the role of pathogen [96]. However, other than in exceptional cases we are not likely to observe disease-associated dysbioses that neatly satisfy all of the criteria that have been proposed to prove causality [42–44]. Nevertheless, these guidelines provide a logical framework for assessing disease etiology that reflects the technological and epistemological developments that have sprung from Koch’s groundwork in clinical microbiology. These investigations have the potential to identify therapeutic targets that can normalize compositional and functional microbial alterations that either primarily or secondarily contribute to disease activity and intensity.
Acknowledgements
This work was supported by Mucosal and Vaccine Research Colorado (DNF), Crohns and Colitis Foundation of America (RBS and EL), Helmsley Foundation (RBS) and NIH grants HG005964 (DNF), DK053347 (RBS), RR018603 (RBS), DK034987 (RBS) and HD059527 (EL). We thank Norman R. Pace for his conception of the microbial community as potential pathogen and Gail Teitzel for her editorial insight.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tamboli CP, et al. Dysbiosis in inflammatory bowel disease. Gut. 2004;53:1–4. doi: 10.1136/gut.53.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flint HJ, et al. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6:121–131. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 3.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 4.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young VB, Schmidt TM. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J Clin Microbiol. 2004;42:1203–1206. doi: 10.1128/JCM.42.3.1203-1206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang JY, et al. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 7.Fredricks DN, et al. Molecular identification of bacteria associated with bacterial vaginosis. N Engl J Med. 2005;353:1899–1911. doi: 10.1056/NEJMoa043802. [DOI] [PubMed] [Google Scholar]
- 8.Oakley BB, et al. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl Environ Microbiol. 2008;74:4898–4909. doi: 10.1128/AEM.02884-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravel J, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Palma G, et al. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2011;10:63. doi: 10.1186/1471-2180-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scanlan PD, et al. Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ Microbiol. 2008;10:789–798. doi: 10.1111/j.1462-2920.2007.01503.x. [DOI] [PubMed] [Google Scholar]
- 12.Sobhani I, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris JK, et al. Molecular identification of bacteria in bronchoalveolar lavage fluid from children with cystic fibrosis. Proc Natl Acad Sci U S A. 2007;104:20529–20533. doi: 10.1073/pnas.0709804104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Gast CJ, et al. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011;5:780–791. doi: 10.1038/ismej.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pei Z, et al. Bacterial biota in reflux esophagitis and Barrett's esophagus. World J Gastroenterol. 2005;11:7277–7283. doi: 10.3748/wjg.v11.i46.7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank DN, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokol H, et al. Analysis of bacterial bowel communities of IBD patients: What has it revealed? Inflamm Bowel Dis. 2008;14:858–867. doi: 10.1002/ibd.20392. [DOI] [PubMed] [Google Scholar]
- 18.Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 2009;22:292–301. doi: 10.1097/QCO.0b013e32832a8a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willing B, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn's disease. Inflamm Bowel Dis. 2009;15:653–660. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- 21.Willing BP, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854. doi: 10.1053/j.gastro.2010.08.049. e1841. [DOI] [PubMed] [Google Scholar]
- 22.Frank DN, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kassinen A, et al. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology. 2007;133:24–33. doi: 10.1053/j.gastro.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Malinen E, et al. Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. Am J Gastroenterol. 2005;100:373–382. doi: 10.1111/j.1572-0241.2005.40312.x. [DOI] [PubMed] [Google Scholar]
- 25.Matto J, et al. Composition and temporal stability of gastrointestinal microbiota in irritable bowel syndrome--a longitudinal study in IBS and control subjects. FEMS Immunol Med Microbiol. 2005;43:213–222. doi: 10.1016/j.femsim.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Codling C, et al. A molecular analysis of fecal and mucosal bacterial communities in irritable bowel syndrome. Dig Dis Sci. 2010;55:392–397. doi: 10.1007/s10620-009-0934-x. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, et al. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009;3:944–954. doi: 10.1038/ismej.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krieger JN, Riley DE. Bacteria in the chronic prostatitis-chronic pelvic pain syndrome: molecular approaches to critical research questions. J Urol. 2002;167:2574–2583. [PubMed] [Google Scholar]
