

Abstract
Mutations in progranulin (PGRN) are associated with frontotemporal dementia with or without parkinsonism. We describe the prominent phenotypic variability within and among eight kindreds evaluated at Mayo Clinic Rochester and/or Mayo Clinic Jacksonville in whom mutations in PGRN were found. All available clinical, genetic, neuroimaging and neuropathologic data was reviewed. Age of onset ranged from 49 to 88 years and disease duration ranged from 1 to 14 years. Clinical diagnoses included frontotemporal dementia (FTD), primary progressive aphasia, FTD with parkinsonism, parkinsonism, corticobasal syndrome, Alzheimer’s disease, amnestic mild cognitive impairment, and others. One kindred exhibited maximal right cerebral hemispheric atrophy in all four affected individuals, while another had maximal left hemisphere involvement in all three of the affected. Neuropathologic examination of 13 subjects revealed frontotemporal lobar degeneration with ubiquitin-positive inclusions plus neuronal intranuclear inclusions in all cases. Age of onset, clinical phenotypes and MRI findings associated with most PGRN mutations varied significantly both within and among kindreds. Some kindreds with PGRN mutations exhibited lateralized topography of degeneration across all affected individuals.
Keywords: Frontotemporal dementia, FTDP-17, Progranulin, PGRN, MRI
1. Introduction
In 1892, Arnold Pick first described behavioral and language abnormalities in association with frontotemporal lobar degeneration. Subsequently, many familial cases of frontotemporal dementia with parkinsonism were linked to chromosome 17 (FTDP-17) (Foster et al. 1997). Some were associated with mutations in the microtubule associated protein tau (MAPT),(Hutton et al. 1998; Ingram and Spillantini 2002) while others were not (Kertesz et al. 2000; Rademakers et al. 2002; Mackenzie et al. 2006b). Many cases of FTDP-17 did not exhibit immunostaining for tau on pathologic examination (Kertesz et al. 2000; Rosso et al. 2001; Savioz et al. 2003; Mackenzie et al. 2006b; van der Zee et al. 2006), and mutations in MAPT were absent in all of these. We recently reported several FTDP-17 kindreds having frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) neuropathology in association with mutations in the gene encoding Progranulin (PGRN) (Baker et al. 2006; Boeve et al. 2006; Gass et al. 2006; Mackenzie et al. 2006b). The eight families described here extend the phenotypic and MRI findings associated with mutations in PGRN and highlight the variability in presentation, clinical course and neuroimaging findings within and among kindreds.
2. Methods
2.1 Subjects
Mutations in PGRN were identified in eight families whose probands were evaluated at Mayo Clinic Rochester (n=6) or Mayo Clinic Jacksonville (n=2). At least one affected individual from each family was enrolled in the Mayo Clinic Alzheimer Disease Research Center, a Mayo Foundation Institutional Review Board-approved program. All available clinical records and neuroimaging studies on affected members of these kindreds were reviewed. Genetic analyses, MRI scans, and autopsies were performed after subjects or appropriate proxies provided written consent.
2.2 Clinical Evaluations
Age of onset was the age at which the subject first demonstrated behavioral or personality change, memory loss, motor changes, or other neurological changes as noted by themselves, family, friends or colleagues. All neurobehavioral clinical data (Members of the Department of Neurology 1998) were reviewed.
2.3 Laboratory Analyses
DNA was extracted from peripheral blood leucocytes and sequence analysis of MAPT and PGRN from patient genomic DNA was performed as previously described (Hutton et al. 1998; Baker et al. 2006).
2.4 Neuroimaging Examinations
MRI was performed using a GE scanner at 1.5 Tesla, and images of the brain were obtained in the sagittal (T1-weighted), axial [proton-density, T2-weighted, and fluid attenuation inversion recovery (FLAIR)], and coronal (T1-weighted and FLAIR) planes.
2.5 Neuropathologic Examination
Sections of neocortex, hippocampus, thalamus, basal ganglia, midbrain, pons, medulla and cerebellum were stained with hematoxylin and eosin and thioflavin-S fluorescent microscopy and immunocytochemistry for phospho-tau. Sections of cortex and hippocampus were stained with Bielschowsky and Luxol fast blue, and immunostained for ubiquitin, neurofilament, Aβ40, and Aβ42. Midbrain and amygdale sections were studied with alpha-synuclein immunostains. Immunohistochemistry for TDP-43 was applied in selected cases (1:8000; Proteintech Group, Chicago, IL).
Neuropathological diagnoses were based on established guidelines (McKhann et al. 2001). A diagnosis of frontotemporal lobar degeneration with ubiquitin-only immunoreactive neuronal changes (FTLD-U) was made if there was neuronal loss and gliosis affecting frontal or temporal lobe, plus ubiquitin-positive; tau, alpha-synuclein and neurofilament negative intraneuronal inclusions or neurites in frontal or temporal neocortex or hippocampal dentate granule cells in the absence of any evidence of motor neuron degeneration (Josephs et al. 2006a). In cases with multiple pathologies, TDP-43 immunohistochemistry was used to confirm FTLD-U.
3. Results
3.1 Family Descriptions
Pedigrees are shown in Figure 1. Clinical, radiologic, and neuropathologic findings are summarized in Table 1, with additional clinical details presented in Table 2. Representative MRI scans are shown in Figure 2, and Figure 3 is a schematic drawing localizing the mutations in PGRN. Descriptions of Kindred 7 (Mesulam et al. 2007), two members of Kindred 1 (Boeve et al. 2002) and the proband of Kindred 5 (Boeve et al. 2006) have previously been reported.
Figure 1.
