

Abstract
HIV/AIDS remains a formidable disease with millions of individuals inflicted worldwide. Although treatment regimens have improved considerably, drug resistance brought on by viral mutation continues to erode their effectiveness. Intense research efforts are currently underway in search of new and improved therapies. This review is concerned with the design of novel HIV-1 protease inhibitors that incorporate heterocyclic scaffolds and which have been reported within the recent literature (2005–2010). Various examples in this review showcase the essential role heterocycles play as scaffolds and bioisosteres in HIV-1 protease inhibitor drug development. This review will hopefully stimulate the widespread application of these heterocycles in the design of other therapeutic agents.
HIV-1 protease inhibitors (PIs) are a critical component of current antiretroviral therapy with nine PIs approved by the US FDA (Figure 1) [1-12]. The effectiveness of the early PIs was limited due to poor pharmacokinetic properties, side effects and the rapid development of viral resistance. The bioavailability problems of early PIs were caused by the presence of peptidic characters that gave rise to rapid metabolic degradation. Later PIs have demonstrated moderately improved absorption profiles but still require co-administration with ritonavir (RTV) to limit their metabolic degradation [1,13]. Identifying new PIs that do not require boosting with RTV remains a central goal of HIV drug research. In addition, the emergence of drug resistance has proven to be a persistent challenge and requires the development of new therapeutic options. Resistance is commonly incurred by an amino acid mutation or collection of mutations that either modifies the steric environment of the binding site, resulting in a loss of fit or a loss of van der Waals’ interactions, or by eliminating a residue involved in a hydrogen bonding interaction with the PI. To combat drug resistance, current strategies focus on the development of new PIs that maximize interactions in the active site particularly, by forming hydrogen bonds with the enzyme’s backbone residues thereby rendering them less susceptible to viral mutations [14].
Figure 1. Current FDA-approved HIV-1 protease inhibitors.

As can be seen in this review, another attractive approach in the design of PIs has been the use of heterocyclic ring systems as bioisosteres for peptide/amide bonds to attenuate pharmacokinetic properties, enhance binding affinity, and provide novel chemical scaffolds to combat drug-resistant viral strains. Such heterocycles offer many desirable properties that make them attractive in molecular design. For example, many heterocycles are chemically stable towards reductive/oxidative conditions and acid/base hydrolysis. Their confined geometries add rigidity to a molecule’s structure and are responsible for maintaining the spatial orientation of the pharmacophoric groups, an attribute that can have a marked effect on binding. In addition, the heterocycle itself may directly bind to the enzyme forming new hydrogen bonds or hydrophobic interactions resulting in increased potency. As a bioisostere, the use of heterocyclic surrogates can also provide an attractive means to modulate a drug’s ADME profile without compromising activity. Consequently, the design and development of novel heterocyclic ligands and their incorporation into PIs have led to a new generation of PIs with marked improvements in potency and pharmacological properties [1,13].
Cyclic ethers
Cyclic ethers are designed to reduce the peptidic features of the first generation PIs. These structural features are inherent to numerous bioactive natural products [15]. One of the intriguing elements of cyclic ethers is that the ether oxygen could conceivably replace a peptide’s carbonyl oxygen and form hydrogen bonds in the active site similar to a peptide carbonyl. The importance of cyclic ether-derived PIs has been demonstrated [16]. Such structural features have been incorporated into the chemical structure of two commercially available PIs, amprenavir (APV) 7 and darunavir (DRV) 9 (Figure 1) [6,15]. In this review, we will focus on recent approaches to identify novel PIs using modified/designed cyclic ether structures.
Tetrahydrofuran
The importance of tetrahydrofuran rings as potent P2 ligands in the design of PIs has been reviewed previously [17]. The potency enhancing effect of the 3-(S)-tetrahydrofuranyl urethane was demonstrated and utilized in the design of APV [6,17]. During further design work, Ghosh et al. developed a novel fused cyclopen cyclopentane–tetrahydrofuran (Cp–THF) ring system as a P2 ligand for a new series of PIs [18]. Initial efforts identified 10 (Figure 2) as a potent inhibitor with Ki = 0.14 nM and EC50 = 8 nM. The significance of the tetrahydrofuran ring oxygen is exemplified by compound 11 (Figure 2), which replaces the oxygen with a methylene group resulting in a drastic loss of antiviral activity (Ki = 5.3 nM and EC50 > 1000 nM). This is likely due to a loss of hydrogen binding within the P2 site. Interestingly, replacing the 4-amino group of the P2′ sulfonamide with a para-hydoxymethyl (12, GRL-06579A) significantly improved its enzyme inhibitory potency more than 30-fold (Ki = 4.5 pM and EC50 = 1.8 nM). The reason for this marked improvement in binding is possibly due to its enhanced hydrogen-bonding interactions with the Asp-29 and Asp-30 backbone NHs and the side chain carboxylates.
Figure 2. Structure–activity relationship of cyclopentane–tetrahydrofuran-based protease inhibitors.

C4 substituted bis-tetrahydrofuran
The use of bis-tetrahydrofuran rings as highly potent P2 ligands has been demonstrated [11,15,16]. As part of an ongoing effort to form novel interactions with the protease backbone, Ghosh and co-workers recently synthesized and evaluated a series of PIs containing C4 ether groups on the bis-THF ligand [19]. The new C4 functionality was intended to form additional hydrogen-bonding interactions with the backbone NH of Gly-48, similar to the aforementioned spirocyclic ether ligands. Within this series of compounds (Table 1), the (R)-methoxy substituted PI 14 was the most active (Ki = 2.9 pM and IC50 = 2.4 nM) having comparable antiviral activity as the non-substituted analog 13 (TMC-126 Ki = 14 pM and IC50 = 1.4 nM). Stereoisomer 15 was significantly less potent (Ki = 35 pM and IC50 = 55 nM). Replacing the methoxy with a benzyl group further reduced antiviral activity (16 & 17 in Table 1 with Ki = 73 and 300 pM, respectively). Interestingly, the activity pattern of the chiral benzyl analogs was the reverse of the pattern that was seen with the methoxy analogs.
Table 1. Structure–activity relationship of C4 substituted bis-tetrahydrofuran protease inhibitors.

| Compound Ligand number |
Ki (nM) |
IC50 (nM) |
|
|---|---|---|---|
|
13 (TMC-126) |

|
0.014 | 1.4 |
| 14 |

|
0.0029 | 2.4 |
| 15 |

|
0.035 | 55 |
| 16 |
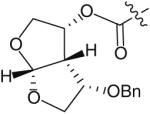
|
0.073 | 335 |
| 17 |
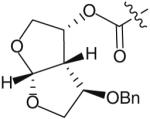
|
0.3 | ND |
Tricyclic tetrahydrofuran
An examination of DRV’s crystal structure by Ghosh and co-workers led to speculation that a third tetrahydrofuran ring attached to the bis-THF P2 ligand could fill additional space within the hydrophobic pocket and lead to new hydrogen-bonding interactions with the enzyme. This hypothesis led to the synthesis of two novel tris-THF PIs (Figure 3) with improved binding and antiviral properties as compared with DRV [20]. Biological evaluations revealed the syn-anti-syn configuration of tris-THF rings (18) was tenfold more potent than the syn-syn-syn equivalent (19). The x-ray crystal structure of inhibitor 18 bound to HIV-1 protease indicated the oxygen of the third THF ring was involved in a network of hydrogen-bonding interactions between two water molecules and Arg-8′, Arg-87, Asp-29, Thr-26, and Gly-27 (Figure 3) . In addition, the hydrogens of the third THF ring appear to be involved in a direct hydrogen bond with the carbonyl of Gly-48 and a water-mediated hydrogen-bond interaction with the amine of Gly-48. During further biological testing, 18 (GRL-0519A) demonstrated a tenfold increase in antiviral activity as compared with DRV against a panel of multidrug-resistant HIV-1 clinical isolates, and was shown to have an enhanced ability (tenfold over DRV) to block protease dimerization [21]. These results are extremely encouraging and suggest 18 may be highly effective in fighting multidrug-resistant viral strains.
Figure 3. Compounds 18 and 19 with hydrogen-binding interactions between the oxatricyclic tetrahydrofuran ligand and the protease enzyme [20].

