

Abstract
Checkpoints block cell cycle progression in eukaryotic cells exposed to DNA damaging agents. We show that several Drosophila homologs of checkpoint genes, mei-41, grapes, and 14-3-3ε, regulate a DNA damage checkpoint in the developing eye. We have used this assay to show that the mutagen-sensitive gene mus304 is also required for this checkpoint. mus304 encodes a novel coiled-coil domain protein, which is targeted to the cytoplasm. Similar to mei-41, mus304 is required for chromosome break repair and for genomic stability. mus304 animals also exhibit three developmental defects, abnormal bristle morphology, decreased meiotic recombination, and arrested embryonic development. We suggest that these phenotypes reflect distinct developmental consequences of a single underlying checkpoint defect. Similar mechanisms may account for the puzzling array of symptoms observed in humans with mutations in the ATM tumor suppressor gene.
Keywords: Cell cycle, checkpoint, DNA damage, Drosophila, development
Signal-transduction pathways known as checkpoints help eukaryotic cells recognize and react to DNA damage. Both budding yeast and fission yeast respond to DNA damage by delaying cell cycle progression (Weinert and Hartwell 1988; Kitazono and Matsumoto 1998; Rhind and Russell 1998; Weinert 1998). This delay provides time for DNA repair prior to replication in S phase or to chromosome segregation in M phase. These checkpoints are initiated by a group of sensor proteins that may directly interact with damaged DNA. These sensor proteins help to activate a kinase of the ATM protein kinase family that is closely related to the human tumor suppressor gene ATM (Savitsky et al. 1995). Downstream components, such as the Chk1 and Cds1 kinases of Schizosaccharomyces pombe, then act on regulators of the cell cycle.
The Cdc2 kinase is the best characterized of the checkpoint targets (Weinert 1997). Dephosphorylation of Cdc2 by Cdc25 and association of Cdc2 with a cyclin subunit are both required for the G2/M transition of the cell cycle. In fission yeast, the Chk1 kinase acts with the 14-3-3 protein Rad24 to inactivate Cdc25 and help prevent Cdc2 dephosphorylation (Lopez-Girona et al.1999). In mammalian cells, both Cdc2 phosphorylation as well as the subcellular localization of Cyclin B are regulated by a DNA damage checkpoint (Jin et al. 1998; Toyoshima et al. 1998).
Whereas yeast checkpoint mutants typically exhibit sensitivity to agents that damage DNA or inhibit replication, disruption of checkpoint function in multicellular organisms may have more pleiotropic effects. Mutation of the human checkpoint gene ATM is responsible for the inherited syndrome ataxia telangiectasia (A-T) (Savitsky et al. 1995; Lavin and Shiloh 1997). ATM regulates DNA damage checkpoints at multiple points in the cell cycle. Some of the targets of ATM during checkpoint control include p53 and the Chk1 and Chk2 kinases (Westphal 1997; Matsuoka et al. 1998). Among the diverse symptoms of A-T, radiation sensitivity and a high incidence of lymphomas are easily accounted for by a cellular defect in responding to DNA damage; however, other features, including neurodegeneration and telangiectasias (dilated blood vessels) are not clearly related to DNA repair defects (Hawley and Friend 1996; Morgan and Kastan 1997; Rotman and Shiloh 1998; Brown and Chakravarti 1999). This range of symptoms may indicate that ATM acts in additional signal-transduction pathways or may reflect aspects of physiology that are unusually sensitive to checkpoint defects.
