

Abstract
p19ARF is a tumor suppressor that is frequently deleted in human cancer. It lies at chromosome 9p21 and shares exons 2 and 3 with p16ink4a, which is also inactivated by these cancer-associated deletions. The “canonical pathway” by which p19ARF is thought to suppress tumorigenesis through activation of the p53 tumor suppressor. In response to hyperproliferative signals, such as expression of oncogenes, p19ARF is induced and binds to the MDM2 ubiquitin ligase, sequestering it in the nucleolus to allow the accumulation of p53. However, p19ARF also has MDM2 and p53 independent functions. In human colon cancer, p19ARF is only rarely deleted, but it is more frequently silenced by DNA promoter methylation. Here we show that inactivation of p19ARF in mice increases the number of cycling cells in the crypts of the colonic epithelium. Moreover, inactivation of p19ARF exacerbated the ulceration of the colonic epithelium caused by dextran sodium sulfate (DSS). These effects were similar to those observed in mice lacking myeloid translocation gene-related-1 (Mtgr1), and mice lacking both of these genes showed an even greater sensitivity to DSS. Surprisingly, inactivation of p19ARF restored the loss of the secretory lineage in mice deficient in Mtgr1, suggesting an additional role for p19ARF in the small intestinal epithelium.
Keywords: p19ARF, Mtgr1, colon, tumor suppressor
In response to oncogenic stimuli, the p19ARF tumor suppressor is transcriptionally induced to high levels. p19ARF binds to and inactivates the MDM2 E3 ubiquitin ligase, thereby allowing p53 to accumulate to trigger cell cycle arrest or apoptosis [Quelle et al., 1995; Palmero et al., 1998; Pomerantz et al., 1998; Zhang et al., 1998; Zindy et al., 1998; Tao and Levine, 1999]. Thus, p19ARF is a tumor suppressor that is typically inactivated in a reciprocal manner to p53 [Sherr, 1998; Vonlanthen et al., 1998; Eischen et al., 1999]. While the tumor studies place these two tumor suppressors in the same genetic pathway, mouse studies have suggested p53-independent functions for p19ARF. Mice lacking p19ARF develop a range of tumors, most frequently sarcomas and lymphomas by 3–9 months of age [Kamijo et al., 1997; Kamijo et al., 1999a], whereas mice lacking p53 develop tumors more rapidly with a preponderance of lymphomas [Donehower et al., 1992; Jacks et al., 1994; Purdie et al., 1994]. These distinct phenotypes may be due to a further role for p53 in the DNA damage response pathway, but the analysis of mice lacking both MDM2 and p53 uncovered functions for p19ARF that are independent of either of these genes [Carnero et al., 2000; Weber et al., 2000]. In fact, expression of p19ARF impaired the growth of MDM2/p53 double knockout cells in vitro and removal of both p19ARF and p53 allowed cells to grow more quickly than cells lacking either p53 or p19ARF alone [Eischen et al., 1999]. Therefore, the p53-independent effects of p19ARF are biologically significant and may contribute to tumorigenesis.
Although mice lacking p19ARF develop tumors, the developmental defects that have been uncovered are modest and appear to be associated with impaired apoptosis. For example, while the breast develops normally, there is a delay in involution of the breast after pregnancy [Yi et al., 2004]. In addition, these mice develop impaired sight due to inefficient regression of the hyaloid vascular system in the eye [McKeller et al., 2002; Silva et al., 2005], which may be similar to persistent hyperplastic primary vitreous in humans. In addition to apoptosis, an internally translated form of p19ARF termed smARF can function in the cytoplasm to dissipate mitochondrial membrane potentials and trigger autophagy [Reef et al., 2006]. Nevertheless, whether via apoptosis or autophagy, the p19ARF-null phenotypes are generally associated with a lack of cell death.
Myeloid translocation gene-related-1 (Mtgr1) is a member of the MTG family of transcriptional co-repressors [Morohoshi et al., 2000; Amann et al., 2005]. This gene family also includes MTG8 and MTG16, which are frequently disrupted by chromosomal translocations leading to acute myeloid leukemia and may be mutated in colorectal carcinoma [Gamou et al., 1998; Gelmetti et al., 1998; Wang et al., 1998; Amann et al., 2001; Davis et al., 2003; Sjoblom et al., 2006]. Mice lacking Mtgr1 are proportionally smaller than wild-type littermates but appear to develop normally [Amann et al., 2005]. However, there is a progressive reduction in the cells of the epithelial secretory lineage within the small intestine. In addition, Mtgr1-null mice display increased cellular proliferation in the colon and hypersensitivity to dextran sodium sulfate (DSS) treatment [Martinez et al., 2006].
