

Abstract
p21 inhibits cyclin-dependent kinase (CDK) activity and proliferating cell nuclear antigen (PCNA)-dependent DNA replication by binding to CDK/cyclin complexes and to PCNA through distinct domains. The human papillomavirus (HPV)-16 E7 oncoprotein (16E7) abrogated a DNA damage-induced cell cycle arrest in vivo, despite high levels of p21. Using cell lysates and purified proteins we show that 16E7 prevented p21 both from inhibiting CDK2/cyclin E activity and PCNA-dependent DNA replication, whereas the nononcogenic HPV-6 E7 had reduced effects. Inactivation of both inhibitory functions of p21 was attained through binding between 16E7 and sequences in the carboxy-terminal end of p21 that overlap with the PCNA-binding site and the second p21 cyclin-binding motif. These data imply that the carboxyl terminus of p21 simultaneously modulates both CDK activity and PCNA-dependent DNA replication and that a single protein, 16E7, can override this modulation to disrupt normal cell cycle control.
Keywords: CDK inhibitors, p21CIP1/WAF1/SDI1, DNA replication, PCNA, cell cycle arrest, HPV-16 E7, HPV-6 E7
Regulation of cyclin-dependent kinase (CDK) activities occurs at several levels, including binding of CDK inhibitors to the CDK/cyclin complex (for review, see Morgan 1995). CDK inhibitors are involved in cell cycle arrest in response to various antimitogenic signals (for review, see Sherr and Roberts 1995). One member of the CIP/KIP family of CDK inhibitors, p21CIP1/WAF1/SDI1 (El-Deiry et al. 1993; Gu et al. 1993; Harper et al. 1993; Xiong et al. 1993a; Noda et al. 1994), is a transcriptional target of p53 and is a critical determinant of the G1 arrest in response to DNA damage (Dulic et al. 1994; Brugarolas et al. 1995; Deng et al. 1995; Waldman et al. 1995). However, p21 also responds to other signals independently of p53 and has been implicated in terminal differentiation and senescence (Noda et al. 1994; Halevy et al. 1995; Macleod et al. 1995; Parker et al. 1995). Under some conditions, p21 can function as an adaptor protein in the assembly of CDK/cyclin complexes (Xiong et al. 1993b; Harper et al. 1995; LaBaer et al. 1997).
p21 is an unusual CDK inhibitor; in addition to the well-defined amino-terminal cyclin and CDK-binding motifs (Chen et al. 1995; Luo et al. 1995; Nakanishi et al. 1995) a second cyclin binding site is located near the carboxyl terminus (Adams et al. 1996; Ball et al. 1996; Chen et al. 1996), partially overlapping the domain that binds to the proliferating cell nuclear antigen (PCNA; Warbrick et al. 1995; Gulbis et al. 1996). PCNA, an auxiliary factor for DNA polymerase δ and ε, facilitates loading of the polymerases onto DNA templates and increases their activity in both DNA replication and repair. Through an interaction with PCNA, p21 inhibits PCNA-dependent DNA replication (long-range DNA synthesis; Flores-Rozas et al. 1994; Waga et al. 1994b), whereas PCNA-dependent nucleotide excision–repair (short-patch DNA synthesis) is not inhibited (Li et al. 1994, 1996; Shivji et al. 1994). Thus, p21 may mediate the coordination between cell cycle progression, DNA replication, and DNA repair (Li et al. 1994; Waga et al. 1994b), but the exact contribution of each of the inhibitory activities of p21 to cell cycle arrest in vivo is unclear.
The E7 oncoprotein of the cancer-associated human papillomaviruses (HPVs) such as HPV-16 (16E7) has been shown to overcome a DNA damage-induced G1 cell cycle arrest, whereas E7 from the generally benign HPV types like HPV-6 (6E7) has not (Demers et al. 1994; Slebos et al. 1994). The mechanisms involved in the abrogation of G1 cell cycle arrest by 16E7 are incompletely understood, and subsequent analysis was mostly focused on the inactivation of the retinoblastoma protein (RB) by 16E7. Binding of 16E7 to RB obviates the requirement for cyclin D-associated kinase activity to phosphorylate RB and leads to the release of E2F transcription factors that activate S-phase genes (DeGregori et al. 1995), including cyclin E (Zerfass et al. 1995), which is necessary for the G1-to-S-phase transition (Ohtsubo et al. 1995; for review, see Reed 1996); cyclin A, which is required during S phase; and PCNA. Elevation of these cell cycle proteins may contribute to bypassing G1 arrest signals. Additionally, 16E7 expression results in reduced amounts of RB protein (Demers et al. 1994; Boyer et al. 1996; Jones and Münger 1997), though the mechanism is unclear. Nevertheless, studies using mutated E7 or adenovirus E1A proteins have pointed to additional activities of the viral oncoproteins, unrelated to RB binding, that are involved in bypassing growth arrest signals (Abraham et al. 1992; Missero et al. 1995; Demers et al. 1996).
In this study we demonstrate that 16E7 blocks the ability of p21 to inhibit CDK activity as well as PCNA-dependent DNA replication through direct binding to the carboxyl terminus of p21. We propose that interactions with the carboxyl terminus of p21 modulate both its CDK- and PCNA-inhibitory functions.
Results
Effects of 16E7 on CDK activity and PCNA-dependent DNA replication
Human keratinocytes were infected with a retroviral vector expressing 16E7, treated with actinomycin D for 24 hr to induce DNA damage, and extracts were prepared. Cell cycle progression was monitored by flow cytometry. Our previous studies (Demers et al. 1994) showed that vector-infected cells arrested with an increased G1 population and depletion of S-phase cells to 3%–15% of the number of S-phase cells in the untreated population, whereas 16E7-expressing cells consistently retained 60%–90% of their S-phase population after DNA damage. Figure 1A demonstrates that DNA damage resulted in induction of p53 and p21 in both populations of cells; however, 16E7-expressing cells remained in the cell cycle despite induction of p21. Cyclin E-associated CDK activity was elevated in untreated 16E7-expressing cells compared to vector-infected cells, and notably actinomycin D-treated, 16E7-expressing cells retained 50% kinase activity of untreated cells (Fig. 1A). These results indicated that 16E7-expressing cells were able to abrogate the CDK-inhibitory function of p21.
Figure 1.