Drosophila homologs of several yeast checkpoint genes have been identified by cloning meiotic and developmental mutants. mei-41 was identified as a gene required for normal meiosis and DNA repair (Baker and Carpenter 1972; Boyd et al. 1976). Molecular cloning of mei-41 revealed homology to the human ATM, budding yeast MEC1, and fission yeast rad3 checkpoint genes (Hari et al. 1995). mei-41 is required for a DNA damage checkpoint in the Drosophila brain, indicating that mei-41 is a functional ATM homolog (Hari et al. 1995). grapes (grp), a homolog of the S. pombe gene chk1, was identified as a maternal effect gene required for normal cell cycle progression during the nuclear divisions preceding cellularization of the embryo (Fogarty et al. 1994, 1997; Sibon et al. 1997; Su et al. 1999). grp and mei-41 are both required in the embryo to delay mitosis following inhibition of DNA replication (Sibon et al. 1997, 1999). These studies provided the first evidence that checkpoint homologs play an essential role during Drosophila embryogenesis. In contrast, another Drosophila checkpoint homolog, 14-3-3ε, was isolated as a component of the Ras signal-transduction pathway, but is not required for either viability or fertility (Chang and Rubin 1997).
In this study, we show that there is a DNA damage checkpoint in the developing Drosophila eye and that mei-41, grp, and 14-3-3ε have defects in this checkpoint. We also describe a new checkpoint gene, mus304, identified from a collection of mutagen-sensitive mutants. mus304 encodes a novel, cytoplasmic protein with a predicted coiled-coil domain. Genetic analysis indicates that mus304 is not required for viability and acts in the mei-41 pathway. mus304 shares several phenotypes with mei-41, including defective chromosome break repair and a high level of genomic instability. Loss of zygotic mus304 function leads to bristle defects and decreased meiotic recombination. Embryos from mus304 mothers undergo a disruption in the early cell cycles leading to chromosome damage and a block in embryonic development. Our results indicate that a DNA damage checkpoint pathway containing mus304 is required for genomic stability and several specific aspects of development.
Results
A DNA damage checkpoint in the developing eye
We examined a G2/M DNA damage checkpoint in the developing Drosophila eye. Differentiation in the eye is particularly sensitive to disruptions in cell cycle control; even mild alterations in cell cycle gene function can result in a rough eye phenotype. The third instar eye disc is an epithelium with three populations of cells: postmitotic, dividing asynchronously, or dividing synchronously in response to a developmental signal (Wolff and Ready 1993; Bonini and Choi 1995). Mitotic cells can be identified by their condensed chromosomes, apical position in the epithelium, or by staining with an antibody to phospho-histone H3 (PH3) (Hendzel et al. 1997).
We determined the number of mitotic cells in untreated or irradiated eye discs. Approximately 100 mitotic cells were stained with the anti-PH3 antibody in a wild-type disc (Fig. 1A). One hour following X-irradiation, very few cells were in M phase (Fig. 1B). Detection of mitotic cells by DAPI staining confirms that the irradiation-induced checkpoint blocks mitosis, not simply the PH3 epitope (not shown). The same experiments were carried out using mei-41 animals. The number of mitotic cells was unchanged following irradiation of mei-41 discs (Fig. 1C) indicating that the DNA damage checkpoint is inactive in these cells. We obtained similar results using the developing wing and legs for the checkpoint assay (data not shown). In unirradiated mei-41 animals, the morphology of the eye disc as well as the adult eye is normal, indicating that this gene is not required for cell cycle progression.
Figure 1.