MTG family members appear to act by bridging DNA binding transcriptional repressors and histone deacetylases to allow these transcription factors to carry out cell fate decisions [Lutterbach et al., 1998; Amann et al., 2001]. For example, MTG family members are recruited by TAL1/Scl to mediate functions in erythroid differentiation [Grosveld et al., 2005; Schuh et al., 2005]. In the gut, MTG family members may be recruited by Gfi1 [McGhee et al., 2003], which is required for the formation of goblet and Paneth cells in the small intestine [Amann et al., 2005; Shroyer et al., 2005]. Moreover, MTG family members associate with members of the T-cell factor (TCF) family [Moore et al., 2008]. Mtgr1 binds to and impairs ß-catenin-mediated TCF4-dependent transcriptional activation, and inactivation of Mtgr1 appears to de-repress specific TCF4 regulated genes such as c-Myc, which may contribute to oncogenesis [Moore et al., 2008].
The human homologue of p19ARF (p14ARF) is frequently mutated in non-intestinal malignancies, but in colorectal carcinoma it is primarily inactivated through DNA methylation [Burri et al., 2001; Lee et al., 2004; Iacopetta et al., 2006]. While there are no obvious alterations of the intestinal epithelium in mice lacking p19ARF [Kamijo et al., 1997; Kamijo et al., 1999b; Zindy et al., 2003], phenotypes can be uncovered by applying stress to a tissue. Therefore, to probe the function of p19ARF in the intestinal epithelium we stressed the colonic epithelium of p19ARF-null mice with DSS, which causes a mild ulceration of the epithelium [Bansal and Sonnenberg, 1996]. Contrary to the expected protection from cell death, loss of this tumor suppressor cooperated with DSS to cause more epithelial cell damage. In addition, we noted an expansion of the proliferating cells in the colonic crypts. Intriguingly, removing both Mtgr1, which also cooperates with DSS causing ulceration [Martinez et al., 2006], and p19ARF exacerbated the effects of DSS, yet inactivation of p19ARF reverted the Mtgr1-null phenotype in the small intestine. Thus, DSS treatment uncovered a key role for p19ARF in the intestinal epithelium.
MATERIALS AND METHODS
Mice
Mtgr1-null mice of the N5 generation of C57BL/6 background were bred with p19ARF-null mice of a mixed SvEv129 × C57BL/6 [Kamijo et al., 1997] genetic background to obtain mice heterozygous for both genes. These mice were then bred to obtain double-null animals. In addition, the Mtgr1-null mice were backcrossed for three generations to SvEv129 mice to examine the effects on the small intestine in this genetic background.
Immunohistochemistry and Histology
Tissues were fixed in buffered formalin overnight at room temperature prior to paraffin embedding and sectioning. Antibodies used for immunohistochemistry include anti-BrdU (Accurate Labs), anti-Ki67 SP6 (Neomarkers, Fremont, CA), and anti-Chromogranin A (Immunostart, Hudson, WI). The Vectastain ABC Elite (Vector Laboratories, Inc) system and diaminobenzidine (Dakocytomation Inc.) were used to visualize staining. Slides were counter-stained with Mayer’s hematoxylin prior to mounting. Hematoxylin and Eosin (H&E) and periodic acid-Schiff (PAS) staining were performed according to standard procedures. For TUNEL staining, paraffin embedded tissue sections were analyzed using the Apop-Tag Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon International). For the analysis of cycling cells, BrdU (5-bromo-2′deoxyuridine) was injected intraperitonally (IP) at a dose of 16.7 μg/g of body weight. The mice were sacrificed 2 h post-injection. Actively replicating cells were visualized using anti-BrdU.
DSS Treatment
A 3% (w/v) DSS (MW 40,000–50,000; USB Corporation) solution was made with water and filtered with a 0.22 μm cellulose acetate filter [Okayasu et al., 1990]. A control group received untreated water for 7 days. The treatment group received DSS-treated water for 4 days, followed by 3 days of untreated water. Mice were sacrificed at day 7, following the recovery period. Daily weights were obtained throughout the treatment and recovery periods. In addition, stools were examined for consistency and the presence of blood. After sacrificing the mice, the colon lengths were measured. The colons were then irrigated with PBS, and cut open longitudinally. The colon was rolled so that the most distal region became the innermost part of the roll. The tissue was fixed in formalin (1:10 dilution buffered) overnight. The solution was then changed to 70% ethanol prior to standard paraffin embedding. Sections were cut and stained with H&E for evaluation.
Statistics
Immunohistochemistry (number of staining cells) was analyzed for statistical significance using the Student’s t-test. Cells were counted from at least 100 villi or glands from a minimum of three independent samples.
RESULTS
p19ARF-Null Colons Display Increased Sensitivity to DSS Treatment
p14ARF is a tumor suppressor that is silenced by DNA methylation in colorectal carcinoma. However, p19ARF-deficient mice do not have a colonic phenotype and do not show a predis-position to colonic tumors, but instead succumb primarily to sarcomas and lymphomas between 3 and 9 months of age [Kamijo et al., 1997, 1999a]. Given that inactivation of p19ARF impairs p53-mediated apoptosis in response to proliferative signals, we hypothesized that removal of p19ARF would protect the colonic epithelium from insults that trigger apoptosis. Low-molecular-weight DSS triggers modest levels of apoptosis in the colonic epithelium, leading to an ulcerative colitis [Cooper et al., 1993; Vetuschi et al., 2002]. When two separate cohorts of p19ARF-null mice were fed water containing 3% DSS, rather than block DSS-induced cell death, these mice displayed hyper-sensitivity to DSS (Fig. 1). The colonic epithelium showed regions of significant erosion with more prominent inflammatory cell infiltration than control mice. The levels of damage observed were comparable to mice lacking the transcriptional co-repressor Mtgr1, which also show increased sensitivity to DSS [Martinez et al., 2006] and were used as a positive control (Fig. 1).