Spirocyclic ethers
Following the development of the Cp–THF ligand in PI 12, efforts in the Ghosh research labs turned towards incorporating new functionalities onto the 3-position of the Cp–THF ligand as a means to create additional interactions with the enzyme’s binding site. In this context, a series of spirocyclic ligands were designed featuring dioxolane, tetrahydrofuran, and oxazolidinone motifs (Table 2) [22]. Unfortunately, this new series of PIs was, in general, significantly less potent than DRV (Ki between 0.2 and 2 nM). Within the series, the oxazolidinone 23 (Table 2) displayed the best antiviral activity with an IC50 of 21 nM.
Table 2. Structure–activity relationship of spirocyclic ether-based protease inhibitors.

| Compound Ligand number |
Ki (nM) | IC50 (nM) |
|
|---|---|---|---|
| 20 |
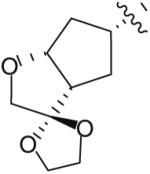
|
0.16 | 280 |
| 21 |
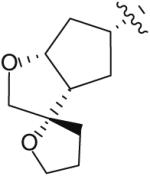
|
2.2 | 170 |
| 22 |

|
0.38 | ND |
| 23 |

|
0.17 | 21 |
Meso-bicyclic ethers
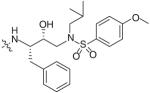
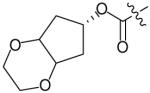
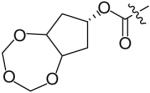
In an effort to reduce the stereochemical complexity of the Cp–THF ligand in 12, the corresponding meso-bicyclic ether moieties were investigated. Conceivably, either oxygen atom of the meso-ligand can form the critical hydrogenbonding interactions with the enzyme similar to Cp–THF. Based on this concept, a series of meso-bicyclic ligands was synthesized and evaluated for their antiviral activity (Table 3) [23]. The dioxolane-based ligand 24 displayed the best antiviral results exhibiting an IC50 of 3.8 nM, whereas larger ether rings such as dioxane 25 and trioxepane 26 resulted in reduced potency (IC50 = 210 and 380 nM, respectively). Further structural modifications at the P2′ site did not result in any appreciable change in potency in stark contrast to the Cp–THF ligand in PI 12.
Table 3. Structure–activity relationship of meso-bicyclic ether P2 ligands.
| Compound Ligand number |
Ki (nM) |
IC50 (nM) |
|
|---|---|---|---|
| 24 |

|
0.11 | 3.8 |
| 25 |
|
0.18 | 210 |
| 26 |
|
0.51 | 380 |
Flexible polyethers
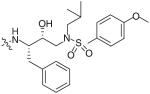
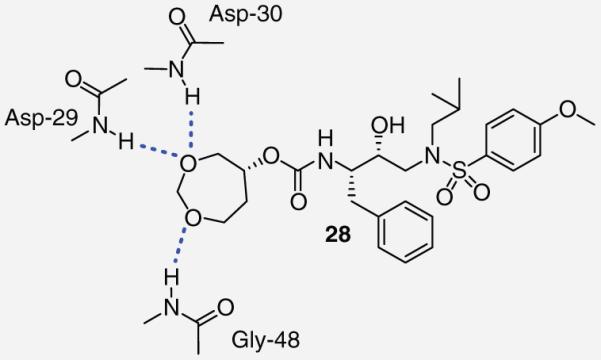
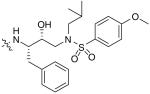
The bis-THF moiety of DRV and its extensive binding interactions with the HIV-1 protease backbone atoms imparts an extremely high binding affinity that contributes to DRV’s high potency. Recently, Ghosh and co-workers investigated alternative cyclic ether ligand designs by using flexible polyethers that would maintain binding interactions but allow ring flexibility to accommodate viral mutations (Table 4) [24]. From these studies, six- and seven-membered polyether rings (27 & 28) were identified as highly potent PIs (IC50 = 3.4 and 4.9 nM, respectively) whereas larger ring sizes resulted in a significant loss of activity. The (R) stereoisomer 28 demonstrated a sixfold increase in antiviral activity as compared with the (S) isomer 29. The importance of the ether’s oxygen atoms within the rings was clearly demonstrated by selective replacement with carbon. Deletion of a single oxygen as in 30 and 31 led to a 30-fold drop in enzyme inhibitory activity whereas removal of both oxygen atoms as in 32 resulted in a 1000-fold loss. An x-ray structure of inhibitor 28 bound to the HIV-1 protease (Figure 4) revealed extensive interactions including hydrogen bonding with the backbone Asp-29 and Asp-30 NHs. Furthermore, the bottom oxygen of the 1,3-dioxocycloheptane ring forms a unique water-mediated interaction with the NH of Gly-48 [24].
Table 4. Structure–activity relationship of flexible polyether P2 ligands.
| Compound Ligand number |
Ki (nM) |
IC50 (nM) |
|
|---|---|---|---|
| 27 |

|
0.041 | 3.4 |
| 28 |

|
0.026 | 4.9 |
| 29 |
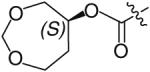
|
0.16 | 30 |
| 30 |

|
0.74 | ND |
| 31 |

|
0.81 | ND |
| 32 |

|
27 | ND |
Figure 4. Proposed binding interactions of 28 with the protease active site.
Tetrahydropyran
Recently, Ghosh and co-workers designed and synthesized a series of PIs that incorporated a fused tetrahydropyran–tetrahydrofuran (Tp–THF) ring as a novel P2 ligand [25]. It has been rationalized that replacing the bis-THF ligand of DRV 9 with a larger Tp–THF ligand would effectively fill the enzyme’s hydrophobic binding pocket and enhance van der Waals’ interactions with the protease [25]. In addition, the larger ring would bring the cyclic ether oxygens closer to the Asp-29 and Asp-30 residues resulting in optimized hydrogen-bonding interactions. Furthermore, the increased flexibility of the tetrahydropyran ring could more easily accommodate enzyme mutations leading to PIs with improved potency against drug-resistant strains. From this work, 33 (GRL-0476) was identified as a potent PI (Ki = 2.7 pM, IC50 = 0.5 nM) with a similar activity profile as 9 against multidrug-resistant HIV-1 strains. The significance of the cyclic ether oxygens is demonstrated in Table 5. Substitution of either oxygen of the Tp–THF ligand with carbon (35 & 36) results in a significant loss of antiviral activity (Ki = 0.005 and 1.4 nM), with the loss of the THF oxygen 36 having the most pronounced effect. Similarly, altering the stereochemistry of the Tp–THF group (34, Ki = 0.068 nM) or changing the position of the attached urethane (37, Ki = 0.11 nM) decreased antiviral activity considerably. Further modifications of the P2′ methoxy did not alter activity by any appreciable amount.
Table 5. Structure–activity relationship of tetrahydropyran–tetrahydrofuran-based protease inhibitors.
| Compound Ligand number |
Ki (nM) |
IC50 (nM) |
|
|---|---|---|---|
| 33 |

|
0.0027 | 0.5 |
| 34 |
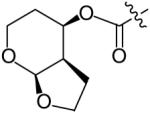
|
0.068 | 19 |
| 35 |

|
0.005 | 8 |
| 36 |

|
1.43 | ND |
| 37 |

|
0.11 | ND |
Triazole, oxadiazole & thiophene ring systems
Triazoles, oxadiazoles and thiophenes have found widespread use as bioisosteres for amide bonds and phenyl groups during the development of drug molecules [26,27]. In addition to supplying heteroatoms that may engage in hydrogen bonding they contain π-bonds that may create π-stacking interactions with the enzyme [27]. The following represents recent examples of their use as novel PI ligands.
1,2,3-triazoles
The facile synthesis of triazole rings via azidealkyne click chemistry denotes a compelling reason to incorporate this isostere into the scaffold of HIV PIs as a replacement for peptidic bonds. Using this methodology, a library of triazole-based small molecules was synthesized and screened for anti-HIV activity leading to the identification of novel triazole-based HIV PIs. Further structural refinements mediated by click chemistry, led to the highly active compound 38 with a Ki value of 8 nM against HIV-1 protease (Figure 5) [28]. An examination of its binding through docking simulations indicated the triazole nitrogen atom (N3) formed a water-mediated hydrogen bond to Ile-50 and Ile-50′ that is further stabilized through hydrogen bonding with the carbonyl oxygen of the carbamate. Interestingly, replacement of the triazole with a two- or three-carbon amide linker greatly decreased potency thereby demonstrating the intrinsic value of the triazole ring.
Figure 5. Proposed binding interactions between 38 and 39 with the protease enzyme.