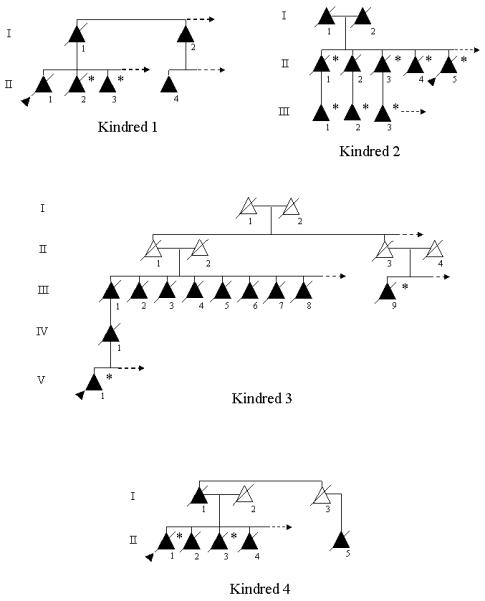
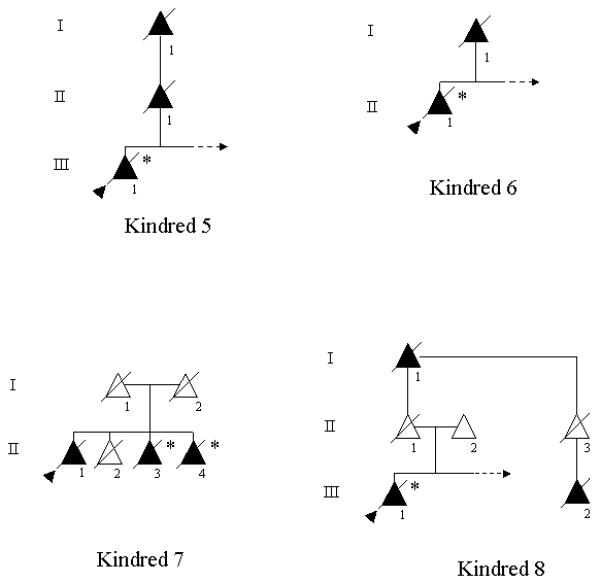
Pedigrees of the eight kindreds. Triangles represent individuals, shaded triangles represent affected individuals. Triangles with diagonal lines through them represent deceased individuals. The proband is indicated by a shaded arrowhead. Individuals with a confirmed progranulin mutation are indicated by an asterisk. An arrow pointing to the right in a sibship represents additional unaffected persons, but they were purposefully excluded to maintain confidentiality.
Table 1.
Demographic, clinical, neuropathologic, and genetic findings of all affected individuals
| Kindred | Case | Duration (years) |
Initial Diagnosis |
Current/Final Diagnosis |
Autopsy Findings |
PGRN Mutation |
Reference |
|---|---|---|---|---|---|---|---|
| Kindred 1 | I.1 | 5 | dementia NOS |
encephalomalacia | |||
| I.2 | * | dementia NOS |
dementia NOS | ||||
| II.1 (P) | 6 | PD | CBS | FTLD-U NII | Boeve et al, 2002 | ||
| II.2 | 7 | FTD | FTDP | FTLD-U NII | c.1145delC | Boeve et al, 2002 | |
| II.3 | >4 | FTD | FTD/CBS | c.1145delC | |||
| II.4 | * | dementia NOS |
dementia NOS | ||||
| Kindred 2 | I.1 | 5 | AD | AD | |||
| I.2 | 3 | dementia NOS |
dementia NOS | ||||
| II.1 | 7 | AD | AD | FTLD-U NII + AD + VD |
c.154delA | ||
| II.2 | * | dementia NOS |
dementia NOS | ||||
| II.3 | 2 | AD | AD + Park | FTLD-U NII | c.154delA | ||
| II.4 | 7 | AD | FTD | FTLD-U NII | c.154delA | ||
| II.5 (P) | 7 | amnestic MCI |
AD | FTLD-U NII | c.154delA | ||
| III.1 | >2 | amnestic MCI + dep |
amnestic MCI + dep |
c.154delA | |||
| III.2 | >7 | FTD/DLB | FTDP | c.154delA | |||
| III.3 | >7 | PPA | PPA/FTD | c.154delA | |||
| Kindred 3 | III.1 | 9 | dementia NOS |
dementia NOS | |||
| III.2 | 7 | dementia, AD |
AD | ||||
| III.3 | 1 (fire) | dementia NOS |
dementia NOS | ||||
| III.4 | 9 | dementia NOS |
dementia NOS | ||||
| III.5 | >2 | dementia NOS |
dementia NOS | ||||
| III.6 | * | dementia NOS |
dementia NOS | ||||
| III.7 | 5 | dementia NOS |
dementia NOS | ||||
| III.8 | 7 | dementia NOS |
dementia NOS | ||||
| III.9 | 3 | AD | AD | FTLD-U NII | c.1477C>T | ||
| IV.1 | 1 (MVA) | not Dx | not Dx | ||||
| V.1 (P) | 6 | FTD | FTD | c.1477C>T | |||
| Kindred 4 | I.1 | 5 | PD | PD | |||
| II.1 (P) | 13 | PD | PD + D | FTLD-U NII + LBD |
c.910_911insTG | ||
| II.2 | 7 | dementia NOS |
dementia NOS | ||||
| II.3 | 8 | FTD | FTD | FTLD-U NII | c.910_911insTG | ||
| II.4 | * | PD | PD + dementia NOS |
||||
| II.5 | * | PD | PD | ||||
| Kindred 5 | I.1 | 13 | dementia NOS |
dementia NOS | |||
| II.1 | 7 | dementia NOS |
dementia NOS | ||||
| III.1 (P) | 7 | AD | FTD | FTLD-U NII | c.138+1G>A | Boeve et al, 2006 | |
| Kindred 6 | I.1 | 5 | dementia NOS |
dementia NOS | |||
| II.1 (P) | 5 | FTD | FTD | FTLD-U NII | c.1395_1396insC | ||
| Kindred 7 | II.1 | 12 | PPA | PPA | Mesulam et al, 2007 | ||
| II.3 | 4 | PPA | PPA | FTLD-U NII | c.998delG | Mesulam et al, 2007 | |
| II.4 (P) | 8 | PPA | PPA | FTLD-U NII | c.998delG | Mesulam et al, 2007 | |
| Kindred 8 | I.1 | 14 | aphasia | aphasic dementia | |||
| III.1 (P) | >4 | PPA | PPA | c.26C>A | |||
| III.2 | * | aphasia | aphasic dementia |
Abbreviations: AD=Alzheimer’s disease; CBS=corticobasal syndrome; dep=depression, DLB=dementia with Lewy bodies; FTD=frontotemporal dementia; FTDP=frontotemporal dementia and parkinsonism; FTLD-U NII=frontotemporal lobar degeneration with ubiquitin positive inclusions and neuronal intranuclear inclusions; LBD=Lewy body disease; MCI=mild cognitive impairment; MVA=motor vehicle accident; NOS=not otherwise specified; not Dx=not diagnosed; park=parkinsonism; PD=Parkinson’s disease; PPA=primary progressive aphasia; VD=vascular dementia; * = unknown, (P)=proband
Table 2.