Fig. 1.

Loss of p19ARF sensitizes the colonic epithelium to DSS. Histological sections of colons from mice of the indicated genotypes were rolled from distal to proximal prior to formalin fixation, paraffin embedding, sectioning and staining with H&E (40× magnification).
That apoptosis was not affected in the absence of p19ARF was confirmed using TUNEL staining. TUNEL-positive cells were abundant in both wild-type and p19ARF-null mice (Fig. 2A). Although the damage was more severe in p19ARF-null mice, we did not observe a dramatic change in the number of TUNEL-positive cells, but this may be due to the nature of the assay.
Fig. 2.

Loss of p19ARF caused no apparent changes in apoptosis, but allowed for an increase in proliferation. A. TUNEL staining was used to detect apoptotic cells in the colonic epithelium of mice of all genotypes indicated (200× magnification). Note that the number of TUNEL-positive cells per crypt could not be quantified due to the lack of crypt structures. B. Inactivation of p19ARF impairs crypt regeneration following DSS treatment. DSS-treated and untreated mice of all genotypes were injected with BrdU (16.7 μg/g body weight) 2 h prior to sacrifice. Immunohistocheminstry was performed on formalin fixed, paraffin embedded sections with anti-BrdU to detect regenerating crypts in the colons of these mice (200× magnification). Mice fed only water served as controls. C. Quantification of the number of BrdU-positive cells per gland in untreated mice of the indicated genotypes. At least 100 glands were counted in a minimum of 3 separate mice from each genotype and the means and standard deviations are shown (as compared to wild-type, p19ARF−/−, P = 0.0018; Mtgr1−/− P = 0.0103).
To further characterize the p19ARF-null sensitivity to DSS, we examined the burst of proliferation that occurs during regeneration of the glands after treatment with DSS (Fig. 2B). BrdU was injected to label the cells actively synthesizing DNA, and the incorporated BrdU was detected by immunohistochemistry. Wild-type mice contained many BrdU-positive cells and mice lacking either p19ARF or Mtgr1 displayed reduced numbers of proliferating cells in the colons, likely owing to the reduced numbers of surviving crypt cells after DSS treatment (Fig. 2B). However, of the surviving glandular structures, there were similar numbers of cycling cells per crypt. Unexpectedly, in the untreated control colons of p19ARF-null mice, roughly 50% more BrdU-positive cells were found than in the wild-type control mice (Fig. 2B and C, P = 0.0018). This level of proliferation was similar to that observed in Mtgr1-null mice (Fig. 2C, P = 0.0103), and was confirmed using anti-Ki-67 (data not shown). Thus, p19ARF is required to suppress the proliferation of cells in the region of the crypts that contain stem and progenitor cells, and loss of this tumor suppressor sensitized this population to the effects of DSS.
Inactivation of Mtgr1 and p19ARF Exacerbates DSS Sensitivity
Both Mtgr1-null and p19ARF-null mice are hypersensitive to DSS. The Mtgr1−/− phenotype correlates with enhanced proliferation and activation of c-Myc, a known trigger of p19ARF [Moore et al., 2008]. Therefore, we cross bred these mice to test whether inactivation of Mtgr1 would cooperate with deletion of p19ARF or whether loss of p19ARF would ameliorate the effects of inactivation of Mtgr1. Two cohorts of double-null mice were treated with 3% (w/v) DSS. As compared to inactivation of either Mtgr1 or p19ARF alone (Figs. 1 and 3), the double-null mice showed an even more dramatic denuding of the colonic epithelium (Fig. 3). Histological analysis showed little to no crypt structure remaining in the double-null mice, leaving behind a heterogeneous cellular mass (Fig. 3). In addition to the inflammatory infiltrate observed in all genotypes, inflammatory cell aggregates were more prevalent in the double-null mice (Fig. 3).
Fig. 3.

Loss of both p19ARF and Mtgr1 causes a greater sensitivity to DSS. Histological sections of the colons from mice of the indicated genotypes were rolled from distal to proximal prior to formalin fixation, paraffin embedding and staining with H&E (40× magnification).