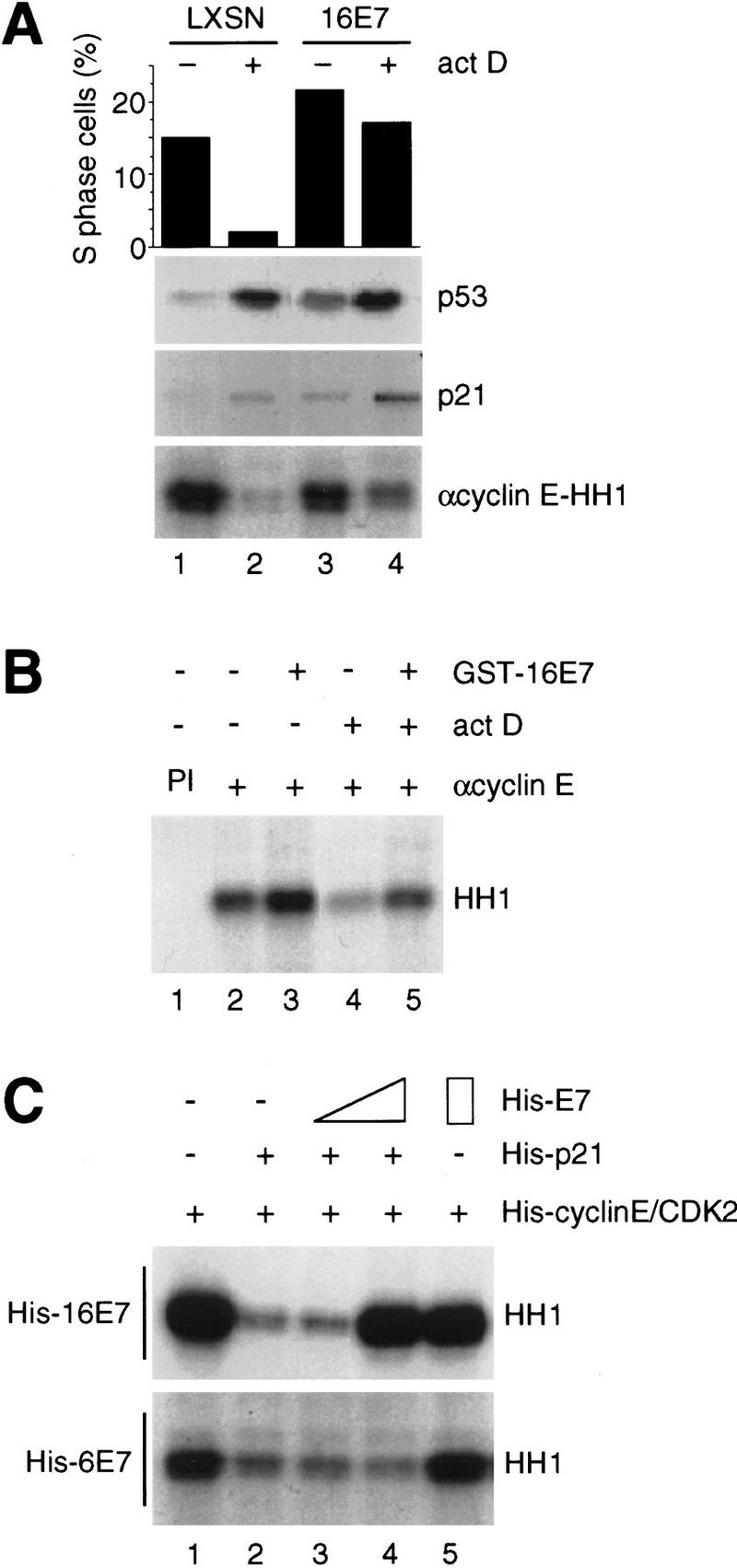
Effect of HPV-16 E7 and HPV-6 E7 on inhibition of CDK activity by p21 in vivo and in vitro. (A) Human keratinocytes were infected with retroviral constructs alone (LXSN) or expressing 16E7 and were treated with actinomycin D (act D) for 24 hr. Cells were harvested, and a fraction was used for cell cycle analysis by flow cytometry to determine the fraction of S-phase cells (top); the remainder was used to prepare extracts for immunoblots with anti-p53 or anti-p21 antibodies (middle) and in vitro kinase assays after anti-cyclin E immunoprecipitations (bottom). (B) Extracts of untreated or actinomycin D-treated (act D) human keratinocytes (200 μg) were coincubated with GST–16E7 (850 ng); then cyclin E was immunoprecipitated [lanes 2–5; (PI) preimmune serum in lane 1] and kinase assays were performed for 20 min at 30°C using histone H1 (HH1) as a substrate. (C) Purified recombinant His–cyclin E/CDK2 complexes, produced in baculovirus-infected cells, were coincubated with His–p21 (10 ng) alone or with His–p21 preincubated with increasing amounts (20 and 200 ng) of His–16E7 (top) or His–6E7 (bottom), and kinase assays were performed.
The mechanism by which 16E7 bypassed p21 inhibition of CDK activity in in vivo systems was not clear and might involve the ability of 16E7 to release E2F and thus elevate cyclin E levels (Zerfass et al. 1995) to titrate the increase in p21, or to reduce levels of RB protein (Demers et al. 1994; Jones and Münger 1997). However, none of these explanations was fully consistent with the observation that some mutants that bind RB remained unable to inactivate p21 in vivo (Demers et al. 1996). The overexpression of CDK2/cyclin E only resulted in 60% of the activity of 16E7 in bypassing a p21-mediated cell cycle arrest (S.A. Foster and D.A. Galloway, unpubl.). Taken together, these data suggested that 16E7 used an additional pathway to inactivate p21.
To determine whether 16E7 could directly affect p21-inhibited CDK complexes, extracts of untreated or actinomycin D-treated keratinocytes were mixed with glutathione S-transferase (GST)–16E7 fusion proteins in vitro. Protein 16E7 effectively restored the inhibited (15% of control; Fig. 1B, lane 4) CDK2/cyclin E activity (90% of control; Fig. 1B, lane 5) and also elevated CDK2 activity slightly in extracts of untreated cells (120% of control; Fig. 1B, lane 3). To analyze whether the reversal of p21 inhibition of CDK activity by 16E7 directly involved the CDK/cyclin/p21 complex or required other 16E7-associated proteins such as p107, purified recombinant, histidine-tagged (His)–cyclin E/CDK2 was inhibited by His–p21 (Fig. 1C, lanes 1,2). Addition of purified His–16E7 restored the p21-inhibited CDK2 activity in a dose-dependent manner, whereas His-6E7 had greatly reduced effects (Fig. 1C).
Reversal of the effect of p21 on CDK activity by 16E7 may contribute to HPV DNA replication by promoting the G1-to-S-phase transition. However, viral DNA replication requires PCNA, and p21 also blocks PCNA-dependent DNA replication. Therefore, the effect of 16E7 on the p21-mediated inhibition of PCNA-dependent DNA replication was examined in vitro. His–16E7 effectively abrogated the inhibition by p21 of PCNA-dependent DNA synthesis (Fig. 2A) and also SV40 DNA replication with crude cell extracts (Fig. 2B) in a dose-dependent manner, whereas His–6E7 had only marginal effects. The effects of the His–E7 proteins on SV40 replication were also analyzed quantitatively [deoxy AMP (dAMP) incorporation] and paralleled the results from the purified replication system (data not shown). The amounts of E7 proteins used were based on this quantitation. Proteins 16E7 and 6E7 did not affect SV40 DNA replication in the absence of p21 (data not shown). Importantly, the process of SV40 DNA replication was fully recovered by 16E7, as completely replicated products (discrete bands of topoisomers) and late replication intermediates (smears above the 4.4-kb marker; Prelich and Stillman 1988; Waga et al. 1994b) were generated (Fig. 2B). In this assay, 6E7 had a detectable, dose-dependent effect on the generation of replication products, but it was greatly reduced compared to the activity of 16E7.
Figure 2.