Similar click chemistry-based synthetic efforts later identified 39 (AB2, Figure 5) as a potent HIV-1 PI (IC50 = 6 nM) with excellent retention of activity against mutant viral strains with known cross resistance to saquinavir (SQV), RTV, and nelfinavir (NFV) [29]. The PI 39 exhibited a slightly different binding profile as compared with 38 with the triazole nitrogen atom and sulfonamide oxygen forming a water-mediated hydrogen bond to Ile-50 and Ile-50′ [30]. In addition, the C5 hydrogen of the triazole appeared to form a weak hydrogen bond to Gly-25. Although still in early development, this triazole-based PI represents a novel chemical scaffold and can be further improved.
In addition to using click chemistry for selective triazole synthesis, it has been shown that biological templates can be used to catalyze the same reaction and selectively synthesize triazole analogs from a pool of alkyne and azide fragments. In this manner, HIV-1 protease was recently used to catalyze the reaction of an azide in solution with a mixture of alkyne fragments to preferentially give a single triazole (39) entity [31]. This methodology offers an efficient means to discover new PIs from lower affinity fragments.
Another example of the use of triazoles in HIV PIs was found with an initial structure–activity relationship (SAR) study on a series of triazolamers [32]. Arora and co-workers replaced all the peptide bonds of the HIV-1 protease substrate with triazoles and modified the side chains (R1–R4) with various hydrophobic groups. Unfortunately, this work provided compounds with only modest activity towards HIV-1 protease (IC50 = 25 μM). Initial docking studies have suggested the R2 and R3 hydrophobic groups of 40 occupy the S1 and S1′ enzyme subsites, respectively (Figure 6). Furthermore, no hydrogen-bonding interactions were detected between the protease and the nitrogens of the triazole rings for this series of compounds, perhaps explaining the low potency of these compounds.
Figure 6. Novel tetratriazole protease inhibitor 40.

Oxazole & oxadiazoles
Researchers at Merck Research Laboratories examined ways to reengineer their PI indinavir (IDV) to combat drug resistance. They explored viable replacements for the P1′ phenyl group, which is found in four of the nine FDA-approved HIV PIs. Initially, oxadiazoles were substituted at this position leading to novel PIs (41), which exhibited excellent activity (picomolar enzyme and nanomolar cell based) against native and IDV-resistant strains (Figure 7) [33]. These results confirmed that P1′ modifications could play an important role in combating drug resistance. In later work, the oxadiazole was replaced with a pyridylthiophene group and further refinements were investigated at P3 [34]. Introduction of an oxazole ring at P3 produced an extremely potent inhibitor (42) with broad-spectrum activity (CIC95 <7.8 nm) across a panel of mutant viruses (Figure 7). Interestingly, replacing the oxazole with a thiazole ring resulted in a loss of activity against the viral mutant V-18C, whereas furan and thiophene substitutions led to a general loss in activity.
Figure 7. Protease inhibitors 41 and 42.

Thiazoles
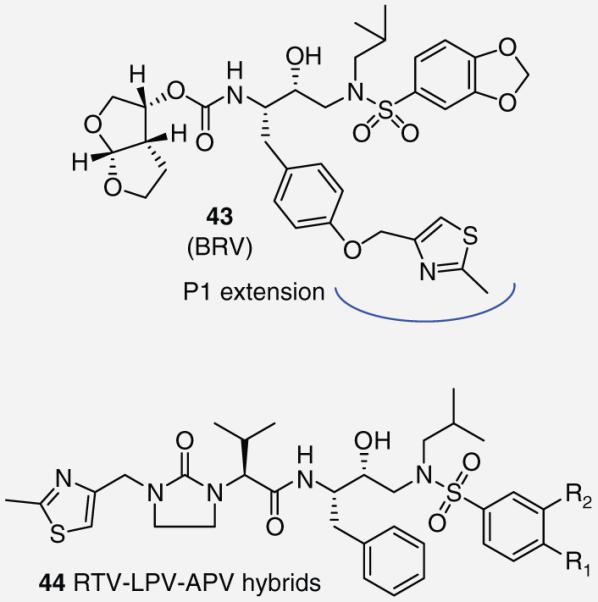
Researchers at GlaxoSmithKline investigated PIs by modifications of the P1 phenyl group through the attachment of various aryl and heteroaryl moieties by a para-oxygen atom (Figure 8). Their SAR efforts identified a PI, 43, which was named brecanavir (BRV) with femtomolar enzyme inhibitory potency. It incorporated a methylated thiazole ring at P1 [35]. The thiazole group is inherent to the commercially available PI 4. Such groups have been subsequently reported in four hybrid molecules (44, Figure 8 ) [36]. However, RTV-based inhibitors place the thiazole at the P3 location whereas BRV incorporates the thiazole as an extension of the P1 subsite. BRV maintained efficacy against a panel of drug-resistant viral strains in the low nM. X-ray crystallography identified a novel interaction between the thiazole nitrogen and Arg-8/Asp-29 side chains. Interestingly, substitution of the thiazole with triazole or imidazole groups significantly decreased potency against the wild-type strain of the HIV-1 protease but maintained potency against drug-resistant mutants. BRV was advanced by GlaxoSmithKline into early clinical trials where it showed promising results, however, its development was later cancelled due to formulation problems [37].
Figure 8. Thiazole-containing protease inhibitors 43 and 44.
Thiophenes
The substitution of sulfur for −CH=CH− within an aromatic ring is a classical example of isosterism with many successfully demonstrated examples. It is not surprising then that De Bonis and co-workers recently reported on NFV and SQV analogs that use thiopene as a P1 phenyl substitute (45–48 Figure 9 ). Initially, the thiophenes were directly attached to the PI scaffold resulting in a loss of potency (45 & 47; IC50 = 5 and 30 μM, respectively, as compared with SQV = 0.0004 and NFV = 0.0019 μM), however, the addition of a methylene spacer greatly restored their inhibitory activity (46 & 48; IC50 = 0.02 and 0.003 μM, respectively) [38,39]. Unfortunately, analogs containing the thiophene ring demonstrated a significant loss in enzyme inhibitory potency when assayed against a V32I mutant enzyme, similar to SQV. However, further structural refinements led to the incorporation of a benzothienyl moiety as a P1 ligand producing 49 (Figure 9), which retained activity against V32I [38,39].
Figure 9. Protease inhibitors 45–50.