DSS-induced colon damage leads to weight loss and shortening of the colon [Vetuschi et al., 2002], which provide independent measures of colon damage. Inactivation of p19ARF caused more weight loss than observed in wild-type control mice and even slightly more than that found in Mtgr1-null mice (Fig. 4A). The double-null mice displayed even greater weight loss, which is consistent with the increased damage observed histologically. Indeed, these mice exhibited severe hematochezia, lost up to 25% of their initial body weight, and demonstrated exaggerated shortening of the colon at the time of sacrifice when compared to wild-type or even Mtgr1-null mice (Fig. 4B). A similar enhancement in inflammation was observed using the Dieleman colitis score, which combines grades for inflammation, extent of damage, regeneration, and crypt damage (Fig. 4C). Though only the double-null mice demonstrated a statistically significant score increase (P = 0.0051), there was a noticeable increase in inflammation when p19ARF-null or Mtgr1-null mice were examined.
Fig. 4.

Quantification of the increased DSS sensitivity in Mtgr1/p19ARF double-null mice. A. Double-null mice lose a larger percent of their initial body weight than the other genotypes when treated with DSS. For simplicity, only the double-null control group is shown here, but the mock treated mice all behaved similarly (N = 8 per group). B. Colon lengths were also significantly reduced in the DSS-treated double-null mice when compared to DSS-treated wild-type mice (P = 0.0307). Only the double-null control group is shown here, but the mock treated mice all behaved similarly (N = 8 per group). C. Using the Dieleman colitis grading system, sections were blindly graded. Genotypes are as follows: WT, wild-type; AN, p19ARF-null; MN, Mtgr1-null, DN, double-null. The double-null group displayed significantly more damage than the control group (P = 0.0051).
To further characterize the double-null mice, we performed TUNEL and BrdU analysis. More TUNEL-positive cells were observed in the Mtgr1/p19ARF double-null mice, which is consistent with the greater DSS-mediated damage (Fig. 5A). Due to the severe damage and lack of crypt structure, we were unable to determine an apoptotic index (TUNEL-positive cells/crypt) for these samples. Additionally, BrdU was injected into experimental animals prior to sacrifice to label cells actively synthesizing DNA, and the incorporated BrdU was detected by immunohistochemistry (Fig. 5B). Wild-type mice contained many BrdU-positive cells and the Mtgr1/p19ARF double-null mice displayed reduced numbers of proliferating cells in the colons, again likely owing to the reduced numbers of surviving crypt cells after DSS treatment, which is consistent with the nearly total denuding of the epithelium by DSS. However, the surviving crypt cells were able to re-enter the cell cycle (Fig. 5B).
Fig. 5.

Double-null mice show severe damage after DSS treatment with increased apoptosis and a decrease in regenerative proliferation. A. TUNEL staining shows increased apoptosis in response to DSS treatment (200× magnification). B. Immunohistochemistry using anti-BrdU to detect regenerating crypts after DSS treatment (200× magnification).
Loss of p19ARF complements the Secretory Lineage Defect in the Small Intestines of Mtgr1-null Mice
Given the expansion of BrdU and Ki-67-positive cells in the crypt region of p19ARF-null colons, we expanded this analysis to the small intestine. As was observed in the colon, inactivation of p19ARF increased the number of cycling cells at the “shoulders” of the small intestinal crypts, where stem and progenitor cells reside (Fig. 6A). This expansion of cycling cells was akin to that found in Mtgr1-null mice and did not increase or decrease in the double knockout mice (Fig. 6B). Thus, the inactivation of p19ARF may allow progenitor cells or even stem cells to enter the cell cycle precociously, but did not cooperate with inactivation of Mtgr1 to further enhance proliferation.
Fig. 6.

p19ARF-null mice display increased intestinal proliferation. A. Immunohistochemical staining for Ki-67 to detect cycling cells, which appear dark brown (200× magnification). B. Quantification of the number of proliferating cells. Proliferating cells were counted per villus in at least 100 villi per mouse. Comparison of control vs. p19ARF−/−, Mtgr1−/−, or Mtgr1−/−/p19ARF−/−, shows thatmicefrom each null genotype have increased proliferation at P = 0.0369, 0.0009, and 0.0004, respectively.
Deletion of Mtgr1 in the germ line of mice leads to an erosion of the secretory lineage of the small intestine beginning after the weaning transition and reaching almost complete loss by 8 weeks of age [Amann et al., 2005]. While examining the small intestines of the Mtgr1/p19ARF double-null mice for Ki-67 expression, we noted that the numbers of secretory cells appeared to be, reduced in the Mtgr1-null small intestines, but normal in the Mtgr1/p19ARF double-null mice (Fig. 7A). PAS staining of the distal small intestine confirmed the presence of an abundance goblet cells (Fig. 7A), as compared to Mtgr1-null small intestines. By contrast, there was no apparent change in the architecture of the villi in the absence of only p19ARF. To extend the analysis of the secretory lineage cells, enteroendocrine cells were stained with anti-Chromagranin A (Fig. 7A). Removal of only p19ARF had no effect on lineage allocation in the small intestine, whereas the double knockout mice had a fully restored secretory lineage (Fig. 7A and 7B).
Fig. 7.