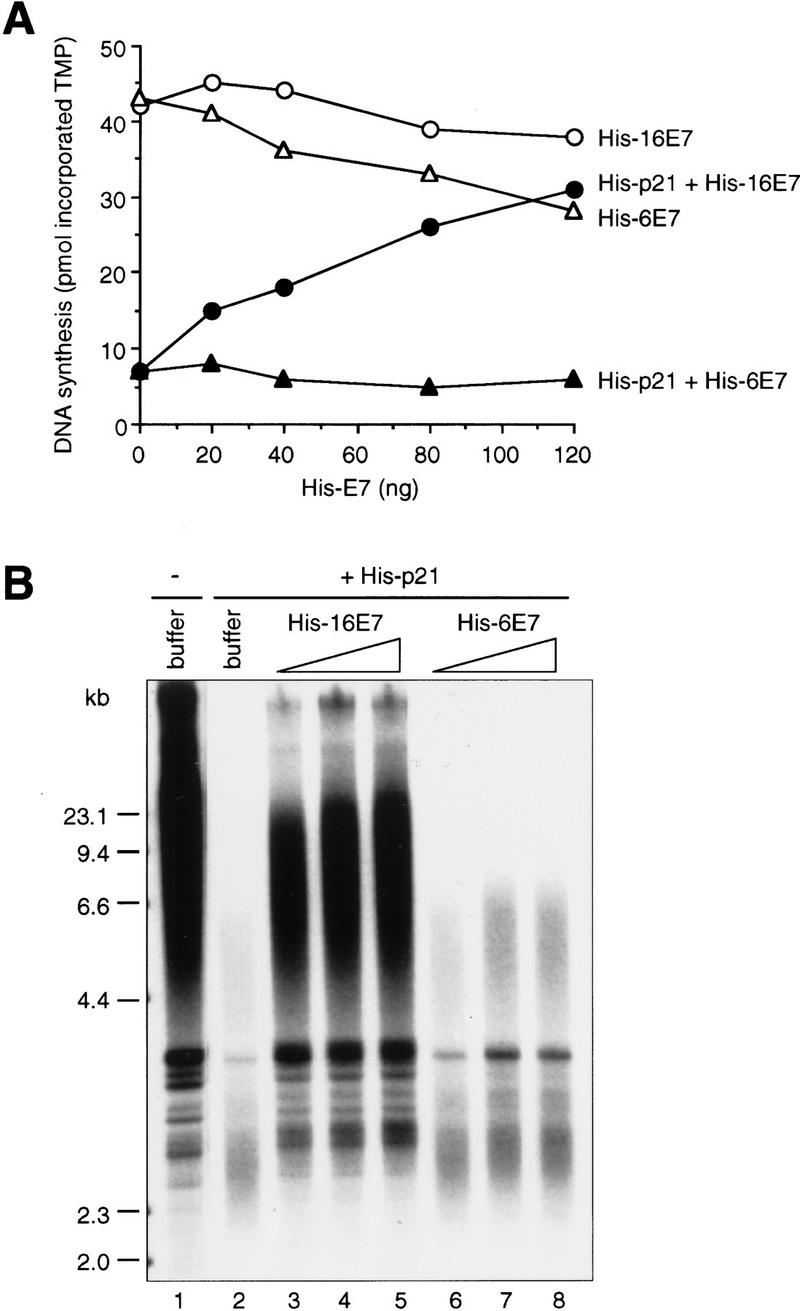
Effect of HPV-16 E7 and HPV-6 E7 on inhibition of PCNA-dependent DNA replication by p21 in vitro. (A) Increasing amounts of His–16E7 or His–6E7 were preincubated with (50 ng; closed symbols) or without (open symbols) His–p21 for 60 min at room temperature, then mixed with PCNA (10 ng) and DNA polymerase δ (∼15 ng) and further incubated for 30 min on ice. DNA synthesis was started by adding [α32P]TTP and oligo(dT)/poly(dA) at 37°C. The amount of DNA synthesis is expressed as picomoles of TMP incorporated in a 25-μl reaction in 30 min. (B) Increasing amounts (250 ng, 500 ng, 1 μg) of His–16E7 or His–6E7 were preincubated with or without His–p21 (330 ng) for 60 min at room temperature, then mixed with a 293 human cell extract (19.5 μg) and further incubated for 30 min on ice. DNA replication was started by adding purified T antigen, an SV40 ori-containing plasmid pSVO11 (Prelich and Stillman 1988), and nucleotides including [α32P]dATP and incubated at 37°C. Analysis by neutral agarose gel electrophoresis of purified replication products is shown. Lane 1 shows SV40 DNA replication in the absence of p21 or E7.
Interaction between p21 and 16E7
The ability of 16E7 to block p21-mediated inhibition of CDK activity and DNA replication in purified systems suggested that 16E7 might interact directly with p21. This possibility was tested in binding assays with purified or in vitro-translated and -labeled (IVT) proteins. GST–16E7 bound His–p21 (Fig. 3A), IVT p21 (Figs. 4A and 5A), IVT p27KIP1, and IVT p57KIP2 (data not shown), but not His–CDK2 or His–cyclin E (Fig. 3A), or IVT p16INK4a (data not shown). Reciprocally, GST–p21 bound His–16E7 (Fig. 5B) and IVT 16E7 but ∼10-fold less well to IVT 6E7 (Fig. 3B). Under these conditions, the amount of p21 bound to 16E7 was usually <10% of the input, but binding was seen even in the presence of 1% NP-40 (data not shown). To examine whether an interaction between 16E7 and p21 occurred in vivo, actinomycin D-treated, 16E7-expressing keratinocytes were metabolically labeled, and extracts were prepared. After a first immunoprecipitation with anti-16E7 or anti-p21 antibodies under low-stringency conditions (data not shown) and elution of the nonimmunoreactive fraction under high-stringency conditions (Fig. 3C, lanes 7,8), the associated proteins were reimmunoprecipitated with a second antibody (anti-p21 and anti-16E7, respectively; Fig. 3C, lanes 9,10). In both cases, p21 and 16E7 were detected following reimmunoprecipitation. This experiment showed that 16E7 and p21 were weakly associated with each other in vivo. Such an interaction was not seen in vector-infected cells nor with an irrelevant antibody (Fig. 3C, lanes 1–6).
Figure 3.

Analysis of the HPV-16 E7–p21 interaction in vitro and in vivo. (A) In vitro binding reactions with GST–16E7 or GST (100, 200, 400 ng) immobilized on glutathione–Sepharose and His–p21, His–cyclin E, or His–CDK2 (200 ng) were performed, and the resulting protein complexes were visualized by Coomassie staining (top) and Western blotting (bottom) with anti-His antibodies. Lane 1 represents 10% of the input. (B) In vitro binding reactions with GST–p21 or GST (1 μg) immobilized on glutathione–Sepharose and IVT 16E7 or IVT 6E7 were performed, and the resulting protein complexes were visualized by Coomassie staining (top) and fluorography (bottom). Lane 1 represents 10% of the input. (C) Actinomycin D-treated, 16E7-expressing keratinocytes were metabolically labeled with [35S]Protein Labeling Mix, and extracts were prepared. After a first immunoprecipitation (IP) of extracts (900 μg; anti-16E7 or anti-p21 antibody) under low-stringency conditions (data not shown), the associated proteins were eluted under high-stringency conditions (IP shown after the elution; lanes 7,8) and reimmunoprecipitated with a second antibody (anti-p21 and anti-16E7, respectively; lanes 9,10). Vector (LXSN)-infected cells (lanes 1–4) or an irrelevant antibody (anti-16L1; lanes 5,6) were used as negative controls. Four percent of the first IP after the high-stringency elution and 100% of the second IP are shown.
Figure 4.
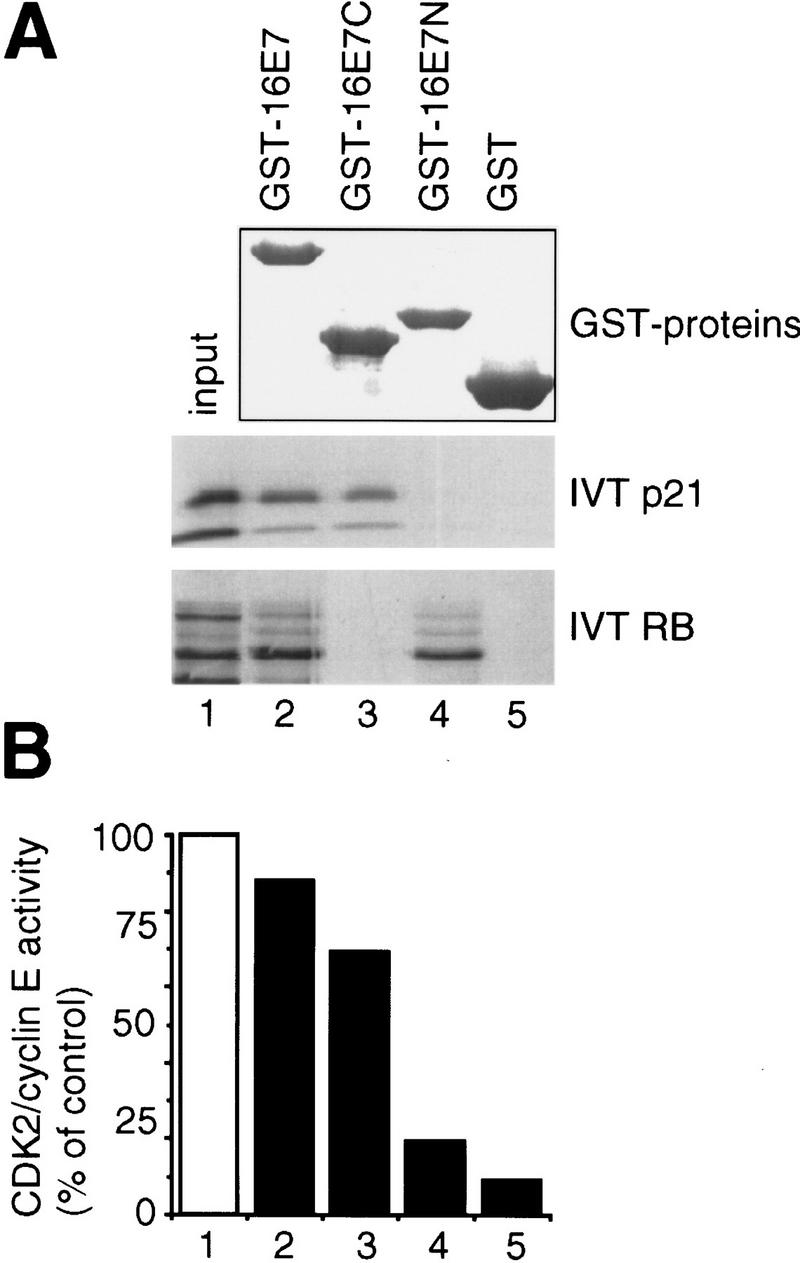
Analysis of binding sites in HPV-16 E7 proteins. (A) In vitro binding reactions with GST–16E7 mutants (1.5 μg) immobilized on glutathione–Sepharose and IVT p21 or IVT RB were performed, and the resulting protein complexes were visualized by Coomassie staining (top) and fluorography (bottom). Lane 1 represents 10% (IVT p21) or 20% (IVT RB) of the input. (B) Purified recombinant His–cyclin E/CDK2 complexes were coincubated with buffer (□) or coincubated with His–p21 (10 ng, ▪) alone or with His–p21 preincubated with GST fusion proteins (300 ng; as indicated in A, lanes 2–5), and kinase assays were performed. The quantitation from PhorphorImager analysis is shown.