The thiophene subunit was recently found in a novel allosteric HIV-1 PI 50 (Figure 9), which was discovered during high-throughput screening of 44,000 compounds of a combinatorial library. This compound contains two thiophene moieties and appears to bind in a non-competitive fashion to the protease enzyme [40]. Docking studies suggests 50 binds in a pocket found on the side of the protease away from the substrate binding site. Although the potency of this compound is modest (IC50-wt = 17 μM), it represents an interesting starting point for an alternative class of PIs with a novel mechanism of action [40].
Unsaturated heterocyclic rings
Pyrrolidine & azepine
A common theme for many HIV PIs to date has been the use of a hydroxyl moiety as a transitionstate mimic that forms strong binding interactions with the catalytic aspartic acid residues. Recently, the Klebe group has explored a pyrrolidine-based platform of HIV-1 PIs that uses a cyclic amine to bind to the aspartic acids. Their work, inspired by a report of a potent piperidine-based renin inhibitor, led to the design of inhibitor 51 (Figure 10) that possessed moderate antiviral activity (IC50 = 2.2 μM) [41,42]. Later efforts to optimize this series of compounds evaluated C2 symmetrical molecules. A series of diesters 52 (Figure 10) was synthesized but failed to appreciably improve on potency; however, replacing the esters with sulfonamides led to PI 53 (Figure 10), which has shown a modest binding affinity for the protease enzyme (Ki = 70 nM) [43,44]. Although both inhibitor 53 and the protease enzyme are symmetrical in nature, x-ray analysis revealed 53 binds in an asymmetric fashion. The oxygens of only one of the sulfonamides interacts with the amines of Ile-50 and Ile-50′ while the two benzamide groups display distinctly different binding profiles (Figure 11) [45].
Figure 10. Development of pyrrolidine-based HIV-1 protease inhibitors 51–53.
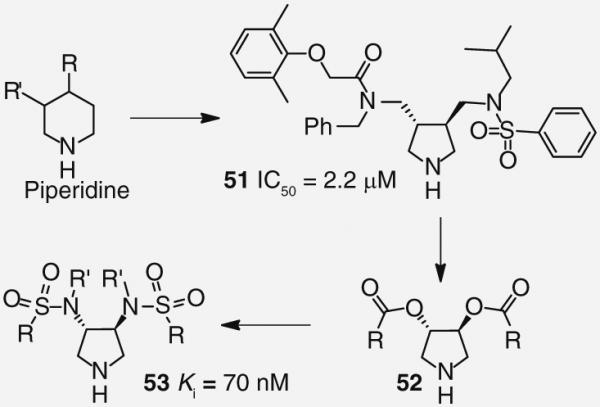
Figure 11. Asymmetric binding of symmetric pyrrolidine 53 within the HIV protease enzyme.

In line with Klebe’s work, a series of substituted azepin rings were synthesized via ring-closing metathesis (RCM) and evaluated as aspartic acid PIs [46]. Although this work is in its early phases, 54 (Figure 12) was identified as a lead molecule possessing moderate antiviral activity (IC50 = 40 μM) against HIV-1.
Figure 12. Recently reported azepine-based HIV-1 protease inhibitor 54.

Thiazolidine & spiro-oxazolidine
Thiazolidine 55 (JE-2147, also referred to as KNI-764) was developed jointly by the Kiso research group and the Japan Energy Corporation (Mimoto, Hayashi and co-workers) with later involvement from Dainippon Sumitomo during clinical development work [47]. It represents a novel class of allophenylnorstatine-based HIV-1 PIs th at retain potency against drug-resistant HIV-1 strains. The incorporation of a thiazolidine (a common proline isostere) into the P1′ site appears to enhance the compounds potency by restricting the overall molecular geometry to a favorable conformation. Subsequently, a number of research groups have engaged in SAR studies aimed at improving its potency and pharmacokinetic profile (Figure 13).
Figure 13. Compound 55 and related allophenylnorstatine analogs.

For example, pharmacokinetic studies on 55 revealed the compound was highly metabolized by human liver microsomes via glucuronidation of the P2 phenolic hydroxyl group and oxidation of the thiazolidine sulfur. Replacement of the phenol with an aniline group in the P2 site eliminates the possibility of glucuronidation. This modifcation led to 56 (SM-309515), which was less potent than 55 in terms of enzyme binding (Ki = 353 and 35 pM, respectively) but displayed more favorable antiviral activity in cells (EC50 = 77 nM) due to increased cell permeability. In addition, 56 displayed a markedly improved pharmokinetic profile as compared with 55 due to its increase resistance to metabolic degradation [48].
Scientists at Merck’s research laboratories developed a hybrid compound, which incorporated the P1–P3 attributes of 55 with the P1′–P3′ elements of IDV producing 57 as a highly potent PI (IC50 = 0.03 nM). Further structural refinements were aimed at identifying suitable surrogates for the thiazolidine ring. There work confirmed the importance of steric bulk introduced by the dimethyl group and led them to identify a spiro-oxazolidine system 58 as a viable alternative to the thiazolidine with comparable activity (IC50 = 0.1 nM) against wild-type and drug-resistant viral strains [49]. Ghosh and co-workers investigated a hybrid compound of 55 and 9. By combining their bis-THF urethane with the allophenylnorstatine they produced 59 (GRL-0355), which exhibited picomolar enzymatic activity (Ki = 5.2 pM) and low nanomolar antiviral activity (IC50 = 9 nM) [50].
Various other research groups have engaged in SAR studies based on JE-2147 with varying success. El-Koussi and co-workers explored variations of the P2 and P2′ ligands but were unable to improve antiviral activity [51]. Ami and co-workers replaced the P2/P3 ligands with novel derivatives of d-cysteine leading to inhibitor 60 with IC50 = 0.18 nM and demonstrated efficacy against drug-resistant strains [52]. Kiso and co-workers investigated P2′ replacements and identified a β-methallyl moiety as a novel small-sized phenyl isostere providing 61 (EC50 = 10 nM) [53]. Their work also demonstrated that modifying the compound’s hydrophobic character could be used to rationally improve membrane permeability and antiviral activity.
Heterocycles possessing a carbonyl
Oxazolidinones
The oxazolidinones are an important class of molecules that are commonly used in antibiotics such as Linezolid [27]. Recent work by Rana and co-workers replaced the P2 THF moiety of APV with N-phenyloxazolidinones to create a novel series of oxazolidinones inhibitors (Figure 14) [54]. Subsequent structural refinements of the P2 and P2′ phenyl rings led to the identification of 63 (Figure 14), which exhibited extremely high enzyme-inhibitory activity (Ki = 0.8 pm). Protein–inhibitor crystal structure analysis identified multiple interactions with the oxazolidinone group including a direct hydrogen-bonding interaction between the carbonyl oxygen and the amine of Asp-29, as well as water-mediated interactions between the oxazolidinone nitrogen and Gly-48, the carbonyl oxygen and the Asp-29 carboxylic acid side chain, and the oxygen in the ring to the amine of Asp-30 (Figure 14) [54]. In-cell antiviral analysis revealed a large drop in potency for 63 warranting further development work. SAR studies were completed to combine the N-phenyloxazolidinones with various dipeptide isosteres (64; Figure 14 ), however, activities decreased with Ki in the low nanomolar range and EC50 in the submicromolar region [55]. An extensive SAR that focused on modifying P2 and P2′ elements to modulate hydrophilic nature and improve antiviral activity revealed a number of insights into this class of molecules (65; Figure 14 ). For instance, the S configuration of the N-phenyloxazolidinone is preferred over the R while para substitutions of the N-phenyl ring were favored over meta substitutions. A variety of polar groups could be tolerated at the para substitution including amine, carbamate and methylsulfonyl providing compounds with antiviral activity comparable to lopinavir (LPV) with better performance against the multi-drug resistant mutant strain (low pM Ki and nM EC50). In addition, substitution of the P2′ ligand with a benzothiazole was generally preferred (65, EC50 = 7.3 nM) [56].
Figure 14. Oxazolidinone-based protease inhibitors.

Pyrrolidinone & pyrrolinone
Building on their previous work, Smith and co-workers recently reported a third generation of novel pyrrolinone-based inhibitors that incorporate a pyrrolinone into the P1′ site. Following a brief SAR study of the P2′ site, they identified 66 as a novel PI with subnanomolar enzyme inhibitory activity (Figure 15). The potency enhancement likely results from an additional hydrogen-bonding interaction formed between the P2′ benzylic carbamate and Asp-29′. Despite the observed improvement in enzyme activity, 66 displayed greatly reduced potency in cellular assays (IC50 = 0.44 nM) suggesting problems with cell permeability. When assayed against a panel of clinically significant HIV-1 mutants, 66 demonstrated comparable activity, thereby indicating the importance of this PI scaffold [57].
Figure 15. Protease inhibitors 66–69 with pyrrolidinones.