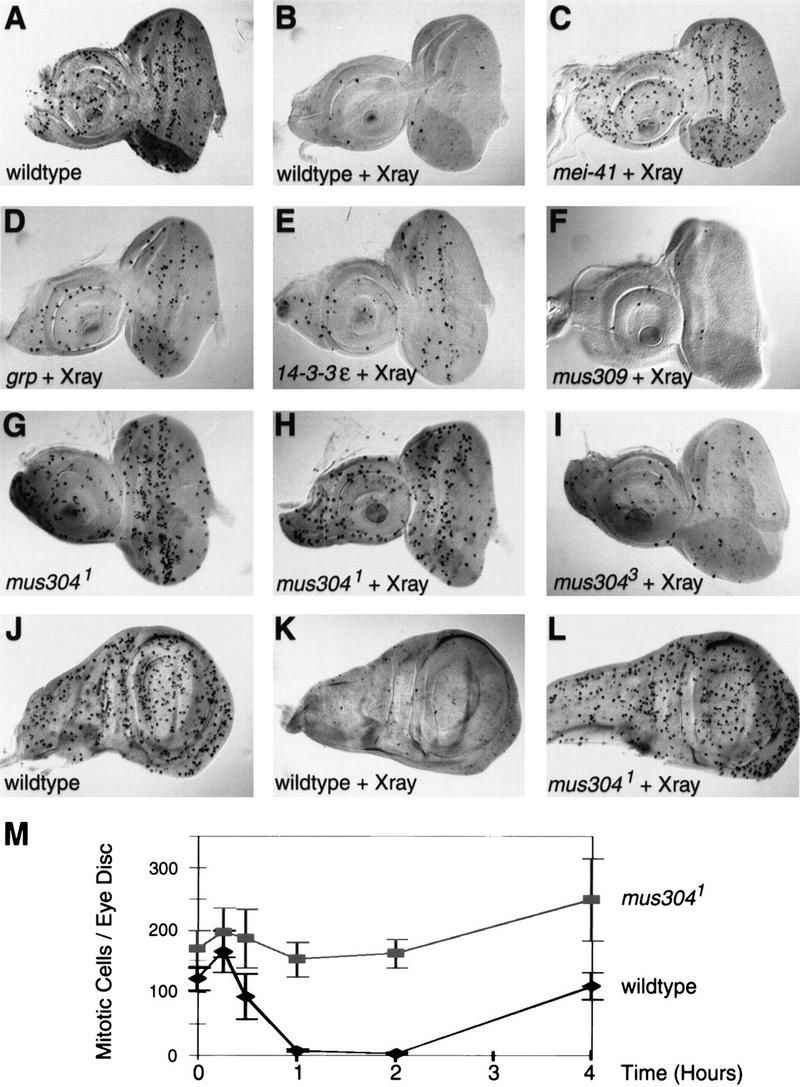
A DNA damage checkpoint in wild-type and mutant imaginal discs. Larvae were either unirradiated (A,G,J) or irradiated with 4000 rads of X-rays (B–F, H–I, K–L). Discs were dissected and fixed 1 hr following irradiation. Histochemical staining with a mitotic-specific antibody reveals the number and distribution of mitotic cells. The genotype of each disc is indicated. Alleles are listed in Materials and methods. (A–I) Eye-antennal imaginal discs with anterior left and dorsal on top. In each disc, the round tissue on the left is the antennal disc and the oval tissue to the right is the eye disc. (J–L) Wing imaginal discs with the future notum to the left and the wing pouch to the right. (M) A time course of mitotic cells in irradiated wild-type and mus3041/Df(3L)WR4 eye discs. Irradiation with 4000 rads was performed at time zero. Error bars, s.e.m..
grp and 14-3-3ε help regulate this DNA damage checkpoint. In the absence of irradiation, grp and 14-3-3ε animals exhibit normal external appearance, normal disc morphology, and normal numbers of mitotic cells (not shown). Following irradiation of grp and 14-3-3ε discs, the number of mitotic cells was greater than found in irradiated wild-type discs, but less than found in irradiated mei-41 discs (Fig. 1D,E). In contrast, mutations in mus309, the Drosophila homolog of Ku p70 (Beall and Rio 1996), do not affect the checkpoint (Fig. 1F).
mus304 is required for the DNA damage checkpoint
We used the eye disc assay to identify a new checkpoint gene, mus304. In budding yeast, checkpoint genes were identified within collections of radiation-sensitive (rad) mutants (Weinert and Hartwell 1988, 1989). Previously, a collection of Drosophila DNA repair mutants was generated in screens for mutagen-sensitive mutants (Boyd et al. 1987). To identify new checkpoint mutants, we examined these mutants for a checkpoint defect (M.H. Brodsky and G.M. Rubin, unpubl.). We found that mutations in a locus designated mutagen-sensitive 304, abbreviated mus304 (Boyd et al. 1981) disrupt the checkpoint.