Figure 5.



Analysis of binding sites in p21 proteins. (A) In vitro binding reactions with GST–16E7 or GST (1 μg) immobilized on glutathione–Sepharose and IVT p21 mutant proteins were performed, and the resulting protein complexes were visualized by fluorography. The mutants used were p21(full-length, 1–164) (lanes 1,2,7,10,11,16), p21(1–90) (lanes 3,4,8), p21(87–164) (lanes 5,6,9), p21(1–148) (lanes 12,13,17), and p21(1–158) (lanes 14,15,18). Ten percent of the input is shown. (B) In vitro binding reactions with GST–p21 mutants (100 ng) immobilized on glutathione–Sepharose and His–16E7 (200 ng) or PCNA (40 ng) were performed, and the resulting protein complexes were visualized by Western blotting with anti-16E7 (top) and anti-PCNA (bottom) antibodies. Lane 1 represents 20% of the input. (C) Schematic representation of p21 and summary of in vitro binding analyses with p21 mutants using purified and IVT proteins. The binding motifs for cyclin (cyclin 1), CDK, and PCNA are shown as solid boxes, the second cyclin binding motif (cyclin 2) overlapping the PCNA binding motif is shown as a shaded box.
Amino- and carboxy-terminal fragments of 16E7 were used to begin to map the p21-interacting domain. The amino terminus of 16E7 contains motifs for RB binding and phosphorylation by casein kinase II and shares homology with the adenovirus E1A-conserved regions I and II and an analogous region in the SV40 large T antigen; the carboxyl terminus has a zinc finger motif (for review, see zur Hausen 1996). Although it does not contain the high-affinity RB-binding motif, 16E7C(Δ1–39) retained the ability to bind to p21, though somewhat reduced in comparison to intact 16E7, whereas 16E7N(Δ40–98) showed no detectable binding (Fig. 4A). Consistent with this finding, 16E7C at least partially restored kinase activity using p21-inhibited purified CDK2/cyclin E complexes (Fig. 4B) and extracts of actinomycin D-treated cells (data not shown). Although these results demonstrate that the ability of 16E7 to bind to p21 resides primarily in the carboxyl terminus, the data may point to an additional role for sequences in the amino terminus. An analysis of p21 binding to various 16E7 proteins carrying deletions or point mutations has so far been inconclusive (data not shown).
Next, mutations in p21 were used to define the region required for 16E7 binding (summarized in Fig. 5C). The carboxy-terminal half of p21 [p21C(87–164)] (Fig. 5A, lanes 5,6), but not the amino-terminal half [p21N(1–90)] (Fig. 5A, lanes 3,4), bound to 16E7. In accordance with this result, mutants defective in the cyclin 1 motif within the amino-terminal half of p21 (Lin et al. 1996) or in the nearby CDK motif (Nakanishi et al. 1995) bound efficiently to 16E7 (data not shown). Several carboxy-terminal mutations in p21 were then tested. Deletion of 6 amino acids [p21(1–158)] did not appreciably reduce 16E7 binding (Fig. 5A, lanes 14,15), but a carboxy-terminal deletion of 16 amino acids [p21(1–148)] eliminated the binding (Fig. 5A, lanes 12,13). Furthermore, p21(139–164) bound to 16E7 (Fig. 5B). Therefore, the region of p21 sufficient for the interaction with 16E7 resides in amino acids 139–158, which interestingly overlaps the PCNA-binding region (Gulbis et al. 1996) and the second cyclin-binding region (Fig. 5C; Adams et al. 1996; Ball et al. 1996; Chen et al. 1996).
Role of the carboxyl terminus of p21 in its inactivation by 16E7
Previous studies have shown that the CDK-inhibitory function of p21 resides mainly in the amino terminus, yet our results indicated that the ability of 16E7 to reverse p21 inhibiton of CDK activity occurred through its interaction with the carboxyl terminus of p21. To test this assumption GST–p21, GST–p21N, and GST–p21C proteins were used to inhibit CDK2/cyclin E complexes. 16E7 and, to a much lesser extent 6E7, reconstituted CDK2 activity in the presence of full-length p21, but not in the presence of p21N, showing that the carboxyl terminus of p21 was required for the 16E7-dependent reversal of CDK inhibition (Fig. 6A, left). p21C did not inhibit CDK2 activity at low concentrations, which were sufficient for inhibition by p21 or p21N (Fig. 6A, left); however, at ∼150-fold higher concentrations, p21(139–164) containing the cyclin 2 site also showed CDK-inhibitory activity (Adams et al. 1996; Ball et al. 1996), and this inhibition was overcome by 16E7 (Fig. 6A, right). Furthermore, p21(139–164), which contains the full PCNA-binding site (Fig. 5B; Warbrick et al. 1995), inhibited PCNA-dependent DNA synthesis and 16E7 abrogated this effect (Fig. 6B).
Figure 6.
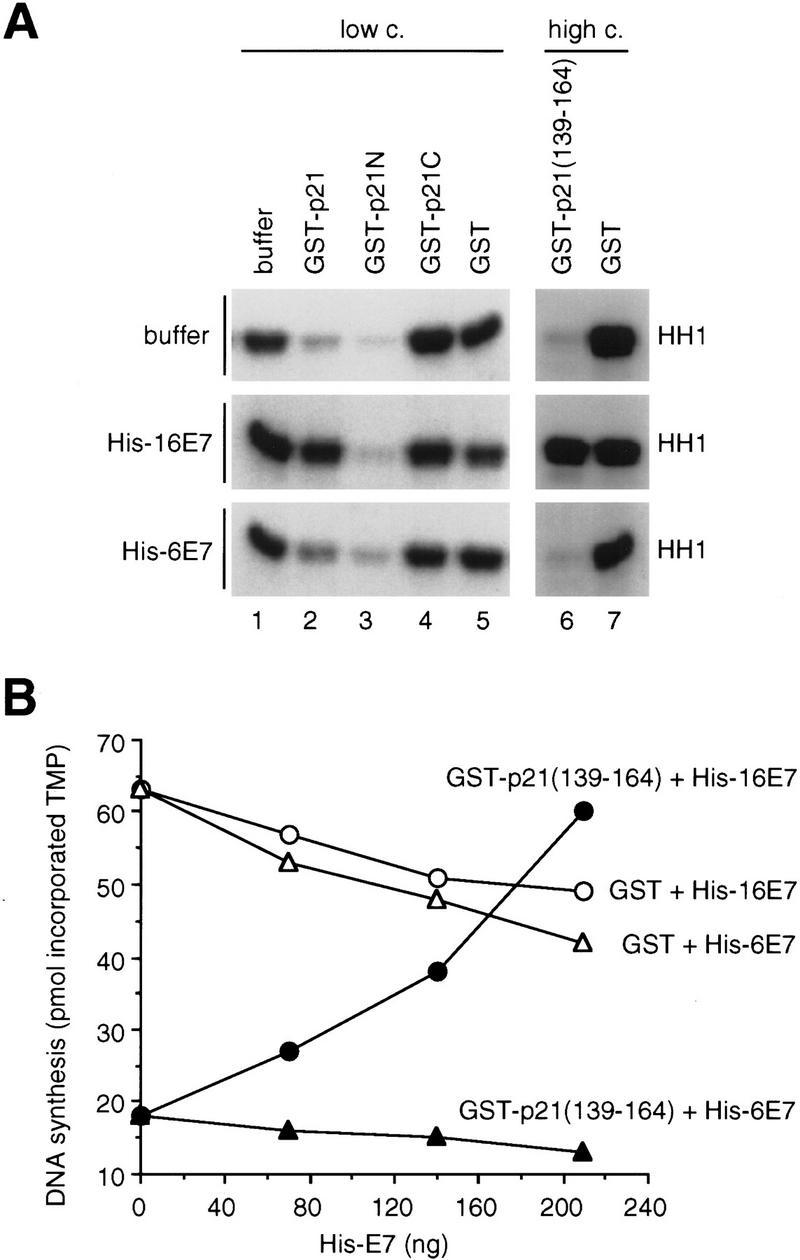
Analysis of the function of the carboxy-terminal domain of p21 in the inactivation by HPV-16 E7. (A) Purified recombinant His–cyclin E/CDK2 complexes were coincubated with buffer (lane 1) or GST proteins at low or high concentration preincubated with buffer (top), His–16E7 (middle), or His–6E7 (bottom). Twenty nanograms of GST or GST–p21 (lanes 2–5), or 2.5 μg of GST or GST–p21(139–164) (lanes 6,7) were used, which had been preincubated with 400 ng (lanes 2–5) or 6 μg (lanes 6,7) of His–16E7 or His–6E7, and kinase assays were performed for 20 min at 30°C. (B) Increasing amounts of His–16E7 or His–6E7 were preincubated with GST or GST–p21(139–164) (120 ng). The subsequent DNA synthesis reaction was carried out as described in Fig. 2A.