Loss of p19ARF restores the secretory lineage in mice lacking Mtgr1. A: H&E stained sections of ilea from adult wild-type (Con.), Mtgr1−/−, p19ARF−/−, and Mtgr1−/−/p19ARF−/− mice (100× magnification). PAS staining of the ilea of adult mice to detect Goblet cells, which stain fuchsia (100× magnification) and immunohistochemical staining with anti-Chromogranin A to detect enteroendocrine cells in dark brown (100× magnification). A larger view of the Mtgr1−/− and double-null panels for PAS can be seen in Supplemental Figure 1. B: Quantification of the results in (A) shown in graphical form. The numbers of-positive cells were counted over at least 100 villi per mouse with mean value indicated with a solid line. For PAS, control mice were compared to Mtgr1−/− mice, P = 0.0001; for Mtgr1−/− compared to Mtgr1−/−/p19ARF−/−, P = 0.0002. For Chromagranin A, comparison of control vs. Mtgr1−/−, P = 0.0067; Mtgr1−/− vs. Mtgr1−/−/p19ARF−/− P = 0.0007.
Although inactivation of p19ARF sensitized the colonic epithelium to the effects of DSS-mediated cell damage and death, in other systems, loss of this tumor suppressor impairs checkpoints that trigger cell cycle arrest or cell death. Consistent with this function, inactivation of p19ARF allowed the accumulation of cycling progenitor cells in the small intestine (Fig. 6). However, the combined loss of Mtgr1 and p19ARF did not enhance the accumulation of cycling cells, suggesting that enhanced pro-liferation was not the mechanistic basis for the replacement of the lost secretory lineage progenitor cells in the double-null mice (Fig. 7A). Therefore, we tested whether inactivation of p19ARF protected cells against apoptosis, which might contribute to the restoration of the secretory cells. Although the numbers of TUNEL-positive cells were very low in the crypts of the small intestines, mice lacking Mtgr1 had increased numbers of apoptotic cells (Fig. 8). This defect was reversed in the Mtgr1/p19ARF-double-null mice, suggesting that enhanced survival contributed to the increase in the secretory lineage progenitors in the Mtgr1/p19ARF double-null mice, which allows normal secretory lineage development (double-null as compared to the Mtgr1-null; P = 0.0066; Fig. 8).
Fig. 8.
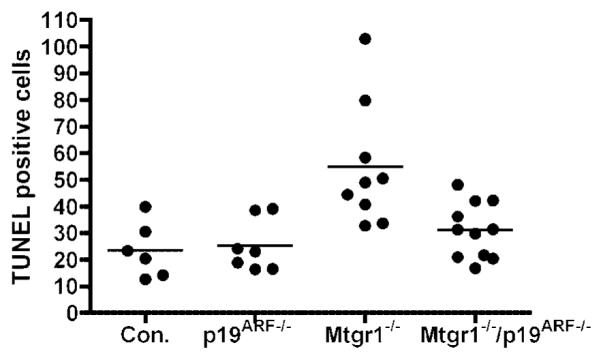
Apoptotic analysis of the small intestine. Graphical representation of TUNEL-positive cells per 100 crypts in the adult ilea shows increased apoptosis in Mtgr1−/− mice, which is not observed in the double-null mice. Comparing control vs. Mtgr1−/−, P = 0.0087; Mtgr1−/− compared to Mtgr1−/−/p19ARF−/−, P = 0.0066.
DISCUSSION
p19ARF is a tumor suppressor that functions upstream of p53 to sense latent oncogenic signals [Chin et al., 1998; Zindy et al., 2003]. Activation of p19ARF expression induces senescence or apoptosis depending on the cellular context. Conversely, the developmental phenotypes observed in mice lacking this tumor suppressor appear to be associated with impaired apoptosis. For example, the delay in involution in the breast after pregnancy, and impaired regression of the hyaloid vascular system in the eye [McKeller et al., 2002; Yi et al., 2004; Silva et al., 2005]. In contrast to the impairment of apoptosis observed in development, inactivation of p19ARF cooperated with DSS and caused more ulceration in the distal colonic epithelium, and rather than ameliorating the effects of inactivation of Mtgr1, the double knockout mice displayed an even greater sensitivity. Thus, inactivation of p19ARF alters the colonic epithelium, perhaps by allowing the accumulation of cycling stem and progenitor cells in the crypts (Fig. 2).
The hypersensitivity of the colons to DSS may suggest that p19ARF has previously unidentified functions outside of triggering apoptosis or autophagy in response to proliferative signals [Chin et al., 1998; Reef et al., 2006]. Other biochemical functions of p19ARF include a role in directing modification of proteins by small ubiquitin like modifiers (SUMO) and ubiquitin [Tago et al., 2005] and the regulation of ribosomal RNA metabolism through association with nucleophosmin [Colombo et al., 2005; Colombo et al., 2006]. Given that DSS may target the stem/progenitor cell compartment to trigger cell death [Vetuschi et al., 2002], our work suggests a role for p19ARF in this compartment. p19ARF-null mice displayed an expansion of the cycling population of cells in the crypts of the colon and small intestine (Figs. 2 and 6), which is consistent with the role of p19ARF in opposing proliferation [Kamijo et al., 1997, 1999a; Pomerantz et al., 1998; Zindy et al., 2003]. That only the proliferative compartment, and not the entire villus or gland, was lengthened may provide clues to the DSS sensitivity if the progenitor cells are the target of DSS. However, in the double-null mice, the proliferative compartment was not expanded in an additive fashion, so simply expanding this population does not explain the exacerbated sensitivity to DSS in the double-null mice. Nevertheless, inactivation of p19ARF may contribute to colorectal carcinogenesis by allowing a highly proliferating cell, perhaps already containing mutations, to further expand and accumulate while also affecting homeostatic maintenance of the colonic epithelium.