We examined mitotic cells in untreated and irradiated mus304 discs. We found that untreated mus304 discs had a wild-type number and distribution of mitotic cells and normal disc morphology (Fig. 1G,M). Following irradiation, the number of mitotic cells was unchanged (Fig. 1H,M). A time course of mitotic cell number following irradiation demonstrated that the presence of mitotic cells in mus304 discs is not a result of either a delayed onset of or a rapid recovery from cell cycle arrest (Fig. 1M). Thus, mus304, like mei-41, is essential for the G2/M checkpoint in the developing eye.
Three alleles of mus304 were isolated in the original mutagen sensitivity screens. On the basis of their sensitivity to mutagen exposure, the mus3041 and mus3042 alleles are stronger than mus3043. Consistent with this result, we found that mus3041 (Fig. 1H) and mus3042 exhibited a strong defect in the eye disc assay, whereas mus3043 (Fig. 1I) only showed a partial loss of the checkpoint. mus304 was also required for DNA damage-induced cell cycle arrest in the wing disc (Fig. 1J–L), leg disc, and other imaginal discs (not shown) examined. Thus, similar to the other checkpoint mutants described above, mus304 is required for the DNA damage checkpoint in all dividing larval cells we have examined.
We used a noncomplementation screen to identify three additional alleles of mus304 with strong loss-of-function phenotypes (see Materials and Methods). Unlike the original alleles of mus304, these new alleles were isolated in a screen that does not require viability. However, all three new alleles (mus3044, mus3045, and mus3046) are homozygous viable, suggesting that zygotic expression of mus304 is not required for viability. Consistent with this conclusion, several previous screens for recessive lethal or visible mutations in the genetic region including mus304 failed to recover any alleles of mus304 (Abbott and Lengyel 1991; Grether et al. 1995).
Genetic Interaction of mus304 with E(mus304) and mei-41
mus304 adults have wild-type eyes (Fig. 2A,B), but animals homozygous for mus304 and heterozygous for a spontaneous mutation in a second locus have rough eyes (Fig. 2C) and abnormal or missing bristles (not shown). This locus, Enhancer of mus304 [E(mus304)] maps to cytological position 93D–F and is required for viability (M.H. Brodsky, unpubl.). Although the origin of the eye and bristle phenotypes are unclear, previous studies (for review, see Ashburner 1989) have reported that irradiation of developing Drosophila with sublethal doses of X-rays causes similar eye (Fig. 2F) and bristle phenotypes. The synthetic phenotype of mus304 with E(mus304) indicates that the Drosophila eye is particularly sensitive to checkpoint defects in some genetic backgrounds.
Figure 2.