Finally, to begin to understand the mechanism by which 16E7 blocked the p21 inhibition of CDK activity, the levels of p21 in anti-CDK2 immunoprecipitates of reconstituted reactions were assessed. Interestingly, no change in the level of p21 was seen (Fig. 7A), suggesting that 16E7 did not appreciably displace p21 from the CDK2/cyclin E complexes. 16E7 was found to be associated with CDK2/cyclin E/p21 complexes, as demonstrated by immunoblotting of anti-cyclin E immunoprecipitates (Fig. 7A). Likewise, 16E7 was detectable in anti-p21 immunoprecipitates of CDK/cyclin complexes and vice versa (data not shown). Next, the effect of 16E7 on PCNA/p21 complex formation in vitro was examined. By adding increasing amounts of 16E7 the levels of PCNA bound to p21 were effectively reduced (Fig. 7B), suggesting that 16E7 and PCNA were competing for p21 binding.
Figure 7.
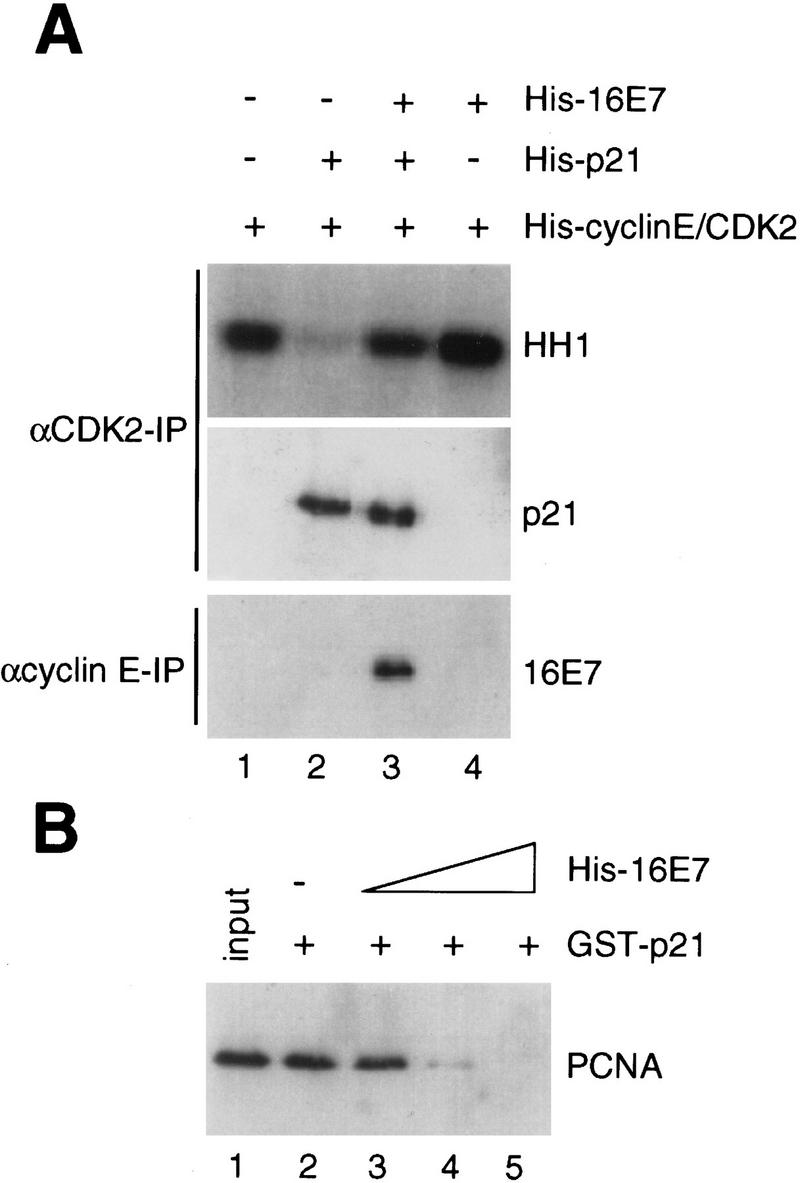
Analysis of protein complexes in the absence and presence of HPV-16 E7. (A) Purified recombinant His–cyclin E/CDK2 complexes were coincubated with His–16E7 and/or His–p21 as described in Fig. 1C, and after an immunoprecipitation (IP) with anti-CDK2 antibodies kinase assays were performed (top). One-half of the anti-CDK2 immunoprecipitate was analyzed by immunoblotting with anti-p21 antibodies (middle). In parallel, an immunoprecipitate with anti-cyclin E antibodies was analyzed by immunoblotting with anti-16E7 antibodies (bottom). (B) Recombinant PCNA (50 ng) was coincubated with GST–p21 (100 ng), immobilized on glutathione–Sepharose alone or in the presence of increasing amounts (200, 400, 800 ng) of His–16E7, and the PCNA bound to GST–p21 beads was analyzed by immunoblotting with anti-PCNA antibodies. Lane 1 represents 10% of the input.
Discussion
These experiments show that the inhibitory functions of p21 on both CDK activity and PCNA-dependent DNA replication can be blocked by 16E7 through mechanisms involving a direct interaction between 16E7 and the carboxyl terminus of p21. Mutagenesis has defined spatially conserved CDK and cyclin-binding motifs in the amino-terminal halves of p21, p27, and p57 (Chen et al. 1995; Luo et al. 1995; Nakanishi et al. 1995), and the crystallographic structure of CDK2/cyclin A/p27N (Russo et al. 1996) has indicated how these motifs interact with CDK/cyclin complexes. However, at odds with the structure is the biochemical evidence that CDK/cyclin complexes containing a single molecule of p21 are catalytically active (Zhang et al. 1994). Unlike other CIP/KIP family members, p21 contains a second cyclin binding motif (Cy2) in the carboxyl terminus, whose core sequence RRLIF is related to the RRLFG motif in the first cyclin-binding site (Cy1; Adams et al. 1996; Ball et al. 1996; Chen et al. 1996). The significance of the Cy2 motif in inhibiting CDK activity is less well understood. The p21 Cy2/cyclin interaction was very weak for cyclins E and A and only detectable indirectly by competition (Chen et al. 1996). Peptides overlapping with Cy2 bound to both CDK4 and cyclin D1, inhibited CDK4/cyclin D1 and CDK2/cyclin E activity in vitro, and caused a G1 arrest in vivo (Ball et al. 1996). Additionally, removal of the amino-terminal 34 amino acids of the Xenopus homolog of p21, p27Xic1, resulted in a protein that was impaired in its ability to inhibit cyclin A-, but not cyclin E-associated CDK activity (Su et al. 1995).