Recently, GlaxoSmithKline reported on a short SAR of P1 pyrrolidinone-based PI inhibitors as an extension of earlier studies [58]. Structural modifications of the P1/P1′ benzyl groups were made leading to the identification of multiple compounds 67 (Figure 15) with antiviral activities that surpass APV. Their most potent compound 68 (Ki = 0.02 nM, IC50 = 63 nM) introduced a morpholinoethoxy ether onto the P1′ benzyl group (X = H, Y = OCH2CON(CH2)O) [58].
An interesting example of pyrrolidinones as novel P1 ligands was reported by Ghosh and co-workers (69; Figure 15 ) [59,60]. The pyrrolidinones and oxazolidinones were incorporated into the P1′ site of DRV in order to form new hydrogen-bonding interactions with the enzyme and increase binding affinity. A preference for R pyrrolidinones was identified and new interactions between the pyrrolidinone amine with the carbonyl of Gly-27′ and a water-mediated interaction between the pyrrolidinone carbonyl with the Arg-8′ side chain were revealed by protein–ligand x-ray crystal structure. The most active compound of the series was 69 (Ki = 0.1 nM, IC50 = 26 μM). Interestlingly, 69 maintained nearly full potency against a panel of multidrug-resistant HIV-1 variants [59,60].
Imidazolidine-2,4-dione & morpholinone
As part of ongoing research into PIs, researchers at Abbott laboratories designed a series of hybrid compounds that incorporated the P1′–P2′ functionality of APV with the P2-P3 groups of LPV. One of the aims of the study was to identify a salvage agent for patients failing current treatments. A focal point of their work was the replacement of the P2 cyclic urea found in LPV. After screening multiple cyclic urea surrogates, they discovered that incorporating an imidazolidine-2,4-dione provided a series of cyclic imides, which displayed higher in-cell antiviral activity against wild-type HIV-1 and maintained potency against the drug-resistant viral strain A17, a mutant NIV-1 virus selected by and resistant to LPV [61,62]. Following an extensive SAR study, 70 (Figure 16) was identified as a promising compound with favorable antiviral activity against wild-type HIV-1 and the A17 mutant strain (EC50 = 1 and 5 nM, respectively) [61].
Figure 16. Protease inhibitors containing cyclic imide and morpholinone ligands.

Researchers at GlaxoSmithKline explored heterocyclic replacements for the P1-P2 ligand site of APV leading them to identify a series of morpholinone-based PIs (Figure 16). Following a brief SAR study that explored substitutions at C2 on the morpholinone ring, two highly active compounds 71 and 72 (Ki = 2 nM) were identified, thereby confirming the viability of this novel isostere [63].
Cyclic ureas, lactams & sulfamides
PIs containing seven-membered ring cyclic ureas were the subject of immense interest over the past decade [1,64]. Recent studies for this class of inhibitors have focused largely on quantitative SARs with relatively few new developments in their structural design. The latest research in this area has produced N-acyl azacylic ureas aimed at improving pharmacokinetic properties, a set of novel lactams synthesized via RCM, and a series of compounds incorporating a sulfamide isostere as a urea surrogate [65-67].
Aza-ureas & lactams
Azacyclic ureas have previously shown to be an interesting class of molecules that displayed good potency against wild-type HIV-1 and many drug-resistant strains [68,69]. However, as a class they have shown poor bioavailability due, in part, to metabolic degradation via oxidation of the carbon α to the aza linkage. In an effort to increase bioavailability, a series of N-acyl azacyclic ureas were synthesized by Abbott laboratories, which were specifically designed to block this metabolic oxidation (73; Figure 17 ) [65]. A brief SAR study identified a variety of acylated analogs possessing comparable cellular antiviral activity as their de-acylated counterparts. Furthermore, the acylated analogs (74; Figure 17; EC50 = 17 nM) maintained potency against drug-resistant strains (A17 and IND). However, low bioavailability was still observed for the acylated compounds [65].
Figure 17. Azacyclic ureas and cyclic lactams.

Recently, a series of novel seven-membered lactams (Figure 17) were reported by Abell and co-workers synthesized by RCM from amino acid derivatives. In general, their compounds demonstrated moderate enzyme inhibitory activity (μM) and further structural refinement is warranted. However, an interesting trend was observed in that their epoxide analogs 75 possessed higher activity than the syn diol equivalents [66].
Cyclic sulfamide
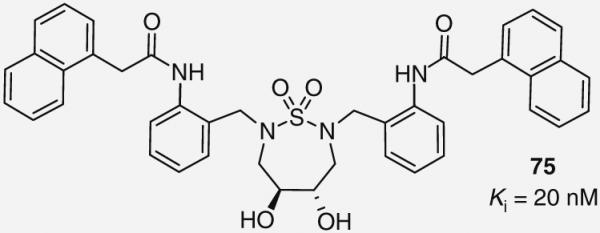
In contrast to cyclic ureas, cyclic sulfamides exhibit a nonsymmetric binding mode with the HIV-1 protease despite the symmetry found in both the ligand and enzyme. As such, Karlen and co-workers recently synthesized a series of cyclic sulfamides with varying ortho substitutions on the P2/P2′ benzyl groups [67]. These compounds were designed based on modeling studies that predicted elongation at the ortho site could provide additional enzyme interactions at S2/S2′ [67]. Although the initial studies failed to improve upon the lead, later work using microwave-based palladium-catalyzed coupling reactions to incorporate ortho amide functionality led to the discovery of a novel inhibitor 75 with Ki = 20 nM (Figure 18) [70]. Building on this success, additional SAR studies have been reported that explore asymmetric analogs. However, no further improvements in activity have been achieved [71].
Figure 18. Cyclic sulfamide protease inhibitor 75.
Benzoheterocycles
Benzodioxane
The most exciting example of benzoheterocycles incorporated into the structure of novel PIs can be found in the discovery of 43 (BRV) by GlakoSmithKline and 76 (GRL-98065), which was reported later by the Ghosh group [16,35]. BRV has been previously reported upon in the thiazole section of this review and as such only 46 will be discussed here. During SAR studies focused on replacement of the P2′ sulfonamide of DRV, Ghosh and co-workers identified the benzodioxane analog 76 as a highly potent PI (Ki < 5 pM and IC50 = 1.1 nM) with a unique profile against drug-resistant viral strains. Extensive biological testing shows that 76 maintains potency against a wide spectrum of clinically relevant drug-resistant strains (EC50 = 3–6 nM) while the commercially available PIs loss activity. Furthermore, the development of resistance to 76 was significantly delayed and the 76 selected strains did not show cross resistance to several commercial PIs [72]. This unique antiviral activity profile is likely related to its distinctive binding profile. X-ray analysis of 76 bound to the enzyme reveals the benzodioxane to be involved in a number of interactions (Figure 19) [73]. The dioxane oxygen atoms form hydrogen bonds with the amine and carboxylic acid of Asp-30′ as well as partake in a water-mediated hydrogen bond with the amine of Gly-48′. Additional hydrogen bonds occur between the carbonyls of Gly-48′ and Asp-30′ and the C-H atoms of the benzene ring. All of these interactions contribute to the high affinity of benzodioxane for the S2′ site and are likely responsible for 76 novel antiviral activity.
Figure 19. Proposed binding interactions of 76 benzodioxane ligand with HIV protease.

Oxyindoles & 2-benzoxazolinone
Ghosh and co-workers investigated the replacement of the P2′ sulfonamide of 9 with a novel oxyindole moiety at the P2′ site [74]. Modeling studies had suggested the oxyindole group would effectively fill the S2′ region and could lead to new hydrogen-bonding interactions with the tightly bound water molecule that coordinates with Ile-50 and Ile-50′. In addition, the basic amine of the oxyindole could have a favorable effect on the compounds absorption profile. A series of oxyindole analogs were prepared with 77 (Figure 20) displaying the most potent enzyme inhibitory effect (Ki = 7 nM). Unfortunately, 77 was much less potent than DRV.
Figure 20. Protease inhibitors 77 and 78.