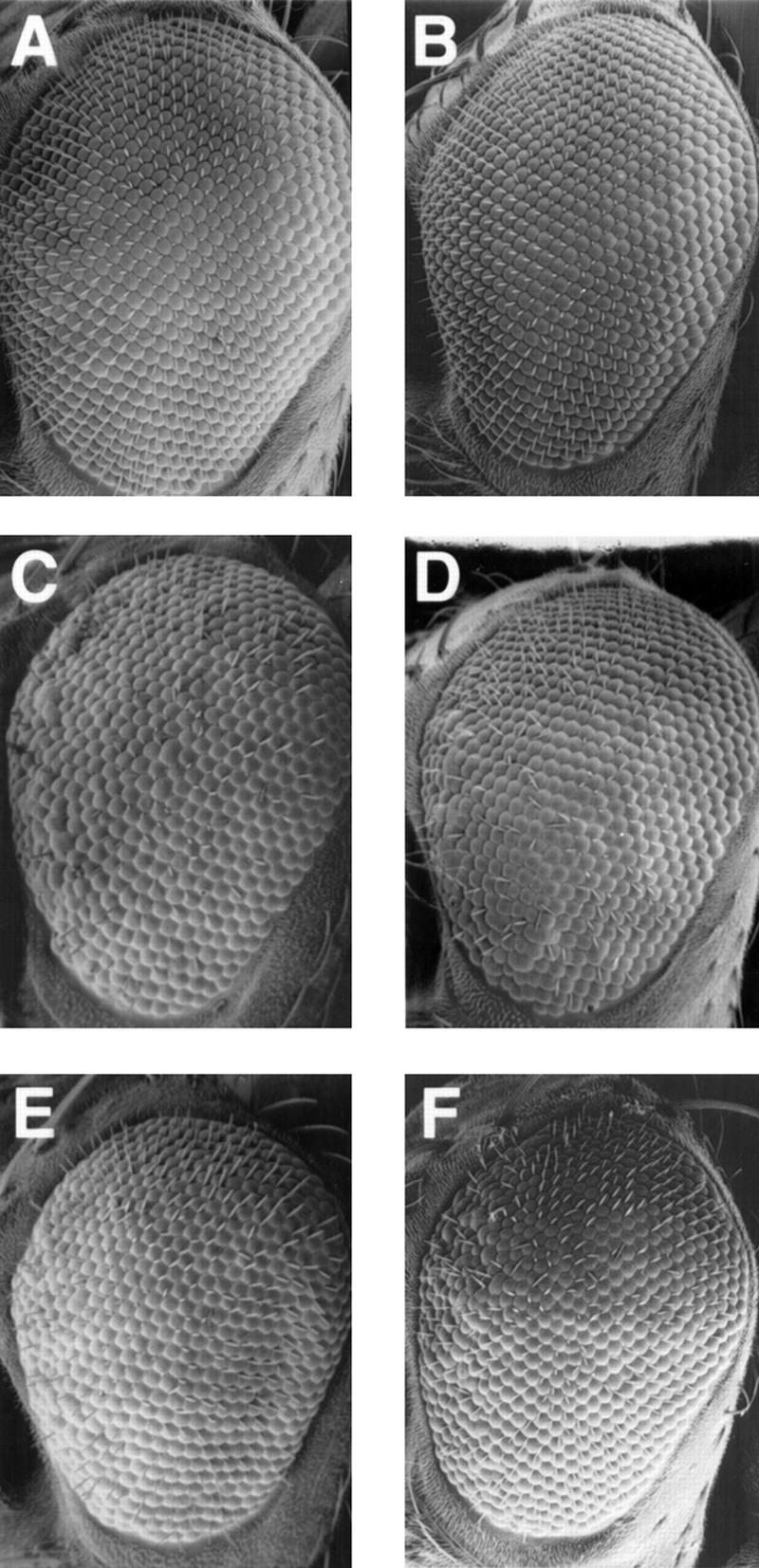
Rough eye phenotypes in checkpoint mutants. Scanning electron micrographs of adult male eyes are shown with anterior to the left and dorsal on top. (A) Wild type. (B) mus3041/mus3042. (C) mus3041, +/mus3042, E(mus304). (D) mei-4129D/Y; +/E(mus304)1. (E) 4129D/Y; mus3041, +/mus3042, E(mus304). (F) Wild type following irradiation with 2000 rads during third instar.
We compared the E(mus304)-dependent rough eye phenotypes among mus304 animals, mei-41 animals, and mei-41; mus304 double-mutant animals. In these experiments, we used genetic null alleles of mei-41 and mus304. As with mus304, animals homozygous for mei-41 mutations have wild-type eyes, whereas animals homozygous for mei-41 and heterozygous for E(mus304) have rough eyes and missing bristles (Fig. 2D). If mei-41 and mus304 act in distinct pathways, the double-mutant phenotype is predicted to be greater than that seen for each single mutant. However, the eye phenotypes of animals with mutations in either mus304 or mei-41 were similar to that of animals with mutations in both genes (Fig. 2C–E). This result combined with the similarity of other mei-41 and mus304 phenotypes suggests that these genes act in the same genetic pathway.
mus304 is required for DNA repair and genomic stability
Checkpoint genes are thought to promote DNA repair by delaying cell cycle progression in the presence of damaged DNA. We used a cytological assay (Gatti et al. 1994) to test whether mus304 is required for chromosome break repair in dividing cells. Metaphase spreads from larval neuroblasts were examined for chromosome aberrations following X-ray irradiation. The frequency of chromosome aberrations in mus304 cells was threefold greater than in wild type (Table 1).