Also unique among the CIP/KIP inhibitors is the ability of p21 to bind PCNA through carboxy-terminal sequences that overlap Cy2. In proliferating normal diploid human cells, the p21 bound to active CDK/cyclin complexes is also bound to PCNA (Zhang et al. 1994). Our results raise the interesting possibility that binding of proteins to the carboxyl terminus of p21, perhaps specifically blocking the Cy2 site, may modulate the activity of p21 on CDK/cyclin complexes. In normal proliferating cells, the Cy2 site is occupied by PCNA; even normal proliferating fibroblasts expressing 16E7 showed no detectable disruption of CDK/cyclin/p21/PCNA complexes (Xiong et al. 1996), consistent with our observations that PCNA bound p21 more avidly than 16E7 (data not shown). In response to DNA damage or epithelial cell differentiation, the levels of p21 may exceed those of PCNA, resulting in inactive CDK/cyclin complexes in normal cells. However, in 16E7-expressing cells, binding of 16E7 to the carboxyl terminus of p21 may promote the accumulation of active CDK/cyclin/p21 complexes, possibly also by enhancing the association between CDK and cyclin subunits (Harper et al. 1995; LaBaer et al. 1997). This model predicts that the 16E7–p21 interaction would be important to overcome G1 arrest and may not normally occur in proliferative conditions. Interestingly, p21 with a single amino acid change at position 94, isolated from a human breast carcinoma, was found to have impaired CDK-inhibitory activity despite unaltered binding to CDK complexes and PCNA (Balbín et al. 1996), suggesting also that alterations in the carboxyl terminus of p21 can modulate CDK activity. It has been shown, for example, that free p21 lacks any stable tertiary structure, but upon binding to a CDK/cyclin complex, p21 adopts an ordered structure (Kriwacki et al. 1996). Thus, it is possible that the 16E7–p21 interaction could result in a structural alteration of p21 that leads to reactivation of the CDK/cyclin complex.
16E7 also impeded the ability of p21 to inhibit PCNA-dependent DNA replication, probably by a competitive mechanism. The structure of the p21–PCNA complex (Gulbis et al. 1996) provided evidence for the possibility that p21 masked elements on PCNA that are required for binding of components of the polymerase assembly, whereas the loading of PCNA onto DNA and the ability of PCNA to move along DNA appeared not to be affected by p21 (Podust et al. 1995). The removal of p21 from PCNA by 16E7 may explain the block of p21 inhibition of PCNA-dependent DNA replication, thereby allowing PCNA to function in viral DNA replication.
In contrast to the effect of 16E7, the nononcogenic 6E7 had a reduced ability to relieve p21 inhibition in vitro, and 6E7 did not overcome a p21-mediated cell cycle arrest in vivo (Demers et al. 1994). In agreement with these data, the organization of 6E7-expressing keratinocytes in organotypic raft cultures was not markedly different from normal epithelium, and only a slight increase in proliferation was observed (Halbert et al. 1992). However, benign warts caused by HPV-6 infection are hyperproliferative, suggesting that 6E7 can block suprabasal quiescence. It is possible that the ability of 6E7 to allow PCNA-dependent DNA replication might be sufficient for a slow increase in proliferation yet too weak to be readily detected in the p21 inhibition assays used in our studies. Because of the difficulty in identifying assays in which 6E7 is equivalent in activity to 16E7, we cannot rule out the possibility that our preparations of 6E7 were suboptimal.
The ability of 16E7 to abrogate growth arrest in previous experiments was shown to be complicated and mapped to three regions of 16E7, all of which have been implicated in the transforming mechanism of 16E7 (Demers et al. 1996). A mutation in the carboxyl terminus of 16E7 did not bypass growth arrest signals; however, 16E7 proteins with an intact carboxyl terminus, but altered conserved regions I or II, also failed to bypass growth arrest (Demers et al. 1996). These data suggest that under the experimental conditions used, either the inactivation of p21 by 16E7 was not sufficient to bypass the G1 arrest or they could provide evidence for multiple domains in 16E7 contributing to the inactivation of p21. In vivo, it is likely that the release of E2F from RB and the inactivation of p21 controls on both CDK/cyclin and PCNA activities are all necessary for the ability of 16E7 to bypass cell cycle arrest signals.
16E7 bypassed a transforming growth factor-β (TGF-β)-induced growth arrest (Pietenpol et al. 1990; Demers et al. 1996), suggesting that 16E7 might also inactivate other CDK inhibitors. Recently, the carboxy-terminal sequences of 16E7 were shown to bind to and inactivate p27 (Zerfass-Thome et al. 1996; data not shown), but the site in p27 to which 16E7 bound and the underlying mechanism have not been analyzed. The adenovirus E1A oncoprotein was also shown to abrogate the CDK-inhibitory function of p27 (Mal et al. 1996). E1A bound to the carboxyl terminus of p27 and prevented the association of p27 with CDK/cyclin complexes. At present, it is difficult to propose a conserved mechanism; the carboxy-termini of p21 and p27 share no obvious homology, and although the domain of E1A that interacts with p27 has not been defined, the carboxyl terminus of 16E7 does not have recognizable homology with E1A. In a broader perspective, these findings suggest that DNA tumor viruses have evolved diverse strategies with the common goal of counteracting CIP/KIP family CDK inhibitors.
Conclusions
We propose a model in which the dual inactivation of p21 in cells by 16E7 is a means to maintain high CDK2/cyclin E activity and allow DNA replication. For HPV-16, whose survival in host cells depends critically on PCNA-dependent DNA replication, this likely represents a central, previously unrecognized pathway. Because p21 is capable of inhibiting E2F activity in the absence of functional RB (Dimri et al. 1996), the inactivation of p21 by 16E7 may be a further safeguard to preserve E2F activity. The positive correlation between p21 inactivation and the transforming ability of different HPV types (16 vs. 6) underscores the significance this mechanism may have to antagonize cell cycle arrest signals and thereby promote genetic instability during HPV-induced cancer progression. Because p21 was also recently shown to be involved in S/M coupling (Waldman et al. 1996) it is tempting to speculate that 16E7 may also affect this property of p21, thus allowing cells to become polyploid.
Finally, our results raise the intriguing possibility that cellular proteins exist that share the ability of 16E7 to interact with the carboxyl terminus of p21 and thereby modulate its dual inhibitory activities. This novel mode of regulation of p21 activities, and consequently of the G1-to-S-phase transition and PCNA-dependent DNA replication, may represent an important mechanism of balancing the effects of p21 in human cells. The abrogation of a p53-induced cell cycle arrest by cellular oncoproteins such as c-Myc (Hermeking et al. 1995) could involve the induction of cellular inhibitors of p21 that might contribute to the development of cancer.
Materials and methods
Cell cultures
Normal human keratinocytes were prepared from infant foreskins and cultured in Keratinocyte Serum-Free Medium (GIBCO BRL) as described (Demers et al. 1994). Retroviruses and generation of retrovirally infected cells were described in detail (Demers et al. 1994, 1996). Subconfluent cells were treated with actinomycin D (2.5 nm) for 24 hr. Cells were harvested, and extracts for kinase assays and flow cytometry were prepared as described (Demers et al. 1994; Hermeking et al. 1995). For metabolic labeling, the cells were grown in cysteine- and methionine-free medium (Clonetics) for 30 min and in the presence of 8.14 MBq/ml of [35S]Protein Labeling Mix (NEN DuPont) for 2 hr directly prior to harvesting.