Recently, a large series of benzoheterocycles were screened as novel P2 ligands by Yang and co-workers [75]. This work focused on using benzoheterocycles as a replacement for the bis-THF of 9. This most potent compound 78 (Figure 20) contains a 2-benzoxazolinone and displays antiviral activity in the low-nanomolar range (IC50 = 5 nM). Modeling studies suggest the carbonyl of the 2-benzoxazolinone is involved in hydrogen-bonding interactions with Asp-29 and Asp-30. In addition, the phenyl group appears to be involved in a π-bonding interaction with Ile-47.
Future perspective
The major obstacles of today’s HIV-1 PIs continue to be their poor pharmacological properties and development of viral resistance. Although recently approved inhibitors, DRV and tipranavir, exhibit marked improvements over the first generation PIs in these regards, there remains room for further improvements. In regards to drug resistance, the discovery of new inhibitor scaffolds targeting the protease backbone atoms will be of critical importance. Heterocyclic rings will play a major role in this work due to their conformational restriction as well as their ability to fill hydrophobic pockets and provide functionality for hydrogenbonding interactions. The bis-THF ligand in 9 or the tris-THF ligand in 18 appear to play an important role in inhibiting dimerization of the protease. Further exploration of protease dimerization inhibitors will likely lead to novel PIs with efficacy against multidrug-resistant strains. Thus, heterocyclic rings and design of molecular scaffolds with novel heterocycles will have a dominant place in future PI design due to their ability to modulate ADME properties without compromising activity. Improvements in pharmacokinetic properties may eventually lead to the discovery of novel PIs that do not require boosting.
Executive summary.
-
■
Cyclic ethers have been shown to play an important role in the development of structurally diverse protease inhibitors (PIs). Since the discovery of the bis-tetrahydrofuran ligand, various other unique cyclic ether P2 ligands have been explored including spirocyclic, meso-bicyclic, flexible polyethers, bicyclic and tricyclic systems. The tricyclic PI 18 has emerged as a promising new entity exhibiting potent antiviral activity against a range of viral strains that may be attributed in part to its ability to inhibit protease dimerization.
-
■
Heterocyclic triazoles, oxazoles and thiophenes are increasingly used as ligands in PI design. A notable achievement includes the identification of a novel thiophene-based allosteric HIV-1 PI from a high-throughput screening program. In addition, a novel series of triazole-based inhibitors have been prepared via click chemistry and offers a unique structural scaffold for further PI development.
-
■
The use of unsaturated heterocycles in PI design has led to two structurally distinct scaffolds. Pyrrolidine- and azepin-based inhibitors have incorporated a nitrogen group as the transition-state mimic in contrast to traditional PI designs that utilize the hydroxyl group. Thiazolidine has been used as a proline isostere in the P1′ position leading to a novel class of inhibitors with a unique viral profile that has attracted significant interest.
-
■
The design of urea-, lactam- and sulfamide-based PIs remains in the early stages of development with significant research effort devoted to improving their potency and bioavailability.
-
■
Oxazolidinones and the corresponding 2-benzoxazolinone derivatives have shown promise as novel P2 ligands leading to potent compounds with good antiviral activities profiles. Benzodioxane is an attractive P2′ ligand with a novel binding profile that contributes to its activity against multidrug-resistant viral strains.
Key Term.
Bioisosteres: Chemical substituents or groups of atoms with comparable chemical and physical properties, which when interchanged, produce broadly similar biological effects.
Acknowledgments
This work was supported by the NIH (GM53386). A significant portion of the authors’ recently published work has been discussed within this review.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Ghosh A, Anderson D, Mitsuya H. The FDA approved HIV-1 protease inhibitors for treatment of HIV/AIDS. In: Abraham DJ, Rotella DP, editors. Burger’s Medicinal Chemistry, Drug Discovery and Development. Vol. 7. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2010. pp. 1–74. ■■ Comprehensive review that describes the design and development of the currently approved HIV-1 protease inhibitors (PIs) in significant detail.
- 2.Roberts N, Martin J, Kinchington D, et al. Rational design of peptide-based HIV proteinase inhibitors. Science. 1990;248:358–361. doi: 10.1126/science.2183354. [DOI] [PubMed] [Google Scholar]
- 3.Kempf D, Sham H, Marsh K, et al. Discovery of ritonavir, a potent inhibitor of HIV protease with high oral bioavailability and clinical efficacy. J. Med. Chem. 1998;41:602–617. doi: 10.1021/jm970636+. [DOI] [PubMed] [Google Scholar]
- 4.Dorsey B, Levin R, McDaniel S, et al. L-735,524: the design of a potent and orally bioaviable HIV protease inhibitor. J. Med. Chem. 1994;37:3443–3451. doi: 10.1021/jm00047a001. [DOI] [PubMed] [Google Scholar]
- 5.Kaldor S, Kalish V, Davies J, et al. Viracept (nelfinavir mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997;40:3979–3985. doi: 10.1021/jm9704098. [DOI] [PubMed] [Google Scholar]
- 6.Tung R, Livingston B, Rao B, et al. Design and synthesis of amprenavir, a novel HIV protease inhibitor. In: Ogden RC, Flexner CW, editors. Protease inhibitors in AIDS therapy. Marcel Dekker, Inc.; New York, NY: 2001. pp. 101–118. [Google Scholar]
- 7.Falcoz C, Jenkins J, Bye C, et al. Pharmacokinetics of GW433908, a prodrug of amprenavir, in healthy male volunteers. J. Clin. Pharmacol. 2002;42:887–898. doi: 10.1177/009127002401102803. [DOI] [PubMed] [Google Scholar]
- 8.Bold G, Fassler A, Capraro H, et al. New aza-dipeptide analogues as potent and orally absorbed HIV-1 protease inhibitors: candidates for clinical development. J. Med. Chem. 1998;41:3387–3401. doi: 10.1021/jm970873c. [DOI] [PubMed] [Google Scholar]
- 9.Sham H, Kempf D, Molla A. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents. Chemother. 1998;42:3218–3224. doi: 10.1128/aac.42.12.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner S, Strohbach J, Tommasi R, et al. Tipranavir (PNU-140690): a potent, orally bioavailable nonpeptidic HIV protease inhibitor of the 5,6-dihydro-4-hydroxy-2-pyrone sulfonamide class. J. Med. Chem. 1998;41:3467–3476. doi: 10.1021/jm9802158. [DOI] [PubMed] [Google Scholar]
- 11.Koh Y, Ghosh A, Mitsuya H, et al. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh A, Krishnan K, Walters D, et al. Structure based design: novel spirocyclic ethers as nonpeptidal P2-ligands for HIV protease inhibitors. Bioorg. Med. Chem. Lett. 1998;8:687–690. doi: 10.1016/s0960-894x(98)00139-5. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh A, Chapsal B. Aspartic acid proteases as therapeutic targets: second-generation approved HIV protease inhibitors for the treatment of HIV/AIDS. In: Ghosh AK, editor. Methods and Principles in Medicinal Chemistry. Vol. 45. Wiley-VCH, Weinheim; Germany: 2010. pp. 169–195. [Google Scholar]
- 14.Ghosh A, Chapsal B, Weber I, Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res. 2008;41:78–86. doi: 10.1021/ar7001232. ■■ Reviews the novel design concept of backbone binding and its use in PI drug design to fight drug resistance.
- 15.Ghosh A, Chapsal B. Aspartic acid proteases as therapeutic targets: darunavir, a new PI with dual mechanism: from a novel drug design concept to new hope against drug-resistant HIV. In: Ghosh AK, editor. Methods and Principles in Medicinal Chemistry. Vol. 45. Wiley-VCH, Weinheim; Germany: 2010. pp. 205–235. [Google Scholar]