Specific clinical features among affected individuals examined at Mayo Clinic
| Kindred | Case | Personality Change |
Hyperoral | Executive Dysfunction |
Memory Dysfunction |
Language Dysfunction |
Parkinsonism |
|---|---|---|---|---|---|---|---|
| 1 | II.1 | mid | mid | mid | mid | early | |
| II.2 | early | late | mid | mid | late | mid | |
| II.3 | early | late | early | early | absent | late | |
| 2 | II.3 | early | early | mid | early | ||
| II.4 | early | early | early | early | late | ||
| II.5 | mid | late | early | early | late | late | |
| III.1 | early | early | early | absent | absent | ||
| III.2 | early | mid | early | mid | late | late | |
| III.3 | late | late | late | early | absent | ||
| 3 | V.1 | early | mid | later | early | early | mid |
| 4 | II.1 | mid | late | mid | late | early | |
| II.3 | early | mid | early | later | absent | ||
| 5 | III.1 | early | mid | early | early | late | late |
| 6 | II.1 | early | mid | early | early | early | absent |
| 7 | II.1 | absent | absent | present | present | early | absent |
| II.3 | present | present | late | absent | early | mid | |
| II.4 | present | absent | mid | absent | early | absent | |
| 8 | III.1 | absent | absent | absent | absent | early | absent |
early=onset within first year of symptoms; mid=onset 1-2 years after symptom onset; late=onset 3 or more years after onset; absent=no evidence in clinical records of this feature being present. Empty fields reflect insufficient details in the clinical record to determine if that feature was present or absent.
Figure 2.
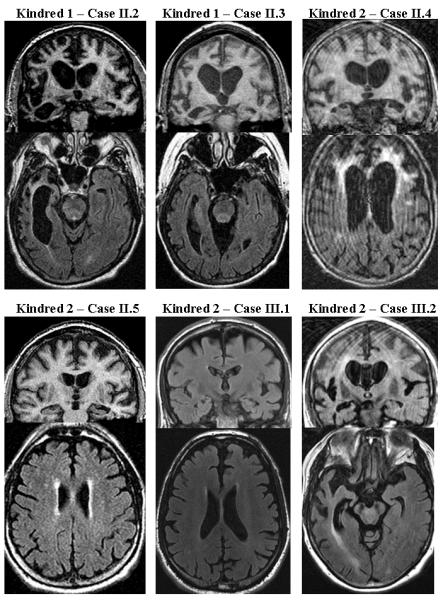
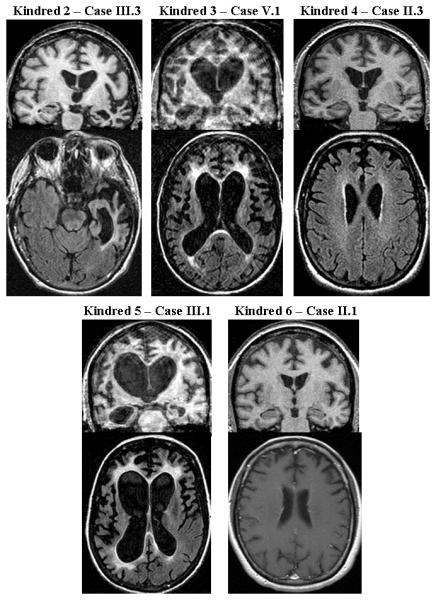
Representative coronal [T1-weighted or fluid attenuation inversion recovery (FLAIR)] (top), and axial T1-weighted or FLAIR (bottom) images for each affected individual for which MRI scans were available. Note the variability in the patterns of cerebral atrophy and degree of signal changes on FLAIR images.
Figure 3.
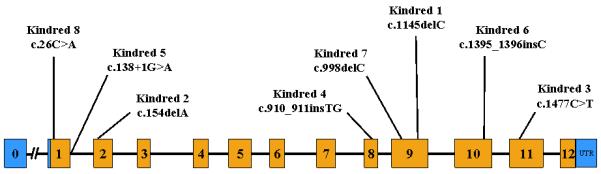
Schematic representation of the progranulin (PGRN) gene illustrating the location of the mutations in the eight kindreds. Mutations are numbered according to GenBank Accession number NM_002087.2.
3.2 Demographics
Symptom onset among 38 individuals ranged from 49 to 88 (mean 64.5 ± 9.6 years). Of 44 individuals reported to be affected, 25 were female. Among the 18 subjects (7 male) with clinical evaluations, age at onset ranged from 49 to 80 years (mean 61.1 ± 8.1 years). In Kindred 2, mean age of symptomatic onset was 75.8 ± 5.0 years in generation II and 60.7 ± 5.5 years in generation III. In Kindred 3, mean age of symptomatic onset was 70.9 ± 10.4 years in generation III and 52 ± 2.8 years in generations IV and V.
Disease duration ranged from 1 year to 14 years, with a mean of 6.6 ± 3.2 years (31 individuals). Two individuals had accidental deaths within 1 year of onset. Excluding these, the range was 2 to 14 years (mean 7.0 ± 3.0 years). Among the 13 subjects clinically evaluated who subsequently died, mean duration was 7.15 ± 2.9 years (range 2 to 13 years).