Mutational analysis and protein purification
pGEX-2T HPV-16 E7 was created by cloning a PCR fragment of HPV-16 coordinates 562–858 into the BamHI and EcoRI sites of pGEX-2T (Pharmacia). pGEX-2T HPV-16 E7N(Δ40–98) and 16E7C(Δ1–39) were created using pGEX-2T HPV-16 E7 as a template for PCR primers that produced HPV-16 fragments with coordinates from 562 to 678 and 679 to 858, repectively. These were cloned into the BamHI and EcoRI sites of pGEX-2T. His–HPV-16 E7 and HPV-6 E7 were generated by PCR using pGEX-2T and pLXSN as templates, respectively. The resulting 16E7 open reading frame (ORF) (562–858) and 6E7 ORF (526–994) were cloned into the BamHI site of pET-16b (Novagen). Preparations of His–p21 and PCNA have been described earlier (Waga et al. 1994a,b). His–cyclin E and His–CDK2 were gifts from J.M. Roberts (FHCRC). The plasmids for GST–p21, GST–p21N(1–90), and GST–p21C(87–164) were gifts from A. Dutta (Chen et al. 1995). To generate the plasmids used for in vitro translation of His–p21 carboxy-terminal truncations the XbaI fragment containing full length His–p21 from the Escherichia coli expression plasmid for His–p21 (Waga et al. 1994b) was cloned into the XbaI site of pRcCMV vector (Invitrogen). Then, the ApaI fragment containing the carboxy-terminal half of p21 in this pRcCMV-based construct was replaced by the PCR-amplified fragment encoding each truncated carboxy-terminal region of p21. The plasmids expressing GST–p21(139–164) and GST–p21(149–164) were constructed by the insertion of the PCR-amplified fragment encoding the respective amino acids of p21 into the BamHI site of pGEX-2TK plasmid (Pharmacia). All other cDNAs encoding wild-type or mutant proteins (Fig. 5C; data not shown) were cloned into pBS. The correct sequences of all constructs were confirmed by dideoxy sequencing. GST proteins were expressed in E. coli [BL21(DE3)] and purified from cell lysates by binding to glutathione–Sepharose 4B (Pharmacia) according to the manufacturer’s instructions. To generate His–cyclin E/CDK2 for kinase assays, Sf9 insect cells were coinfected with baculovirus constructs encoding His–cyclin E/CDK2 (gift from J.M. Roberts), harvested 2 days postinfection, and the pellet was lysed in 10 mm HEPES (pH 7.4), 10 mm NaCl, 1 mm EDTA, 2 mm DTT, and 0.1 mm PMSF, followed by centrifugation at 14000g for 20 min. The other His-tagged proteins (except p21) were expressed in E. coli [BL21(DE3)]. All His-tagged proteins were purified from the insoluble fraction using a Ni–NTA column (Qiagen) according to the manufacturer’s instructions. The GST fusion proteins were dialyzed against a buffer containing 5 mm Tris-Cl (pH 7.5), 10 mm MgCl2, 70 mm NaCl, and the His-tagged proteins were dialyzed against a buffer containing 25 mm Tris-Cl (pH 7.5), 10% glycerol, 1 mm EDTA, 1 mm DTT, 0.01% NP-40, and 100 mm NaCl. All purified proteins were analyzed by SDS-PAGE and Coomassie or silver staining.
Binding assays, immunoprecipitations, and immunoblotting
For in vitro binding reactions, the GST proteins immobilized on glutathione–Sepharose 4B were incubated with His or IVT proteins for 1 hr at room temperature in binding buffer (20 mm HEPES at pH 7.4, 150 mm NaCl, 10% glycerol, 0.05% NP-40) and washed extensively with the binding buffer. The bound proteins were separated by SDS-PAGE, and one half of the sample was analyzed by Coomassie staining and the other half by immunoblotting or fluorography. In vitro transcription and translation of pBS plasmids was performed with the T3/T7 Ribonuclease Combination System (Promega) in wheat germ extracts (Promega) according to the manufacturer’s instructions. Immunoblotting was done as described (Hermeking et al. 1995) using the following antibodies: anti-p53 mAb (Ab-6, Oncogene Sciences), anti-p21 mAb (Ab-1, Calbiochem), anti-His pAb (H-15, Santa Cruz), anti-16E7 mAb (8C9, gift from Zymed Laboratories), and anti-PCNA mAb (PC10). To determine the in vivo association of 16E7 and p21 immunoprecipitations of metabolically labeled cells using anti-16E7 polyclonal antibodies (Demers et al. 1996) or anti-p21 polyclonal antibodies (gift from D. Beach, Cold Spring Harbor Laboratory) were carried out under consecutive low- and high-stringency conditions as described in detail (Blackwood et al. 1992).
Kinase assays
In vitro kinase assays with cell extracts were performed essentially as described in detail (Hermeking et al. 1995) using polyclonal anti-cyclin E antibodies (gift from J.M. Roberts) or polyclonal anti-CDK2 antibodies (M2, Santa Cruz) for immunoprecipitations. For in vitro restoration of CDK activity, GST–16E7 was added to cell extracts prior to the immunoprecipitation and incubated for 1 hr at room temperature; then kinase reactions were performed after extensive washing of the precipitate. For kinase reactions with purified CDK2/cyclin E complexes (2 ng/reaction), p21 and E7 proteins were preincubated for 1 hr at room temperature, then mixed and further preincubated with His–cyclin E/CDK2 in kinase buffer for 20 min on ice prior to kinase reactions. In all reactions histone H1 (2.5 μg; Boehringer) was used as the substrate. The reactions were analyzed by SDS-PAGE followed by autoradiography and PhosphorImager (Molecular Dynamics) analysis.
Replication assays
PCNA-activated DNA synthesis with oligo(dT)/poly(dA) as primer template was carried out as described (Tsurimoto and Stillman 1991), except that 30 mm HEPES–NaOH (pH 6.6) and 80 μm (in nucleotide) oligo(dT)/poly(dA) (19:1) were used. SV40 DNA replication with the extracts of 293 cells was performed as described (Stillman and Gluzman 1985). Measurement of incorporated nucleotides and analysis of replication products were done as described previously (Tsurimoto and Stillman 1991; Waga et al. 1994a). Preincubation of E7 and p21 prior to DNA synthesis was carried out in a buffer containing 25 mm Tris-HCl (pH 7.5), 10% glycerol, 1 mm EDTA, 0.01% NP-40, 1 mm DTT, and 100 mm (Fig. 2A) or 60 mm (Figs. 2B and 6B) NaCl. Preparations of DNA polymerase δ have been described (Waga et al. 1994a,b).
Acknowledgments
We thank J.M. Roberts and members of his laboratory for reagents and advice, D. Beach, A. Dutta, S.J. Elledge, M.L. Harter, G.J. Hannon, T. Hunter, A.J. Levine, R. Li, J. Massagué, O.M. Perreira-Smith, J.R. Smith, and Y. Xiong for generous gifts of plasmids, antibodies, or proteins, K. Münger for sharing results prior to publication, and members of the Galloway and Stillman laboratories, especially S.A. Foster, for fruitful discussions. This research was supported by grants from the National Institutes of Health to D.A.G. (CA64795) and B.S. (CA13106). J.O.F. is supported by a fellowship from the Deutsche Forschungsgemeinschaft (Fu 342/1-1).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL dgallowa@fhcrc.org; FAX (206) 667-5815.