In the small intestine, inactivation of p19ARF complemented the Mtgr1-deficient defect in lineage allocation. p19ARF inactivation also expanded the proliferating cell compartment in the small intestinal crypts (Fig. 6). Similar to the effects in the colon, co-deletion of Mtgr1 and p19ARF did not further expand this population and did not trigger polyp formation or increase villus length. Though the number of TUNEL-positive cells was small, there was a statistically significant decrease in apoptotic cells in the double-null mice, which may suggest that the restoration of the secretory lineage in the double-null mice was due to the impairment of apoptosis within the secretory lineage progenitor cells. In addition to its role in triggering apoptosis, p19ARF is also required for cellular senescence in some contexts, with loss of p19ARF being associated with cellular immortalization [Lundberg et al., 2000; Randle et al., 2001; Shamanin and Androphy, 2004; Nagy et al., 2005; Benanti et al., 2007]. Given that cellular differentiation is generally associated with cells leaving the cell cycle, the restoration of the secretory lineage is contrary to the expected immortalization/proliferation in the absence of p19ARF, but would explain the expansion of the proliferating compartment.
The p19ARF promoter is methylated in colorectal carcinoma [Burri et al., 2001; Lee et al., 2004; Iacopetta et al., 2006]. Our results suggest that silencing of the p19ARF promoter may contribute to colorectal carcinogenesis by both expanding the proliferating cells of the colonic crypts and making the epithelium more sensitive to injury with the accompanying inflammatory processes. Although Mtgr1 has yet to be linked to the development of colorectal cancer, MTG family members can act as co-repressors for TCF4 and can oppose the action of β-catenin [Moore et al., 2008]. However, inactivation of both Mtgr1 and p19ARF did not shorten the latency for tumorigenesis and no polyps were observed in the intestines of double-null mice over the course of 18 months (data not shown). Thus, while the closely related MTG8 is mutated in colorectal carcinoma [Sjoblom et al., 2006] and may act as a “driver” of carcinogenesis [Wood et al., 2007], Mtgr1 inactivation does not appear to contribute to tumorigenesis. Nevertheless, inactivation of Mtgr1 alone, or in the context of p19ARF deficiency, sensitizes the colonic epithelium to the effects of DSS indicating that its loss can contribute to inflammatory bowel diseases and, thus, indirectly to tumorigenesis.
ACKNOWLEDGMENTS
We thank the members of the Hiebert Lab for helpful discussions and encouragement, and the Vanderbilt-Ingram Cancer Center and Vanderbilt Digestive Diseases Research Center for support and the use of shared resources for DNA sequencing, Histology, and Immunohistochemistry. This work was supported by National Institutes of Health grants RO1-CA64140, RO1-CA112005 (SWH), and Center grants from the NCI (CA68485), and NIDDK (5P30DK58404-03).
Grant sponsor: National Institutes of Health; Grant numbers: RO1-CA64140, RO1-CA112005; Grant sponsor: NCI; Grant number: CA68485; Grant sponsor: NIDDK; Grant number: 5P30DK58404-03.
Footnotes
This article contains supplementary material, which may be viewed at the Journal of Cellular Biochemistry website at http://www.interscience.wiley.com/jpages/0730-2312/suppmat/index.html.