3.3 Clinical Features
As Table 2 indicates, personality change was an initial or early symptom (developing within one year of onset) in 10 of the 18 patients evaluated clinically (55%), and later developed in another 7 patients (cumulative 94%). Hyperoral behaviors developed in 8 of the 12 patients from whom this history was collected. Executive dysfunction (evidenced on neuropsychological testing) was an initial or early feature in 7 of 18 patients for whom this data was available (39%). It later developed in all but one of these patients (94.1%). Eleven of the 18 patients evaluated clinically (61%) had personality changes and/or executive dysfunction features within the first year of symptomatic disease. Initial memory impairment was present in 8 of the 18 patients seen clinically (44%), developing prior to personality change or executive dysfunction in 2 unrelated patients. Memory impairment was prominent enough in six members of Kindred 2 that their initial diagnoses were AD or amnestic MCI, and AD remained the final diagnosis in four individuals. Language impairment eventually developed in 16 of the 18 individuals for whom this data was available, and was an initial or early symptom in 8 (44%).
Parkinsonism was seen in 11 of 18 patients (61%). It was an initial or early symptom/sign in 3 patients (17%). In three patients, parkinsonism developed four or more years after onset.
No patient exhibited features of motor neuron disease.
3.4 Neuroimaging Findings
MRI was available for review in 14 patients and brief descriptions of the MRI findings in these kindreds are presented in Table 3. Representative images highlighting the variable patterns of atrophy and the prominent subcortical white matter FLAIR hyperintensities are presented in Figure 2. Atrophy was identified in all patients, with frontotemporal predominance in 13. Parietal cortical atrophy was present in four. Subcortical white matter signal changes with a frontal predominance were identified in six individuals, and in four subjects (cases II.4 and III.2 of Kindred 2, case V.1 of Kindred 3, and case III.1 of Kindred 5), striking signal changes were present. These subcortical signal changes were most prominent in the regions of maximal cortical atrophy, and the extent of signal change increased with disease duration (Boeve et al. 2006). In two cases with focal temporal lobe atrophy (Kindred 1 case II.1 and Kindred 2 case III.3), milder subcortical signal changes were present. Longitudinal MRI scans spanning two or more years were available in six patients, which all showed progression of atrophy, ventricular, and subcortical white matter signal changes.
Table 3.
Descriptive MRI findings in affected individuals
| Case | MRI Findings |
|---|---|
| Kindred 1 II.2 |
Asymmetric right greater than left frontotemporal atrophy, and lesser right parietal and occipital atrophy. Subcortical white matter changes were associated with this atrophy. Subsequent imaging confirmed progression of these findings. |
| Kindred 1 II.3 |
Mild temporal and hippocampal atrophy, and subtle subcortical white matter changes, greater on the right than the left |
| Kindred 2 II.4 |
Prominent frontotemporal atrophy and bilateral frontal white matter T2 hyperintensity with extension into the cortex |
| Kindred 2 II.5 |
Mild atrophy along the falx anteriorly, more obvious atrophy in the left mesial and inferolateral temporal lobe and mild leukoaraiosis |
| Kindred 2 III.1 |
Mild diffuse atrophy, more obvious along the anterior falx, without significant mesial temporal atrophy |
| Kindred 2 III.2 |
Moderately severe generalized cerebral atrophy, most prominent in the right temporal lobe. Mild subcortical white matter T2 hyperintensity evident in the right temporal lobe. |
| Kindred 2 III.3 |
Initial MRI showed marked atrophy of the left anterior temporal lobe and mild atrophy of the left frontal opercular region. Subsequent MRI examinations demonstrated progressive left hemispheric atrophy, most severely in the temporal lobe |
| Kindred 3 V.1 |
Moderately severe atrophy, most prominent in the frontal and temporal lobes. Increased T2 signal in the frontal subcortical white matter adjacent to the lateral ventricles |
| Kindred 4 II.3 |
Moderate anterior falcine atrophy without white matter signal changes |
| Kindred 5 III.1 |
Marked bilateral frontotemporoparietal atrophy particularly on the right, with subcortical white matter changes |
| Kindred 6 II.1 |
Mild diffuse cortical atrophy, slightly more prominent along the falx anteriorly |
| Kindred 7 II.3 |
Progressive left temporal atrophy with less severe frontal atrophy apparent on subsequent neuroimaging |
| Kindred 7 II.4 |
Progressive left temporal and parietal atrophy with enlargement of the Sylvian fissure |
| Kindred 8 III.1 |
Mild left frontotemporal atrophy |
In Kindred 2, the topography of cortical atrophy was variable. Kindred 1 showed right cerebral hemisphere predominance in the three affected siblings, and the same can be presumed given their father’s left-sided motor features. The opposite was apparent in Kindred 7, with all three siblings having left cerebral hemisphere atrophy.
3.5 Genetic Findings
As shown in Figure 3, a different PGRN mutation was identified in each family. (Baker et al. 2006; Gass et al. 2006). The specific mutations are listed in Table 1, indicating which individuals were genetically tested. Mutations in Kindreds 1, 2, 3, 6 and 7 cause frameshifts which create a premature stop codon, resulting in a null allele through nonsense-mediated decay. Similarly, the nonsense mutation in Kindred 3 induces nonsense-mediated decay. The mutation in family Kindred 5 destroys the 5′ splice site of exon 1 thereby removing the start methionine codon, completely blocking PGRN protein from being generated (Boeve et al. 2006; Gass et al. 2006). Kindred 8 carried a mutation that disrupts the signal peptide sequence. RT-PCR transcript analyses for this mutation showed strongly reduced levels of mutant RNA, confirming the generation of a null allele (Gass et al. 2006).
At least one mutation carrier remains asymptomatic more than 15 years past the age of onset of their affected siblings.
3.6 Neuropathological Findings
Neuropathological data was available for 13 affected individuals; all exhibited FTLD-U with NII. Two cases had additional pathologies present; Kindred 2 case II.1 had coexistent Alzheimer pathology with moderate to frequent neuritic plaques (NIA-Reagan high likelihood) and Braak stage V neurofibrillary tangle distribution (The National Institute on Aging 1997) as well as vascular pathology, and Kindred 4 case II.1 had coexistent diffuse Lewy body pathology (McKeith et al. 2005). Prominent areas of hyperintensity on FLAIR imaging were noted in two individuals who had tissue available for review. Representative photomicrographs of these regions are shown in Figure 4, demonstrating marked microglial activation throughout the white matter as well as myelin loss and gliosis.