- 16.Ghosh A, Sridhar P, Kumaragurubaran N, et al. Bis-tetrahydrofuran: a privileged ligand for Darunavir and a new generation of HIV protease inhibitors that combat drug resistance. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. ■ Highlights the importance of the bis-tetrahydrofuran ligand and how it revolutionized PI design.
- 17.Ghosh A, Thompson J, McKee S, et al. Potent HIV protease inhibitors: the development of 3′-tetrahydrofuranglycine as P2-ligands and substituted pyrazine derivatives as P3-ligands. J. Med. Chem. 1993;36:2300–2310. doi: 10.1021/jm00068a006. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh A, Sridhar P, Leshchenko S, et al. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J. Med. Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh A, Martyr C, Mitsuya H, et al. Design of substituted bis-tetrahydrofuran (bis-THF)-derived potent HIV-1 protease inhibitors, protein-ligand X-ray structure, and convenient syntheses of bis-THF and substituted bis-THF ligands. Med. Chem. Lett. 2011;2(4):298–302. doi: 10.1021/ml100289m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A, Xu C, Rao K, et al. Probing multidrug-resistance and protein-ligand interactions with oxatricyclic designed ligands in HIV-1 protease inhibitors. ChemMedChem. 2010;5:1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh Y, Ghosh A, Mitsuya H, et al. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 2007;282:28709–28720. doi: 10.1074/jbc.M703938200. ■ Discovery of a novel protease inhibition mechanism that is only observed for certain PIs.
- 22.Ghosh A, Chapsal B, Baldridge A, et al. Design and synthesis of stereochemically defined novel spirocyclic P2-ligands for HIV-1 protease inhibitors. Org. Lett. 2008;10:5135–5138. doi: 10.1021/ol8020308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh A, Gemma S, Takayama J, et al. Potent HIV-1 protease inhibitors incorporating meso-bicyclic urethanes as P2-ligands: structure-based design, synthesis, biological evaluation and protein-ligand x-ray studies. Org. Biomol. Chem. 2008;6:3703–3713. doi: 10.1039/b809178a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh A, Gemma S, Baldridge A, et al. Flexible cyclic ethers/polyethers as novel P2-ligands for HIV-1 protease inhibitors: design, synthesis, biological evaluation, and protein-ligand x-ray studies. J. Med. Chem. 2008;51:6021–6033. doi: 10.1021/jm8004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh A, Chapsal B, Mitsuya H, et al. Design and synthesis of potent HIV-1 protease inhibitors incorporating hexahydrofuropyranol-derived high affinity P2 ligands: structure–activity studies and biological evaluation. J. Med. Chem. 2011;54(2):622–634. doi: 10.1021/jm1012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahn J, Boyle N, MacDonald M, Janda K. Peptidomimetics and peptide backbone modifications. Mini-Rev. Med. Chem. 2002;2:463–473. doi: 10.2174/1389557023405828. [DOI] [PubMed] [Google Scholar]
- 27.Ciapetti P, Giethlen B. Molecular variations based on isosteric replacements. In: Wermuth CG, editor. The Practice of Medicinal Chemistry. Third Edition Academic Press; St Louis, MO, USA: 2008. pp. 290–342. [Google Scholar]
- 28.Whiting M, Tripp J, Fokin V, et al. Rapid discovery and structure–activity profiling of novel inhibitors of human immunodeficiency virus type 1 protease enabled by the copper(I)-catalyzed synthesis of 1,2,3-triazoles and their further functionalization. J Med Chem. 2006;49:7697–7710. doi: 10.1021/jm060754+. [DOI] [PubMed] [Google Scholar]
- 29.Giffin M, Heaslet H, Torbett B, et al. A copper(I)-catalyzed 1,2,3-triazole azide-alkyne click compound is a potent inhibitor of a multidrug-resistant HIV-1 protease variant. J. Med. Chem. 2008;51:6263–6270. doi: 10.1021/jm800149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brik A, Wlodawer A, Goodsell D, et al. 1,2,-triazole as a peptide surrogate in the rapid synthesis of HIV-1 protease inhibitors. ChemBioChem. 2005;6:1167–1169. doi: 10.1002/cbic.200500101. [DOI] [PubMed] [Google Scholar]
- 31.Whiting M, Elder J, Fokin V, et al. Inhibitors of HIV-1 protease by using in situ click chemistry. Angew. Chem. Int. Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. ■ Describes a novel utilization of the protease enzyme to selectively synthesize PIs from a mix of fragments via click chemistry.
- 32.Jochim A, Miller S, Angelo N, Arora P. Evaluation of triazolamers as active site inhibitors of HIV-1 protease. Bioorg. Med. Chem. Lett. 2009;19:6023–6026. doi: 10.1016/j.bmcl.2009.09.049. [DOI] [PubMed] [Google Scholar]
- 33.Kim R, Rouse E, Chapman K, et al. P1′ oxadiazole protease inhibitors with excellent activity against native and protease inhibitor-resistant HIV-1. Bioorg. Med. Chem. Lett. 2004;14:4651–4654. doi: 10.1016/j.bmcl.2004.06.092. [DOI] [PubMed] [Google Scholar]
- 34.Lu Z, Bohn J, Rano T, et al. Orally bioavailable highly potent HIV protease inhibitors against PI-resistant virus. Bioorg. Med. Chem. Lett. 2005;15:5311–5314. doi: 10.1016/j.bmcl.2005.08.072. [DOI] [PubMed] [Google Scholar]
- 35.Miller J, Andrews C, Sherrill R, et al. Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg. Med. Chem. Lett. 2006;16:1788–1794. doi: 10.1016/j.bmcl.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 36.Yeung C, Klein L, Flentge C, et al. Oximinoarylsulfonamides as potent HIV protease inhibitors. Bioorg. Med. Chem. Lett. 2005;15:2275–2278. doi: 10.1016/j.bmcl.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 37.GlaxoSmithKline GlaxoSmithKline discontinues clinical development of investigational protease inhibitor brecanavir. Press release. 2006 December 18; [Google Scholar]
- 38.Bonini C, Chiummiento L, De Bonis M, et al. Synthesis, biological activity and modeling studies of two novel anti HIV PR inhibitors with a thiophene containing hydroxyethylamino core. Tetrahedron. 2005;61:6580–6589. [Google Scholar]
- 39.Bonini C, Chiummiento L, De Bonis M, et al. Synthesis of new thienyl ring containing HIV-1 protease inhibitors: promising preliminary pharmacological evaluation against recombinant HIV-1 proteases. J. Med. Chem. 2010;53:1451–1457. doi: 10.1021/jm900846f. [DOI] [PubMed] [Google Scholar]
- 40.Chang M, Giffin M, Savage J, et al. Identification of broad-based HIV-1 protease inhibitors from combinatorial libraries. Biochem. J. 2010;429:572–532. doi: 10.1042/BJ20091645. ■■ Describes the first reported allosteric-type PI.
- 41.Specker E, Bottcher J, Klebe G, et al. An old target revisited: two new privileged skeletons and an unexpected binding mode for HIV-protease inhibitors. Angew. Chem. Int. Ed. 2005;44:3140–3144. doi: 10.1002/anie.200462643. [DOI] [PubMed] [Google Scholar]
- 42.Specker E, Bottcher J, Klebe G, et al. Unexpected novel binding mode of pyrrolidine-based aspartyl protease inhibitors: design, synthesis and crystal structure in complex with HIV protease. ChemMedChem. 2006;1:106–117. doi: 10.1002/cmdc.200500008. [DOI] [PubMed] [Google Scholar]
- 43.Bottcher J, Blum A, Klebe G, et al. Targeting the open-flap conformation of HIV-1 protease with pyrrolidine-based inhibitors. ChemMedChem. 2008;3:1337–1344. doi: 10.1002/cmdc.200800113. [DOI] [PubMed] [Google Scholar]
- 44.Blum A, Bottcher J, Diederich W, et al. Structure-guided design of C2-symmetric HIV-1 protease inhibitors based on a pyrrolidine scaffold. J. Med. Chem. 2008;51:2078–2087. doi: 10.1021/jm701142s. [DOI] [PubMed] [Google Scholar]
- 45.Bottcher J, Blum A, Heine A, Diederich W, Klebe G. Structural and kinetic analysis of pyrrolidine-based inhibitors of the drug-resistant Ile84Val mutant of HIV-1 protease. J. Mol. Biol. 2008;383:347–357. doi: 10.1016/j.jmb.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 46.Brass S, Chan N, Diederich W, et al. Synthesis of 2,3,4,6-tetrahydro-1H-azepines as privileged ligand scaffolds for the design of aspartic protease inhibitors via a ring-closing metathesis approach. J. Orgmet. Chem. 2006;691:5406–5422. [Google Scholar]