- 29.Tanner MA, et al. Prevalence of corynebacterial 16S rRNA sequences in patients with bacterial and "nonbacterial" prostatitis. J Clin Microbiol. 1999;37:1863–1870. doi: 10.1128/jcm.37.6.1863-1870.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mshvildadze M, et al. Intestinal microbial ecology in premature infants assessed with non-culture-based techniques. J Pediatr. 2010;156:20–25. doi: 10.1016/j.jpeds.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ley RE, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ley RE, et al. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009;106:2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komanduri S, et al. Dysbiosis in pouchitis: evidence of unique microfloral patterns in pouch inflammation. Clin Gastroenterol Hepatol. 2007;5:352–360. doi: 10.1016/j.cgh.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 35.McLaughlin SD, et al. The bacteriology of pouchitis: a molecular phylogenetic analysis using 16S rRNA gene cloning and sequencing. Ann Surg. 2010;252:90–98. doi: 10.1097/SLA.0b013e3181e3dc8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zella GC, et al. Distinct microbiome in pouchitis compared to healthy pouches in ulcerative colitis and familial adenomatous polyposis. Inflamm Bowel Dis. 2011;17:1092–1100. doi: 10.1002/ibd.21460. [DOI] [PubMed] [Google Scholar]
- 37.Paulino LC, et al. Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. J Clin Microbiol. 2006;44:2933–2941. doi: 10.1128/JCM.00785-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pryde SE, et al. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002;217:133–139. doi: 10.1111/j.1574-6968.2002.tb11467.x. [DOI] [PubMed] [Google Scholar]
- 39.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 40.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 41.Sartor RB. Genetics and environmental interactions shape the intestinal microbiome to promote inflammatory bowel disease versus mucosal homeostasis. Gastroenterology. 2010;139:1816–1819. doi: 10.1053/j.gastro.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 42.Hill AB. The Environment and Disease: Association or Causation? Proc R Soc Med. 1965;58:295–300. [PMC free article] [PubMed] [Google Scholar]
- 43.Evans AS. Causation and disease: the Henle-Koch postulates revisited. Yale J Biol Med. 1976;49:175–195. [PMC free article] [PubMed] [Google Scholar]
- 44.Fredricks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch's postulates. Clin. Microbiol. Rev. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Backhed F, et al. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 46.Rautava S, Walker WA. Commensal bacteria and epithelial cross talk in the developing intestine. Curr Gastroenterol Rep. 2007;9:385–392. doi: 10.1007/s11894-007-0047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faith JJ, et al. Creating and characterizing communities of human gut microbes in gnotobiotic mice. ISME J. 2011;4:1094–1098. doi: 10.1038/ismej.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodman AL, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A. 2011;108:6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng T, Elson CO. Adaptive immunity in the host-microbiota dialog. Mucosal Immunol. 2011;4:15–21. doi: 10.1038/mi.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wehkamp J, et al. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc Natl Acad Sci U S A. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cadwell K, et al. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Konrad A, et al. Tight mucosal compartmentation of the murine immune response to antigens of the enteric microbiota. Gastroenterology. 2006;130:2050–2059. doi: 10.1053/j.gastro.2006.02.055. [DOI] [PubMed] [Google Scholar]
- 54.Slack E, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shroff KE, et al. Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect Immun. 1995;63:3904–3913. doi: 10.1128/iai.63.10.3904-3913.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cebra JJ. Influences of microbiota on intestinal immune system development. Am J Clin Nutr. 1999;69:1046S–1051S. doi: 10.1093/ajcn/69.5.1046s. [DOI] [PubMed] [Google Scholar]
- 58.Hooper LV, et al. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 59.Hamm CM, et al. NOD2 status and human ileal gene expression. Inflamm Bowel Dis. 2010;16:1649–1657. doi: 10.1002/ibd.21208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noble CL, et al. Characterization of intestinal gene expression profiles in Crohn's disease by genome-wide microarray analysis. Inflamm Bowel Dis. 2010;16:1717–1728. doi: 10.1002/ibd.21263. [DOI] [PubMed] [Google Scholar]
- 61.Berndt U, et al. Proteomic analysis of the inflamed intestinal mucosa reveals distinctive immune response profiles in Crohn's disease and ulcerative colitis. J Immunol. 2007;179:295–304. doi: 10.4049/jimmunol.179.1.295. [DOI] [PubMed] [Google Scholar]
- 62.M'Koma AE, et al. Proteomic profiling of mucosal and submucosal colonic tissues yields protein signatures that differentiate the inflammatory colitides. Inflamm Bowel Dis. 2011;17:875–883. doi: 10.1002/ibd.21442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meuwis MA, et al. New biomarkers of Crohn's disease: serum biomarkers and development of diagnostic tools. Expert Rev Mol Diagn. 2008;8:327–337. doi: 10.1586/14737159.8.3.327. [DOI] [PubMed] [Google Scholar]
- 64.Marchesi JR, et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res. 2007;6:546–551. doi: 10.1021/pr060470d. [DOI] [PubMed] [Google Scholar]
- 65.Saric J, et al. Species variation in the fecal metabolome gives insight into differential gastrointestinal function. J Proteome Res. 2008;7:352–360. doi: 10.1021/pr070340k. [DOI] [PubMed] [Google Scholar]