Figure 4.
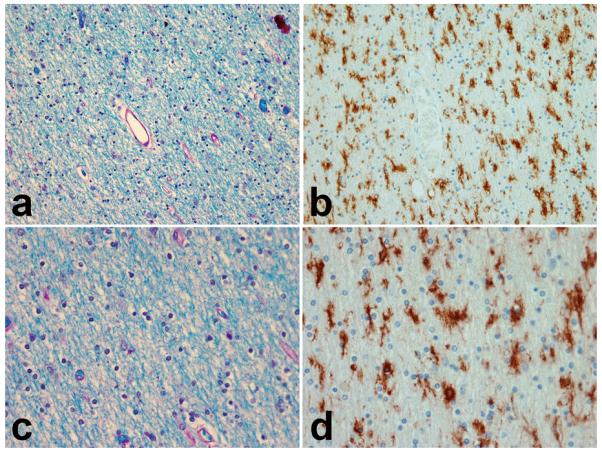
Photomicrograghs showing myelin rarefaction and vacuolation, worse around blood vessels. Microglial stain (B and D) shows marked microglial activation throughout the white matter. Adjacent Luxol fast blue stain (A and C) shows myelin loss and gliosis.
4. Discussion
4.1 Clinical Considerations
Our data suggest a wide spectrum of cognitive, behavioral, and motor features in FTDP-17 associated with mutations in PGRN. Clinical findings characteristic of FTDP-17 - cognitive impairment (executive dysfunction and/or aphasia), behavioral changes and parkinsonism – comprise the core phenotypic features, but specific clinical and radiologic features varied widely. Prominent early memory impairment –typical of amnestic MCI and Alzheimer’s disease (and an exclusionary criterion for the diagnosis of FTD) (Neary et al. 1998) – was present in several cases. Further, personality change was an early finding in only about half of the clinically evaluated patients, and was a late or absent finding in three. Levodopa-responsive parkinsonism was present in some affected individuals. A few had both visual hallucinations and parkinsonism, suggestive of Lewy body disease (McKeith et al. 2005). Some had progressive asymmetric rigidity and apraxia characteristic of the corticobasal syndrome (Boeve et al. 2003). These findings extend the range of phenotypes beyond those of previously published kindreds having PGRN mutations (Cruts et al. 2006; Huey et al. 2006; Masellis et al. 2006; Mukherjee et al. 2006; Snowden et al. 2006; Mesulam et al. 2007; Spina et al. 2007).
Phenotypic heterogeneity also exists within kindreds. To underscore this point, Kindred 2 had several members with primarily amnestic difficulties, one individual with PPA, another with FTDP, and one individual presented with amnestic complaints which quickly evolved into FTD. Phenotypic heterogeneity has been seen both within families and among families having identical mutations in MAPT as well (Bird et al. 1999).
Initial amnestic complaints occurred more frequently than would be expected in frontotemporal lobar degeneration. The proband of Kindred 5 presented with initial memory impairment suggestive of AD. Four members of Kindred 2 presented with amnestic complaints, following a course typical of AD. Another member of Kindred 2 presented with memory impairment, later following a course resembling FTD. Visuospatial deficits accompanied attention/executive impairment in three individuals clinically diagnosed with FTD. This pattern is also uncommon in FTD, but has been previously reported in a case associated with FTLD-U pathology (Meiner et al. 2005).
One sibling in Kindred 1 followed a clinical course resembling corticobasal syndrome (CBS), as did his father. His brother exhibited hemiparkinsonism late in his course, as did his sister. It has been proposed that frontotemporal lobar degeneration, CBS and progressive supranuclear palsy (PSP) represent different points of a disease spectrum on the basis of overlapping neuropathologic findings (Josephs et al. 2006b), and clinical characterization in another clinicopathologic series depended upon when in the course the subjects were evaluated (Kertesz et al. 2005). Families harboring both FTD and CBS phenotypes in association with MAPT mutations have been reported (Bugiani et al. 1999; Baba et al. 2007). Others have reported families having FTD and CBS phenotypes associated with FTLD-U pathology,(Brown et al. 1998) but this report predates the association of mutations in PGRN with FTLD-U. Recently, familial CBS was associated with the c.708+1G>A splice donor site mutation in PGRN (Masellis et al. 2006) as well as a deletion in exon 7 (Benussi et al. 2006).
Parkinsonism was the predominant phenotype in Kindred 4, and CBS-like findings were present in many members of Kindred 1. Similar clinical features were seen in two other kindreds (Benussi et al. 2006; Masellis et al. 2006). It is unclear whether parkinsonism is more predominant in association with certain PGRN mutations, analogous to the parkinsonian predominant phenotype associated with the N279K mutation in MAPT. Presumably, this reflects more prominent extrapyramidal system pathology, but how specific mutations may direct the topography of pathology is not known. Two previous PGRN case-control studies reported higher frequencies of parkinsonism than this series (Josephs et al. 2007; Whitwell et al. 2007). Those series studied autopsy-confirmed cases associated with PGRN mutations, and the lower frequency in this series may reflect that some subjects of this study are still living and may later develop parkinsonism.
The apparent hemispheric predilection in some kindreds raises similar questions. The right cerebral hemisphere was maximally affected in four members of Kindred 1, and the left hemisphere was maximally affected in our Kindred 7, as well as the PPA3 and HDDD2 kindreds (Mukherjee et al. 2006; Mesulam et al. 2007). This hemispheric predilection has not been described in kindreds with mutations in MAPT. As previously suggested (Boeve et al. 2002), some genetic factors may dictate the development of neurologic disease, while others may determine the neurodegenerative topography, analogous to prion disease (Goldfarb et al. 1992).