- 47.Mimoto T, Terashima K, Nojima S, et al. Structure-activity and structure-metabolism relationships of HIV protease inhibitors containing the 3-hydroxy-2-methylbenzoyl-allophenylnorstatine structure. Biorg. Med. Chem. 2004;12:281–293. doi: 10.1016/j.bmc.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 48.Mimoto T, Nojima S, Terashima K, et al. Structure-activity relationships of novel HIV-1 protease inhibitors containing the 3-amino-2-chlorobenzoyl-allophenylnorstatine structure. Bioorg. Med. Chem. Lett. 2008;16:1299–1308. doi: 10.1016/j.bmc.2007.10.062. [DOI] [PubMed] [Google Scholar]
- 49.Raghavan S, Lu Z, Beeson T, et al. Synthesis of novel HIV protease inhibitors (PI) with activity against PI-resistant virus. Bioorg. Med. Chem. Lett. 2007;17:5432–5436. doi: 10.1016/j.bmcl.2007.07.040. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh A, Gemma S, Simoni E, et al. Synthesis and biological evaluation of novel allophenynorstatine-based HIV-1 protease inhibitors incorporating high affinity P2-ligands. Bioorg. Med. Chem. Lett. 2010;20:1241–1246. doi: 10.1016/j.bmcl.2009.11.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abdel-Rahman H, El-Koussi N, Alkaramany G, et al. Allophenylnorstatine-containing HIV-1 protease inhibitors: desing, synthesis and structure-activity relationships for selected P2 ligands. Bull. Pharm. Sci. 2005;28:95–103. [Google Scholar]
- 52.Ami E, Nakahara K, Kiso Y, et al. Synthesis and antiviral property of allophenylnorstatine-based HIV protease inhibitors incorporating d-cysteine derivatives as P2/P3 moieties. Bioorg. Med. Chem. Lett. 2007;17:4213–4217. doi: 10.1016/j.bmcl.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 53.Hidaka K, Kimura T, Kiso Y, et al. Small-sized human immunodeficiency virus type-1 protease inhibitors containing allophenylnorstatine to explore the S2′ pocket. J. Med. Chem. 2009;52:7604–7617. doi: 10.1021/jm9005115. [DOI] [PubMed] [Google Scholar]
- 54.Ali A, Reddy G, Rana T, et al. Discovery of HIV-1 protease inhibitors with picomolar affinities incorporating N-aryl-oxazolidinone-5-carboxamides as novel P2 ligands. J. Med. Chem. 2006;49:7342–7356. doi: 10.1021/jm060666p. [DOI] [PubMed] [Google Scholar]
- 55.Reddy G, Ali A, Rana T, et al. Design and synthesis of HIV-1 protease inhibitors incorporating oxazolidinones as P2/P2′ ligands in pseudosymmetric dipeptide isosteres. J. Med. Chem. 2007;50:4316–4328. doi: 10.1021/jm070284z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali A, Schiffer C, Rana T, et al. Structure-based design, synthesis, and structure–activity relationship studies of HIV-1 protease inhibitors incorporating phenyloxazolidinones. J. Med. Chem. 2010;53:7699–7708. doi: 10.1021/jm1008743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith A, Charnley A, Harada H, et al. Design, synthesis, and biological evaluation of monopyrrolinone-based HIV-1 protease inhibitors possessing augmented P2′ side chains. Bioorg. Med. Chem. Lett. 2006;16:859–863. doi: 10.1016/j.bmcl.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 58.Sherrill R, Andrews C, Bock W. Optimization of pyrrolidinone based HIV protease inhibitors. Bioorg. Med. Chem. Lett. 2005;15:81–84. doi: 10.1016/j.bmcl.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 59.Koh Y, Das D, Mitsuya H, et al. GRL-02031, a novel nonpeptidic protease inhibitor (PI) containing a Stereochemically defined fused cyclopentanyltetrahydrofuran potent against multi-PI-resistant human immunodeficiency virus type 1 in vitro. Antimicrob. Agents Chemother. 2009;52:3902–3914. doi: 10.1128/AAC.00689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghosh A, Leshchenko-Yashchuk S, Anderson D, et al. Design of HIV-1 protease inhibitors with pyrrolidinones and oxazolidinones as novel P1′-ligands to enhance backbone-binding interactions with protease: synthesis, biological evaluation, and protein–ligand x-ray studies. J. Med. Chem. 2009;52:3902–3914. doi: 10.1021/jm900303m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flosi W, DeGoey D, Grampovnik D, et al. Discovery of imidazolidine-2,4-dione-linked HIV protease inhibitors with activity against lopinavir-reistant mutant HIV. Bioorg. Med. Chem. 2006;14:6695–6712. doi: 10.1016/j.bmc.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 62.Randolph J, Huang P, Flosi W, et al. Synthesis, antiviral activity, and pharmacokinetic evaluation of P3 pyridylmethyl analogs of oximinoarylsulfonyl HIV-1 protease inhibitors. Bioorg. Med. Chem. 2006;14:4035–4046. doi: 10.1016/j.bmc.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 63.Kazmierksi W, Furfine E, Spaltenstein A, Wright L. New, potent P1/P2-morpholinone-based HIV-protease inhibitors. Bioorg. Med. Chem. Lett. 2006;16:5226–5230. doi: 10.1016/j.bmcl.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 64.Chrusciel R, Nicholas J, Thaisrivongs S. HIV protease inhibitors in early development. In: Ogden RC, Flexner CW, editors. Protease Inhibitors in AIDS Therapy. Marcel Dekker, Inc.; NY, USA: 2001. pp. 119–138. [Google Scholar]
- 65.Zhao C, Sham H, Sun M, et al. Synthesis and activity of N-acyl azacyclic urea HIV-1 protease inhibitors with high potency against multiple drug resistant viral strains. Bioorg. Med. Chem. Lett. 2005;15:5499–5503. doi: 10.1016/j.bmcl.2005.08.093. [DOI] [PubMed] [Google Scholar]
- 66.Zaman S, Campaner P, Abell A. Synthesis of amino acid derived seven-membered lactams by RCM and their evaluation against HIV protease. Bioorg. Med. Chem. Lett. 2006;14:8323–8331. doi: 10.1016/j.bmc.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 67.Ax A, Schaal W, Vrang L, Samuelsson B, Hallberg A, Karlen A. Cyclic sulfamide HIV-1 protease inhibitors, with sidechains spanning from P2/P2′ to P1/P1′. Bioorg. Med. Chem. 2005;13:755–764. doi: 10.1016/j.bmc.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 68.Sham H, Zhao C, Marsh K, et al. Novel azacyclic ureas that are potent inhibitors of HIV-1 protease. Biochem. Biophys. Res. Commun. 1996;225:436–440. doi: 10.1006/bbrc.1996.1191. [DOI] [PubMed] [Google Scholar]
- 69.Sham H, Zhao C, Stewart K, et al. A novel, picomolar inhibitor of human immunodeficiency virus type 1 protease. J. Med. Chem. 1996;39:392–397. doi: 10.1021/jm9507183. [DOI] [PubMed] [Google Scholar]
- 70.Gold H, Ax A, Larhed M, et al. Fast and selective synthesis of novel cyclic sulfamide HIV-1 protease inhibitors under controlled microwave heating. Tetrahedron. 2006;62:4671–4675. [Google Scholar]
- 71.Ax A, Joshi A, Larhed M, et al. Synthesis of a small library of non-symmetric cyclic sulfamide HIV-1 protease inhibitors. Tetrahedron. 2010;66:4049–4056. [Google Scholar]
- 72.Amano M, Koh Y, Mitsuya H, et al. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 2007;51:2143–2155. doi: 10.1128/AAC.01413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Tie Y, Weber I, et al. Potent new antiviral compounds shows similar inhibition and structural interactions with drug resistant mutants and wild type HIV-1 protease. J. Med. Chem. 2007;50:4509–4515. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghosh A, Schiltz G, Perali R, et al. Design and synthesis of novel HIV-1 protease inhibitors incorporating oxyindoles as the P′2-ligands. Bioorg. Med. Chem. Lett. 2006;16:1869–1873. doi: 10.1016/j.bmcl.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 75.He M, Zhang H, Yang M, et al. Design, biological evaluation, and SAR of novel pseudo-peptide incorporating benzheterocycles as HIV-1 protease inhibitors. Chem Biol. Drug Des. 2010;76:174–180. doi: 10.1111/j.1747-0285.2010.00995.x. [DOI] [PubMed] [Google Scholar]