Table 1.
Spontaneous and induced chromosome aberrations in wild-type and mus304 mutant larvae
| Genotype
|
X-raya
|
Aberration frequencyb
|
|---|---|---|
| +/+ | − | 0.7 (2/264) |
| mus3041/Df(3L)WR4 | − | 9.6 (28/291) |
| +/+ | + | 19 (49/262) |
| mus3041/Df(3L)WR4 | + | 60 (192/318) |
Larvae were treated with either 0 or 220 rads of X-rays. Four hours later, larval neuroblasts were squashed and examined for chromosome aberrations.
The average number of chromosome aberrations found per 100 metaphase cells. The total number of breaks scored over the total number of metaphase cells examined are indicated in parentheses.
We also measured the frequency of spontaneous chromosome breaks in mus304 animals. Metaphase spreads from unirradiated wild-type and mus304 larval neuroblasts were examined for chromosome aberrations. The spontaneous frequency of chromosome aberrations in mus304 animals was >10-fold greater than in wild-type animals (Table 1). The majority of aberrations (26/28) were chromatid or isochromatid breaks.
The cytological assay described above measures the steady-state level of mitotic cells with chromosome breaks. To measure the frequency of post-mitotic cells that lost genetic material during development, we used a loss-of-heterozygosity (LOH) assay based on the multiple wing hair (mwh) gene (Baker et al. 1978; Fig. 3A–D). LOH is greatly elevated in mus304 mutants (Fig. 3E). In a wild-type background, the number of mwh cells per wing was between zero and two (mean = 0.4, s.e.m. = 0.2). This is likely an overestimate of the rate of wild-type LOH, as a small number of cells with multiple hairs appear in animals that have two copies of the wild-type mwh gene (Baker et al. 1978). In the strongest alleles of mus304, there was a nearly 200-fold increase in the level of LOH. In mus3041/Df(3L)WR4 wings there were 75 (s.e.m. = 3) clones of cells with LOH at the mwh locus (Fig. 3E), representing a total of 106 (s.e.m. = 6) mwh cells. In the weakest allele, mus3043, there was a sixfold increase in LOH compared with wild type.
Figure 3.
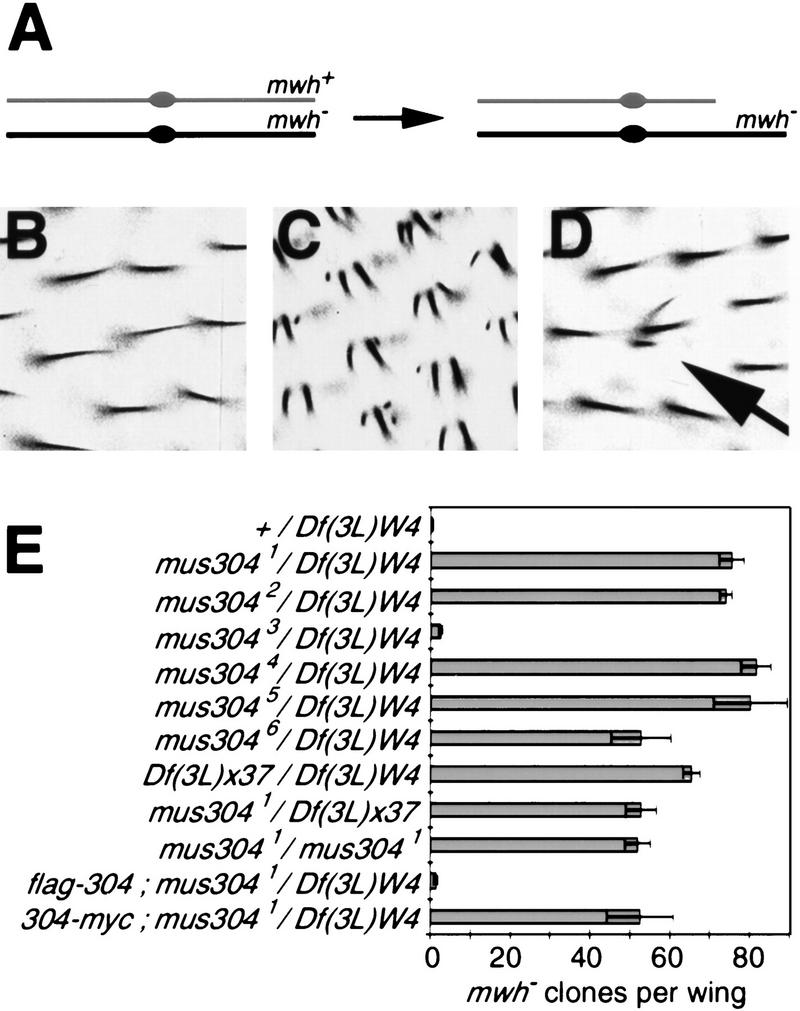