No case had motor neuron disease (MND), and only one report has suggested the involvement of PGRN mutations in the development of ALS (Spina et al. 2007). In that case, a FTD patient with a strong family history of ALS was found to have a PGRN missense mutation (R433W). This individual had clinical manifestations suggestive of motor neuron disease as well. Neuropathologic examination identified FTLD-U, but without NII as well as pathological changes suggestive of motor neuron disease. The relationship between this family’s ALS and the PGRN mutation is not entirely clear. These data suggest that in contrast to MAPT, MND associated with mutations in PGRN may be quite rare (Mackenzie et al. 2006a; Josephs et al. 2007).
Sparse detail was available regarding the first generations in most families, and some later-onset cases may represent incident AD cases. However, one individual with onset at age 79 had neuropathological FTLD-U with NII, and one mutation carrier is asymptomatic at age 73. Given the high penetrance, with 90% of subjects estimated to be symptomatic by age 70 (Gass et al. 2006), as well as the late age of onset in some individuals, it is possible that some late-onset cases had underlying FTLD-U with NII pathology.
In aggregate, age of onset varied, with some individuals presenting in their 50’s and others becoming symptomatic in their 80’s. This was also true within two large kindreds (Kindreds 2 and 3), although in both of these kindreds, age of onset in the earlier generation was over 10 years older than that in subsequent generations. Generations II and III of Kindred 2 were particularly well characterized, and the mean age of onset in generation III was 15 years later than that of generation II (Table 2). In comparison to familial FTD associated with mutations in MAPT, the mean age of onset in our kindreds was older (64.5 years versus 45 years) and the mean disease duration somewhat shorter (6.6 years versus 8.4 years) (Baba et al. 2005).
We did not identify any gender-specificity in phenotype, but limited data regarding previous generations was available. Progranulin’s role in gender-specific brain development (Suzuki and Nishiahara 2002) may suggest gender-based differences in presentation and/or pattern of atrophy, warranting further study.
4.2 Neuroimaging Considerations
The MRI findings in FTD linked to charged multivesicular body protein 2B (CHMP2B) on chromosome 3 (Brown et al. 2004; Skibinski et al. 2005), to the valosin-containing protein (VCP) on chromosome 9 (Vance et al. 2006) and to MAPT on chromosome 17 have been frontotemporal cortical abnormalities varying from symmetric to markedly asymmetric atrophy (Basun et al. 1997; Rosso et al. 2001; Boeve et al. 2005). Most descriptions of MRI findings in sporadic and familial FTD have not reported subcortical white matter signal changes. In FTD-MND associated with chromosome 9, MRI has demonstrated “striking frontal lobe atrophy and an absence of signal change in the white matter of both hemispheres”(Vance et al. 2006). Subtle signal changes of medial temporal regions were noted in the pallidopontal nigral degeneration (PPND) kindred (Frank et al. 2007). Four of our cases with PGRN mutations had striking increased signal in the subcortical white matter adjacent to the cortical regions where atrophy was maximal, and in two other cases with focal temporal lobe atrophy, milder degrees of subcortical signal changes were present. Few FTDP cases with PGRN mutations and MRI scans have been reported, and the MR images and data presented in this report, along with our other reports (Boeve et al. 2002; Boeve et al. 2005), provide far more detail than other reports of PGRN mutation families. The relative sensitivity and specificity of these subcortical signal changes for cases harboring a PGRN mutation remains to be determined.
A recent volumetric analysis comparing individuals having FTLD-U neuropathology suggested a more severe and widespread pattern of atrophy involving the frontal, temporal and even parietal cortex in those having PGRN mutations (Whitwell et al. 2007) compared to those without PGRN mutations. Additional analyses are warranted to further investigate the neuroimaging correlates in FTLD-U with and without associated PGRN mutations and in the tauopathies with and without associated MAPT mutations.
4.3 Neuropathologic Considerations
Many cases of “dementia lacking distinctive histology” (Knopman et al. 1990) have subsequently been shown to exhibit positive immunostaining for ubiquitin (Josephs et al. 2004; Lipton et al. 2004; Mackenzie et al. 2006c). In our series, brain tissue was available in 13 cases. Neuropathologic examination in all 13 cases demonstrated features of FTLD-U with NII. The presence of NII is a feature of FTLD-U with PGRN mutations, but is not specific for this diagnosis as NII have been observed in FTLD-U cases without PGRN mutations (Josephs et al. 2007) and in FTLD due to mutations in VCP (Forman et al. 2006). None of our cases had pathological evidence of motor neuron disease. We confirm that FTLD-U pathology can underlie the CBS, as previously reported (Grimes et al. 1999; Benussi et al. 2006; Masellis et al. 2006).
Two cases had mixed pathologies; one had FTLD-U with Alzheimer and vascular pathology, and the other FTLD-U plus Lewy body disease. A recent study suggested that anti-TDP-43 antibodies are specific to inclusions in FTLD-U (Neumann et al. 2006), which we used to confirm the diagnosis of FTLD-U in our two cases with mixed pathologies. Therefore, we suggest that TDP-43 immunohistochemistry be considered in cases with clinical features of FTD, but pathological diagnosis other than FTLD. In two cases with striking FLAIR abnormalities, myelin loss and gliosis with associated microglial activation was demonstrated in these regions.
4.4 Disease Mechanism Considerations
All PGRN mutations reported in this study are expected to create functional null alleles, predominantly as a result of reading frame shifts and the creation of premature termination codons, such that no progranulin protein is expressed. This is proposed to lead to haploinsufficiency, whereby the single normal copy of the gene is incapable of producing sufficient protein to assure normal function. We suspect this lack of progranulin causes neurodegeneration, but the precise neurodegenerative mechanisms are unknown. Transgenic mouse models will soon enable investigators to test whether replacement of progranulin, or agents that increase progranulin production or secretion, can modulate neurodegeneration.
Acknowledgments
This research is supported by National Institute on Aging grants AG06786, AG16574, AG11378 and AG07216, and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation. RR is a postdoctoral fellow from the Fund for Scientific Research Flanders (FWO-F). We thank the Mayo Clinic Alzheimer’s Disease Research Center staff for their assistance in evaluating the subjects. We particularly thank the members of these eight families for participating in neurodegenerative disease research.
Footnotes
Disclosure The authors report no potential conflicts of interest.