- 66.Jansson J, et al. Metabolomics reveals metabolic biomarkers of Crohn's disease. PLoS One. 2009;4:e6386. doi: 10.1371/journal.pone.0006386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bloom SM, et al. Commensal bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hansen J, et al. The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr Opin Gastroenterol. 2010;26:564–571. doi: 10.1097/MOG.0b013e32833f1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sartor RB. Key questions to guide a better understanding of host-commensal microbiota interactions in intestinal inflammation. Mucosal Immunol. 2011;4:127–132. doi: 10.1038/mi.2010.87. [DOI] [PubMed] [Google Scholar]
- 70.Rath HC, et al. Normal luminal bacteria, especially bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human B2 microglobulin transgenic rats. J.Clin. Invest. 1996;98:945–953. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Veltkamp C, et al. Continuous stimulation by normal luminal bacteria is essential for the development and perpetuation of colitis in Tg(epsilon26) mice. Gastroenterology. 2001;120:900–913. doi: 10.1053/gast.2001.22547. [DOI] [PubMed] [Google Scholar]
- 72.Kim SC, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 73.Petnicki-Ocwieja T, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garrett WS, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garrett WS, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1131. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 77.Vijay-Kumar M, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Turnbaugh PJ, et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1 doi: 10.1126/scitranslmed.3000322. 6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sauer U, et al. Genetics. Getting closer to the whole picture. Science. 2007;316:550–551. doi: 10.1126/science.1142502. [DOI] [PubMed] [Google Scholar]
- 80.Kim J, et al. Unified structural equation modeling approach for the analysis of multisubject, multivariate functional MRI data. Hum Brain Mapp. 2007;28:85–93. doi: 10.1002/hbm.20259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang T, et al. Cluster analysis of genome-wide expression differences in disease-unaffected ileal mucoas in inflammatory bowel diseases; 1st IEEE International Conference on Computational Advances in Bio and medical Sciences (ICCABS); 2011. [Google Scholar]
- 82.Kline RB. Principles and Practice of Structural Equation Modeling. Guilford Press; 2011. [Google Scholar]
- 83.Joreskog KG. Testing a simple structure hypothesis in factor analysis. Psychometrika. 1966;31:165–178. doi: 10.1007/BF02289505. [DOI] [PubMed] [Google Scholar]
- 84.Bentler PM, Dudgeon P. Covariance structure analysis: statistical practice, theory, and directions. Annu Rev Psychol. 1996;47:563–592. doi: 10.1146/annurev.psych.47.1.563. [DOI] [PubMed] [Google Scholar]
- 85.MacCallum RC, Austin JT. Applications of structural equation modeling in psychological research. Annu Rev Psychol. 2000;51:201–226. doi: 10.1146/annurev.psych.51.1.201. [DOI] [PubMed] [Google Scholar]
- 86.McArdle JJ. Latent variable modeling of differences and changes with longitudinal data. Annu Rev Psychol. 2009;60:577–605. doi: 10.1146/annurev.psych.60.110707.163612. [DOI] [PubMed] [Google Scholar]
- 87.Tomarken AJ, Waller NG. Structural equation modeling: strengths, limitations, and misconceptions. Annu Rev Clin Psychol. 2005;1:31–65. doi: 10.1146/annurev.clinpsy.1.102803.144239. [DOI] [PubMed] [Google Scholar]
- 88.Bryan A, et al. Mediational analysis in HIV/AIDS research: estimating multivariate path analytic models in a structural equation modeling framework. AIDS Behav. 2007;11:365–383. doi: 10.1007/s10461-006-9150-2. [DOI] [PubMed] [Google Scholar]
- 89.Buhi ER, et al. Structural equation modeling: a primer for health behavior researchers. Am J Health Behav. 2007;31:74–85. doi: 10.5555/ajhb.2007.31.1.74. [DOI] [PubMed] [Google Scholar]
- 90.Tu YK. Commentary: Is structural equation modelling a step forward for epidemiologists? Int J Epidemiol. 2009;38:549–551. doi: 10.1093/ije/dyn346. [DOI] [PubMed] [Google Scholar]
- 91.Florin TH, et al. Shared and unique environmental factors determine the ecology of methanogens in humans and rats. Am J Gastroenterol. 2000;95:2872–2879. doi: 10.1111/j.1572-0241.2000.02319.x. [DOI] [PubMed] [Google Scholar]
- 92.Lin H, et al. Streptococcal upper respiratory tract infections and psychosocial stress predict future tic and obsessive-compulsive symptom severity in children and adolescents with Tourette syndrome and obsessive-compulsive disorder. Biol Psychiatry. 2011;67:684–691. doi: 10.1016/j.biopsych.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sikes BA, et al. Deciphering the relative contributions of multiple functions within plant-microbe symbioses. Ecology. 2011;91:1591–1597. doi: 10.1890/09-1858.1. [DOI] [PubMed] [Google Scholar]
- 94.Wright S. Correlation and causation. J Agric Res. 1921;20:557–585. [Google Scholar]
- 95.Rabe-Hesketh S, et al. Correcting for covariate measurement error in logistic regression using nonparametric maximum likelihood estimation. Stat Modelling. 2003;3 2150232. [Google Scholar]
- 96.Foster JA, et al. Application of ecological network theory to the human microbiome. Interdiscip Perspect Infect Dis. 2008;2008 doi: 10.1155/2008/839501. 839501. [DOI] [PMC free article] [PubMed] [Google Scholar]