REFERENCES
- Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N, Downing JR, Meyers S, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21:6470–6483. doi: 10.1128/MCB.21.19.6470-6483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann JM, Chyla BJ, Ellis TC, Martinez A, Moore AC, Franklin JL, McGhee L, Meyers S, Ohm JE, Luce KS, Ouelette AJ, Washington MK, Thompson MA, King D, Gautam S, Coffey RJ, Whitehead RH, Hiebert SW. Mtgr1 is a transcriptional corepressor that is required for maintenance of the secretory cell lineage in the small intestine. Mol Cell Biol. 2005;25:9576–9585. doi: 10.1128/MCB.25.21.9576-9585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal P, Sonnenberg A. Risk factors of colorectal cancer in inflammatory bowel disease. Am J Gastroenterol. 1996;91:44–48. [PubMed] [Google Scholar]
- Benanti JA, Wang ML, Myers HE, Robinson KL, Grandori C, Galloway DA. Epigenetic down-regulation of ARF expression is a selection step in immortalization of human fibroblasts by c-Myc. Mol Cancer Res. 2007;5:1181–1189. doi: 10.1158/1541-7786.MCR-06-0372. [DOI] [PubMed] [Google Scholar]
- Burri N, Shaw P, Bouzourene H, Sordat I, Sordat B, Gillet M, Schorderet D, Bosman FT, Chaubert P. Methylation silencing and mutations of the p14ARF and p16INK4a genes in colon cancer. Lab Invest. 2001;81:217–229. doi: 10.1038/labinvest.3780230. [DOI] [PubMed] [Google Scholar]
- Carnero A, Hudson JD, Price CM, Beach DH. p16INK4A and p19ARF act in overlapping pathways in cellular immortalization. Nat Cell Biol. 2000;2:148–155. doi: 10.1038/35004020. [DOI] [PubMed] [Google Scholar]
- Chin L, Pomerantz J, DePinho RA. The INK4a/ARF tumor suppressor: one gene—two products—two pathways. Trends Biochem Sci. 1998;23:291–296. doi: 10.1016/s0968-0004(98)01236-5. [DOI] [PubMed] [Google Scholar]
- Colombo E, Bonetti P, Lazzerini Denchi E, Martinelli P, Zamponi R, Marine JC, Helin K, Falini B, Pelicci PG. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol Cell Biol. 2005;25:8874–8886. doi: 10.1128/MCB.25.20.8874-8886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo E, Martinelli P, Zamponi R, Shing DC, Bonetti P, Luzi L, Volorio S, Bernard L, Pruneri G, Alcalay M, Pelicci PG. Delocalization and destabilization of the Arf tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 2006;66:3044–3050. doi: 10.1158/0008-5472.CAN-05-2378. [DOI] [PubMed] [Google Scholar]
- Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- Davis JN, McGhee L, Meyers S. The ETO (MTG8) gene family. Gene. 2003;303:1–10. doi: 10.1016/s0378-1119(02)01172-1. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamou T, Kitamura E, Hosoda F, Shimizu K, Shinohara K, Hayashi Y, Nagase T, Yokoyama Y, Ohki M. The partner gene of AML1 in t(16;21) myeloid malignancies is a novel member of the MTG8(ETO) family. Blood. 1998;91:4028–4037. [PubMed] [Google Scholar]
- Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18:7185–7191. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosveld F, Rodriguez P, Meier N, Krpic S, Pourfarzad F, Papadopoulos P, Kolodziej K, Patrinos GP, Hostert A, Strouboulis J. Isolation and characterization of hematopoietic transcription factor complexes by in vivo biotinylation tagging and mass spectrometry. Ann NY Acad Sci. 2005;1054:55–67. doi: 10.1196/annals.1345.008. [DOI] [PubMed] [Google Scholar]
- Iacopetta B, Russo A, Bazan V, Dardanoni G, Gebbia N, Soussi T, Kerr D, Elsaleh H, Soong R, Kandioler D, Janschek E, Kappel S, Lung M, Leung CS, Ko JM, Yuen S, Ho J, Leung SY, Crapez E, Duffour J, Ychou M, Leahy DT, O’Donoghue DP, Agnese V, Cascio S, Di Fede G, Chieco-Bianchi L, Bertorelle R, Belluco C, Giaretti W, Castagnola P, Ricevuto E, Ficorella C, Bosari S, Arizzi CD, Miyaki M, Onda M, Kampman E, Diergaarde B, Royds J, Lothe RA, Diep CB, Meling GI, Ostrowski J, Trzeciak L, Guzinska-Ustymowicz K, Zalewski B, Capella GM, Moreno V, Peinado MA, Lonnroth C, Lundholm K, Sun XF, Jansson A, Bouzourene H, Hsieh LL, Tang R, Smith DR, Allen-Mersh TG, Khan ZA, Shorthouse AJ, Silverman ML, Kato S, Ishioka C. Functional categories of TP53 mutation in colorectal cancer: Results of an International Collaborative Study. Ann Oncol. 2006;17:842–847. doi: 10.1093/annonc/mdl035. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999a;59:2217–2222. [PubMed] [Google Scholar]
- Kamijo T, van de Kamp E, Chong MJ, Zindy F, Diehl JA, Sherr CJ, McKinnon PJ. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res. 1999b;59:2464–2469. [PubMed] [Google Scholar]
- Lee S, Hwang KS, Lee HJ, Kim JS, Kang GH. Aberrant CpG island hypermethylation of multiple genes in colorectal neoplasia. Lab Invest. 2004;84:884–893. doi: 10.1038/labinvest.3700108. [DOI] [PubMed] [Google Scholar]
- Lundberg AS, Hahn WC, Gupta P, Weinberg RA. Genes involved in senescence and immortalization. Curr Opin Cell Biol. 2000;12:705–709. doi: 10.1016/s0955-0674(00)00155-1. [DOI] [PubMed] [Google Scholar]
- Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld MG, Glass C, Seto E, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18:7176–7184. doi: 10.1128/mcb.18.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JA, Williams CS, Amann JM, Ellis TC, Moreno-Miralles I, Washington MK, Gregoli P, Hiebert SW. Deletion of mtgr1 sensitizes the colonic epithelium to dextran sodium sulfate-induced colitis. Gastroenterology. 2006;131:579–588. doi: 10.1053/j.gastro.2006.06.009. [DOI] [PubMed] [Google Scholar]
- McGhee L, Bryan J, Elliott L, Grimes HL, Kazanjian A, Davis JN, Meyers S. Gfi-1 attaches to the nuclear matrix, associates with ETO (MTG8) and histone deacetylase proteins, and represses transcription using a TSA-sensitive mechanism. J Cell Biochem. 2003;89:1005–1018. doi: 10.1002/jcb.10548. [DOI] [PubMed] [Google Scholar]
- McKeller RN, Fowler JL, Cunningham JJ, Warner N, Smeyne RJ, Zindy F, Skapek SX. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc Natl Acad Sci USA. 2002;99:3848–3853. doi: 10.1073/pnas.052484199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AC, Amann JM, Williams CS, Tahinci E, Farmer TE, Martinez JA, Yang G, Luce KS, Lee E, Hiebert SW. Myeloid translocation gene family members associate with TCFs and influence TCF-dependent transcription. Mol Cell Biol. 2008;28:977–987. doi: 10.1128/MCB.01242-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morohoshi F, Mitani S, Mitsuhashi N, Kitabayashi I, Takahashi E, Suzuki M, Munakata N, Ohki M. Structure and expression pattern of a human MTG8/ETO family gene, MTGR1. Gene. 2000;241:287–295. doi: 10.1016/s0378-1119(99)00481-3. [DOI] [PubMed] [Google Scholar]
- Nagy E, Veress G, Szarka K, Csoma E, Beck Z. Frequent methylation of p16INK4A/p14ARF promoters in tumorigenesis of Epstein-Barr virus transformed lymphoblastoid cell lines. Anticancer Res. 2005;25:2153–2160. [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, Cordon-Cardo C, DePinho RA. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Purdie CA, Harrison DJ, Peter A, Dobbie L, White S, Howie SE, Salter DM, Bird CC, Wyllie AH, Hooper ML, et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 1994;9:603–609. [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Randle DH, Zindy F, Sherr CJ, Roussel MF. Differential effects of p19(Arf) and p16(Ink4a) loss on senescence of murine bone marrow-derived preB cells and macrophages. Proc Natl Acad Sci USA. 2001;98:9654–9659. doi: 10.1073/pnas.171217498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A. short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22:463–475. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Schuh AH, Tipping AJ, Clark AJ, Hamlett I, Guyot B, Iborra FJ, Rodriguez P, Strouboulis J, Enver T, Vyas P, Porcher C. ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol Cell Biol. 2005;25:10235–10250. doi: 10.1128/MCB.25.23.10235-10250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamanin VA, Androphy EJ. Immortalization of human mammary epithelial cells is associated with inactivation of the p14ARF-p53 pathway. Mol Cell Biol. 2004;24:2144–2152. doi: 10.1128/MCB.24.5.2144-2152.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- Shroyer NF, Wallis D, Venken KJ, Bellen HJ, Zoghbi HY. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 2005;19:2412–2417. doi: 10.1101/gad.1353905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RL, Thornton JD, Martin AC, Rehg JE, Bertwistle D, Zindy F, Skapek SX. Arf-dependent regulation of Pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 2005;24:2803–2814. doi: 10.1038/sj.emboj.7600751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Tago K, Chiocca S, Sherr CJ. Sumoylation induced by the Arf tumor suppressor: a p53-independent function. Proc Natl Acad Sci USA. 2005;102:7689–7694. doi: 10.1073/pnas.0502978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Levine AJ. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci USA. 1999;96:6937–6941. doi: 10.1073/pnas.96.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetuschi A, Latella G, Sferra R, Caprilli R, Gaudio E. Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig Dis Sci. 2002;47:1447–1457. doi: 10.1023/a:1015931128583. [DOI] [PubMed] [Google Scholar]
- Vonlanthen S, Heighway J, Tschan MP, Borner MM, Altermatt HJ, Kappeler A, Tobler A, Fey MF, Thatcher N, Yarbrough WG, Betticher DC. Expression of p16INK4a/p16alpha and p19ARF/p16beta is frequently altered in non-small cell lung cancer and correlates with p53 overexpression. Oncogene. 1998;17:2779–2785. doi: 10.1038/sj.onc.1202501. [DOI] [PubMed] [Google Scholar]
- Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci USA. 1998;95:10860–10865. doi: 10.1073/pnas.95.18.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000;14:2358–2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Yi Y, Shepard A, Kittrell F, Mulac-Jericevic B, Medina D, Said TK. p19ARF determines the balance between normal cell proliferation rate and apoptosis during mammary gland development. Mol Biol Cell. 2004;15:2302–2311. doi: 10.1091/mbc.E03-11-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Williams RT, Baudino TA, Rehg JE, Skapek SX, Cleveland JL, Roussel MF, Sherr CJ. Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc Natl Acad Sci USA. 2003;100:15930–15935. doi: 10.1073/pnas.2536808100. [DOI] [PMC free article] [PubMed] [Google Scholar]