References
- Baba Y, Baker MC, Le Ber I, Brice A, Maeck L, Kohlhase J, Yasuda M, Stoppe G, Bugiani O, Sperfeld AD, Tsuboi Y, Uitti RJ, Farrer MJ, Ghetti B, Hutton ML, Wszolek ZK. Clinical and genetic features of families with frontotemporal dementia and parkinsonism linked to chromosome 17 with a p301s tau mutation. J Neural Transm. 2007 doi: 10.1007/s00702-007-0632-9. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Baba Y, Tsuboi Y, Baker MC, Uitti RJ, Hutton ML, Dickson DW, Farrer M, Putzke JD, Woodruff BK, Ghetti B, Murrell JR, Boeve BF, Petersen RC, Verpillat P, Brice A, Delisle MB, Rascol O, Arima K, Dysken MW, Yasuda M, Kobayashi T, Sunohara N, Komure O, Kuno S, Sperfeld AD, Stoppe G, Kohlhase J, Pickering-Brown S, Neary D, Bugiani O, Wszolek ZK. The effect of tau genotype on clinical features in ftdp-17. Parkinsonism Relat Disord. 2005;11(4):205–208. doi: 10.1016/j.parkreldis.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Basun H, Almkvist O, Axelman K, Brun A, Campbell TA, Collinge J, Forsell C, Froelich S, Wahlund LO, Wetterberg L, Lannfelt L. Clinical characteristics of a chromosome 17-linked rapidly progressive familial frontotemporal dementia. Arch Neurol. 1997;54(5):539–544. doi: 10.1001/archneur.1997.00550170021010. [DOI] [PubMed] [Google Scholar]
- Benussi L, Binetti G, Sina E, Gigola L, Bettecken T, Meitinger T, Ghidoni R. A novel deletion in progranulin gene is associated with ftdp-17 and cbs. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.10.028. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Bird TD, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, Peskind E, Lampe TH, Nemens E, Boyer PJ, Schellenberg GD. A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (p301l) Brain. 1999;122(Pt 4):741–756. doi: 10.1093/brain/122.4.741. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Baker M, Dickson DW, Parisi JE, Giannini C, Josephs KA, Hutton M, Pickering-Brown SM, Rademakers R, Tang-Wai D, Jack CR, Jr., Kantarci K, Shiung MM, Golde T, Smith GE, Geda YE, Knopman DS, Petersen RC. Frontotemporal dementia and parkinsonism associated with the IVS1+1G→A mutation in progranulin: A clinicopathologic study. Brain. 2006;129(Pt 11):3103–3114. doi: 10.1093/brain/awl268. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S15–19. doi: 10.1002/ana.10570. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Maraganore DM, Parisi JE, Ivnik RJ, Westmoreland BF, Dickson DW, Hutton M, Hardy J, Caselli RJ, Petersen RC. Corticobasal degeneration and frontotemporal dementia presentations in a kindred with nonspecific histopathology. Dement Geriatr Cogn Disord. 2002;13(2):80–90. doi: 10.1159/000048638. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Tremont-Lukats IW, Waclawik AJ, Murrell JR, Hermann B, Jack CR, Jr., Shiung MM, Smith GE, Nair AR, Lindor N, Koppikar V, Ghetti B. Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the s305n tau mutation. Brain. 2005;128(Pt 4):752–772. doi: 10.1093/brain/awh356. [DOI] [PubMed] [Google Scholar]
- Brown J, Gydesen S, Johannsen P, Gade A, Skibinski G, Chakrabarti L, Brun A, Spillantini M, Yancopoulou D, Thusgaard T, Sorensen A, Fisher E, Collinge J. Frontotemporal dementia linked to chromosome 3. Dement Geriatr Cogn Disord. 2004;17(4):274–276. doi: 10.1159/000077153. [DOI] [PubMed] [Google Scholar]
- Brown J, Lantos PL, Rossor MN. Familial dementia lacking specific pathological features presenting with clinical features of corticobasal degeneration. J Neurol Neurosurg Psychiatry. 1998;65(4):600–603. doi: 10.1136/jnnp.65.4.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B. Frontotemporal dementia and corticobasal degeneration in a family with a p301s mutation in tau. J Neuropathol Exp Neurol. 1999;58(6):667–677. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu PH, Watts GD, Markesbery WR, Smith CD, Kimonis VE. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65(6):571–581. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: A consensus conference. Conference participants. Ann Neurol. 1997;41(6):706–715. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- Frank AR, Wszolek ZK, Jack CR, Jr., Boeve BF. Distinctive MRI findings in pallidopontonigral degeneration (PPND) Neurology. 2007;68(8):620–621. doi: 10.1212/01.wnl.0000254614.39759.3d. [DOI] [PubMed] [Google Scholar]
- Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Brown SP, Graff-Radford N, Uitti R, Dickson D, Wzsolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL, 3rd, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers R. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15(20):2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: Disease phenotype determined by a DNA polymorphism. Science. 1992;258(5083):806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- Grimes DA, Bergeron CB, Lang AE. Motor neuron disease-inclusion dementia presenting as cortical-basal ganglionic degeneration. Mov Disord. 1999;14(4):674–680. doi: 10.1002/1531-8257(199907)14:4<674::aid-mds1019>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Huey ED, Grafman J, Wassermann EM, Pietrini P, Tierney MC, Ghetti B, Spina S, Baker M, Hutton M, Elder JW, Berger SL, Heflin KA, Hardy J, Momeni P. Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann Neurol. 2006;60(3):374–380. doi: 10.1002/ana.20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ingram EM, Spillantini MG. Tau gene mutations: Dissecting the pathogenesis of FTDP-17. Trends Mol Med. 2002;8(12):555–562. doi: 10.1016/s1471-4914(02)02440-1. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66(2):142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Holton JL, Rossor MN, Godbolt AK, Ozawa T, Strand K, Khan N, Al-Sarraj S, Revesz T. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol. 2004;30(4):369–373. doi: 10.1111/j.1365-2990.2003.00545.x. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Dickson DW. Clinically undetected motor neuron disease in pathologically proven frontotemporal lobar degeneration with motor neuron disease. Arch Neurol. 2006a;63(4):506–512. doi: 10.1001/archneur.63.4.506. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, Parisi JE, Dickson DW. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and psp. Neurology. 2006b;66(1):41–48. doi: 10.1212/01.wnl.0000191307.69661.c3. [DOI] [PubMed] [Google Scholar]