References
- Abraham SE, Carter MC, Moran E. Transforming growth factor β1 (TGFβ1) reduces cellular levels of p34cdc2, and this effect is abrogated by adenovirus independently of the E1A-associated pRB binding activity. Mol Cell Biol. 1992;3:655–665. doi: 10.1091/mbc.3.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Sellers WR, Sharma SK, Wu AD, Nalin CM, Kaelin WG. Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol Cell Biol. 1996;16:6623–6633. doi: 10.1128/mcb.16.12.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbín M, Hannon GJ, Pendás AM, Ferrando AA, Vizoso F, Fueyo A, López-Otín C. Functional analysis of a p21WAF1,CIP1,SDI1 mutant (Arg94 → Trp) identified in a human breast carcinoma. Evidence that the mutation impairs the ability of p21 to inhibit cyclin-dependent kinases. J Biol Chem. 1996;271:15782–15786. doi: 10.1074/jbc.271.26.15782. [DOI] [PubMed] [Google Scholar]
- Ball KL, Lain S, Fåhraeus R, Smythe C, Lane DP. Cell-cycle arrest and inhibition of cdk4 activity by small peptides basd on the carboxy-terminal domain of p21WAF1. Curr Biol. 1996;7:71–80. doi: 10.1016/s0960-9822(06)00029-7. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Lüscher B, Eisenman RN. Myc and Max associate in vivo. Genes & Dev. 1992;6:71–80. doi: 10.1101/gad.6.1.71. [DOI] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteosome pathway. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- Chen J, Jackson PK, Kirschner MW, Dutta A. Separate domains of p21 involved in the inhibition of cdk kinase and PCNA. Nature. 1995;374:386–388. doi: 10.1038/374386a0. [DOI] [PubMed] [Google Scholar]
- Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A. Cyclin-binding motifs are essential for the function of p21CIP1. Mol Cell Biol. 1996;16:4673–4682. doi: 10.1128/mcb.16.9.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers GW, Foster SA, Halbert CL, Galloway DA. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc Natl Acad Sci. 1994;91:4382–4386. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers GW, Espling E, Harry JB, Etscheid BG, Galloway DA. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70:6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Nakanishi M, Desprez P-Y, Smith JR, Campisi J. Inhibition of E2F activity by the cyclin-dependent protein kinase inhibitor p21 in cells expressing or lacking a functional retinoblastoma protein. Mol Cell Biol. 1996;16:2987–2997. doi: 10.1128/mcb.16.6.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer E, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Flores-Rozas H, Kelman Z, Dean FB, Pan Z-Q, Harper JW, Elledge SJ, O’Donnell M, Hurvitz J. Cdk-interacting protein 1 directly binds with proliferating cell nuclear antigen and inhibits DNA replication catalyzed by the DNA polymerase δ holoenzyme. Proc Natl Acad Sci. 1994;91:8655–8659. doi: 10.1073/pnas.91.18.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Turck CW, Morgan DO. Inhibition of CDK2 activity in vivo by an associated 20K regulatory subunit. Nature. 1993;366:707–710. doi: 10.1038/366707a0. [DOI] [PubMed] [Google Scholar]
- Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Structure of the carboxy-terminal region of p21WAF1/CIP1 complexed with human PCNA. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol. 1992;66:2125–2134. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai L-H, Zhang P, Dobrowolski S, Bai C, Connel-Crowley L, Swindell E, Fox MP, Wei N. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Funk JO, Reichert M, Ellwart JW, Eick D. Abrogation of p53-induced cell cycle arrest by c-Myc: Evidence for an inhibitor of p21WAF1/CIP1/SDI1. Oncogene. 1995;11:1409–1415. [PubMed] [Google Scholar]
- Jones DL, Münger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997;71:2905–2912. doi: 10.1128/jvi.71.4.2905-2912.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc Natl Acad Sci. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes & Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- Li R, Hannon GJ, Beach D, Stillman B. Subcellular distribution of p21 and PCNA in normal and repair-deficient cells following DNA damage. Curr Biol. 1996;6:189–199. doi: 10.1016/s0960-9822(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Lin J, Reichner C, Wu X, Levine AJ. Analysis of wild-type and mutant p21WAF-1 gene activities. Mol Cell Biol. 1996;16:1786–1793. doi: 10.1128/mcb.16.4.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Hurwitz J, Massagué J. Cell cycle inhibition mediated by functionally independent CDK and PCNA binding domains in p21CIP1. Nature. 1995;375:159–161. doi: 10.1038/375159a0. [DOI] [PubMed] [Google Scholar]
- Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes & Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- Mal A, Poon RYC, Howe PH, Toyoshima H, Hunter T, Harter ML. Inactivation of p27KIP1 by the viral E1A oncoprotein in TGFβ-treated cells. Nature. 1996;380:262–265. doi: 10.1038/380262a0. [DOI] [PubMed] [Google Scholar]
- Missero C, Calautti E, Eckner R, Chin J, Tsai LH, Livingston DM, Dotto GP. Involvement of the cell-cycle inhibitor Cip1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc Natl Acad Sci. 1995;92:5451–5455. doi: 10.1073/pnas.92.12.5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Robetorge RS, Pereira-Smith OM, Smith JR. The carboxy-terminal region of p21SDI1/WAF1/CIP1 is involved in proliferating cell nuclear antigen binding but does not appear to be required for growth inhibition. J Biol Chem. 1995;270:17060–17063. doi: 10.1074/jbc.270.29.17060. [DOI] [PubMed] [Google Scholar]
- Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1 to S phase transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SB, Eichele G, Zhang P, Rawls A, Sands AT, Bradley A, Olson EN, Harper JW, Elledge SJ. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, Pittelkow MR, Münger K, Howley PM, Moses HL. TGF-β 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming protiens with pRB binding domains. Cell. 1990;61:777–785. doi: 10.1016/0092-8674(90)90188-k. [DOI] [PubMed] [Google Scholar]
- Podust VN, Podust LM, Goubin F, Ducommun B, Hübscher U. Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry. 1995;34:8869–8875. doi: 10.1021/bi00027a039. [DOI] [PubMed] [Google Scholar]
- Prelich G, Stillman B. Coordinated leading and lagging strand synthesis during SV40 DNA replication in vitro requires PCNA. Cell. 1988;53:117–126. doi: 10.1016/0092-8674(88)90493-x. [DOI] [PubMed] [Google Scholar]
- Reed SI. Cyclin E: In mid-cycle. Biochim Biophys Acta. 1996;1287:151–153. doi: 10.1016/0304-419x(96)00006-6. [DOI] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP. Crystal structure of the p27KIP1 cyclin-dependent-kinase inhibitor bound to the cyclin A-cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Shivji MKK, Grey SJ, Strausfeld UP, Wood RD, Blow JJ. Cip1 inhibits DNA replication but not PCNA-dependent nucleotide excision-repair. Curr Biol. 1994;4:1062–1068. doi: 10.1016/s0960-9822(00)00244-x. [DOI] [PubMed] [Google Scholar]
- Slebos RJC, Lee MH, Plunkett BS, Kessis TD, Williams BO, Jacks T, Hedrick L, Kastan MB, Cho KR. p53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proc Natl Acad Sci. 1994;91:5320–5324. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman BW, Gluzman Y. Replication and supercoiling of simian virus 40 DNA in cell extracts from human cells. Mol Cell Biol. 1985;5:2051–2060. doi: 10.1128/mcb.5.8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J-Y, Rempel R, Erikson E, Maller JL. Cloning and characterization of the Xenopus cyclin-dependent kinase inhibitor p27XIC1. Proc Natl Acad Sci. 1995;92:10187–10191. doi: 10.1073/pnas.92.22.10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurimoto T, Stillman B. Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase α and δ during initiation of leading and lagging strand synthesis. J Biol Chem. 1991;266:1961–1968. [PubMed] [Google Scholar]
- Waga S, Bauer G, Stillman B. Reconstitution of complete SV40 DNA replication with purified replication factors. J Biol Chem. 1994a;269:10923–10934. [PubMed] [Google Scholar]
- Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994b;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- Waldman T, Lengauer C, Kinzler KW, Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature. 1996;381:713–716. doi: 10.1038/381713a0. [DOI] [PubMed] [Google Scholar]
- Warbrick E, Lane DP, Glover DM, Cox LS. A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr Biol. 1995;5:275–282. doi: 10.1016/s0960-9822(95)00058-3. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993a;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes & Dev. 1993b;7:1572–1583. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Kuppuswamy D, Li Y, Livanos EM, Hixon M, White A, Beach D, Tlsty TD. Alteration of cell cycle kinase complexes in human papillomavirus E6- and E7-expressing fibroblasts precedes neoplastic transformation. J Virol. 1996;70:999–1008. doi: 10.1128/jvi.70.2.999-1008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerfass K, Schulze A, Spitkovsky D, Friedman V, Henglein B, Jansen-Dürr P. Sequential activation of cyclin E and cyclin A gene expression by human papillomavirus type 16 E7 through sequences necessary for transformation. J Virol. 1995;69:6389–6399. doi: 10.1128/jvi.69.10.6389-6399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz JW, Jansen-Dürr P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996;13:2323–2330. [PubMed] [Google Scholar]
- Zhang H, Hannon GJ, Beach D. p21-containing cyclin kinases exist in both active and inactive states. Genes & Dev. 1994;8:1750–1758. doi: 10.1101/gad.8.15.1750. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomavirus infections—A major cause of human cancers. Biochim Biophys Acta. 1996;1288:F55–F78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]