- Kertesz A, Kawarai T, Rogaeva E, St George-Hyslop P, Poorkaj P, Bird TD, Munoz DG. Familial frontotemporal dementia with ubiquitin-positive, tau-negative inclusions. Neurology. 2000;54(4):818–827. doi: 10.1212/wnl.54.4.818. [DOI] [PubMed] [Google Scholar]
- Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Mastri AR, Frey WH, 2nd, Sung JH, Rustan T. Dementia lacking distinctive histologic features: A common non-alzheimer degenerative dementia. Neurology. 1990;40(2):251–256. doi: 10.1212/wnl.40.2.251. [DOI] [PubMed] [Google Scholar]
- Lipton AM, White CL, 3rd, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol (Berl) 2004;108(5):379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006a;129(Pt 11):3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Baker M, West G, Woulfe J, Qadi N, Gass J, Cannon A, Adamson J, Feldman H, Lindholm C, Melquist S, Pettman R, Sadovnick AD, Dwosh E, Whiteheart SW, Hutton M, Pickering-Brown SM. A family with tau-negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain. 2006b;129(Pt 4):853–867. doi: 10.1093/brain/awh724. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Shi J, Shaw CL, Duplessis D, Neary D, Snowden JS, Mann DM. Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol (Berl) 2006c;112(5):551–559. doi: 10.1007/s00401-006-0123-3. [DOI] [PubMed] [Google Scholar]
- Masellis M, Momeni P, Meschino W, Heffner R, Jr., Elder J, Sato C, Liang Y, St George-Hyslop P, Hardy J, Bilbao J, Black S, Rogaeva E. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129(Pt 11):3115–3123. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: Report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58(11):1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- Meiner Z, Newman JP, Rosenman H, Soffer D, Steiner I. Frontotemporal dementia with ubiquitinated neuronal inclusions and visuospatial impairment. Neurology. 2005;65(3):478–480. doi: 10.1212/01.wnl.0000171347.28558.56. [DOI] [PubMed] [Google Scholar]
- Members of the Department of Neurology, M. C. Clinical examinations in neurology. Mosby; St. Louis: 1998. [Google Scholar]
- Mesulam M, Johnson N, Krefft TA, Gass JM, Cannon AD, Adamson JL, Bigio EH, Weintraub S, Dickson DW, Hutton ML, Graff-Radford NR. Progranulin mutations in primary progressive aphasia: The ppa1 and ppa3 families. Arch Neurol. 2007;64(1):43–47. doi: 10.1001/archneur.64.1.43. [DOI] [PubMed] [Google Scholar]
- Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S, Behrens MI, Budde J, Hinrichs AL, Norton J, Levitch D, Taylor-Reinwald L, Gitcho M, Tu PH, Grinberg L. Tenenholz, Liscic RM, Armendariz J, Morris JC, Goate AM. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60(3):314–322. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Rademakers R, Cruts M, Dermaut B, Sleegers K, Rosso SM, Van den Broeck M, Backhovens H, van Swieten J, van Duijn CM, Van Broeckhoven C. Tau negative frontal lobe dementia at 17q21: Significant finemapping of the candidate region to a 4. Mol Psychiatry. 2002;7(10):1064–1074. doi: 10.1038/sj.mp.4001198. [DOI] [PubMed] [Google Scholar]
- Rosso SM, Kamphorst W, de Graaf B, Willemsen R, Ravid R, Niermeijer MF, Spillantini MG, Heutink P, van Swieten JC. Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q21-22. Brain. 2001;124(Pt 10):1948–1957. doi: 10.1093/brain/124.10.1948. [DOI] [PubMed] [Google Scholar]
- Savioz A, Riederer BM, Heutink P, Rizzu P, Tolnay M, Kovari E, Probst A, Riederer IM, Bouras C, Leuba G. Tau and neurofilaments in a family with frontotemporal dementia unlinked to chromosome 17q21-22. Neurobiol Dis. 2003;12(1):46–55. doi: 10.1016/s0969-9961(02)00011-6. [DOI] [PubMed] [Google Scholar]
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J. Mutations in the endosomal escrtiii-complex subunit CHMP2b in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AM, Varma A, Neary D, Mann DM. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain. 2006;129(Pt 11):3091–3102. doi: 10.1093/brain/awl267. [DOI] [PubMed] [Google Scholar]
- Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Baraibar MA, Barbeito AG, Troncoso JC, Vidal R, Ghetti B, Grafman J. Clinicopathologic features of frontotemporal dementia with progranulin sequence variation. Neurology. 2007;68(11):820–827. doi: 10.1212/01.wnl.0000254460.31273.2d. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Nishiahara M. Granulin precursor gene: A sex steroid-inducible gene involved in sexual differentiation of the rat brain. Mol Genet Metab. 2002;75(1):31–37. doi: 10.1006/mgme.2001.3274. [DOI] [PubMed] [Google Scholar]
- The National Institute on Aging. Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18(4 Suppl):S1–S2. [PubMed] [Google Scholar]
- van der Zee J, Rademakers R, Engelborghs S, Gijselinck I, Bogaerts V, Vandenberghe R, Santens P, Caekebeke J, De Pooter T, Peeters K, Lubke U, Van den Broeck M, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C, Dermaut B. A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain. 2006;129(Pt 4):841–852. doi: 10.1093/brain/awl029. [DOI] [PubMed] [Google Scholar]
- Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, Shaw CE. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129(Pt 4):868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Jr., Baker M, Rademakers R, Adamson J, Boeve BF, Knopman DS, Parisi JF, Petersen RC, Dickson DW, Hutton ML, Josephs KA. Voxel-based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch Neurol. 2007;64(3):371–376. doi: 10.1001/archneur.64.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]