High rates of LOH in mus304 animals. (A) The genetic basis of the LOH assay. The fertilized egg is heterozygous for a mutation in the mwh gene. If the wild-type copy of the gene is lost during the proliferation of a wing cell precursor, that cell's progeny will exhibit the recessive mwh phenotype. Loss of the wild-type gene can occur because of a chromosome break (shown here), point mutation, chromosome loss, or nondisjunction. (B) Individual wing hairs in an animal heterozygous for mwh. Each hair cell has one hair. There are ∼20,000 hair cells per wing. (C) In an animal homozygous for mwh, each hair cell has two or more hairs. (D) In an animal heterozygous for mwh and homozygous for a mutation that increases genomic instability, individual cells that lost the wild-type mwh gene (arrow) can be seen within a field of heterozygous cells. (E) The frequency of mwh clones per wing in wild-type and mus304 animals. Error bars, s.e.m..
We used this assay to ask whether the strong alleles behave like deficiencies for the locus (Fig. 3E). We found that the level of LOH of mus3041 varied somewhat in combination with different deficiencies, possibly because of other genes contained within the deficiency. The smallest available deficiency including mus304 is Df(3L)x37 (Segraves 1988). The phenotype of mus3041 when homozygous was similar to mus3041/Df(3L)x37. In addition, Df(3L)x37/Df(3L)WR4 animals and mus3041/Df(3L)WR4 animals also have the mus3041similar phenotypes. These results indicate that this allele behaves as a genetic null mutation.
Bristle defects in mus304 adults
Despite a high frequency of genetic loss, mus304 adults have largely normal external morphology. However, close examination of these animals revealed a significant fraction of the large bristles, known as macrochaetae, were missing or defective (Fig. 4). Normally, each macrochaete has a characteristic position and length (Fig. 4B). In mus304 animals, some macrochaetae were entirely missing, whereas others were shorter and thinner (Fig. 4C), resembling macrochaetae in animals mutant for a Minute locus (Ashburner 1989). The frequency of defective or missing macrochaetae was scored for the head and thorax (see Materials and Methods). No defective bristles were found in >50 wild-type animals examined. In strong alleles of mus304, 5%–10% of macrochaetae are defective (Fig. 4D). The frequency approached 30% in mus304 animals that were also heterozygous for E(mus304). The bristle defects in checkpoint mutants appear to reflect the loss of genetic material at haploinsufficient loci (see Discussion; Fig. 4A).
Figure 4.
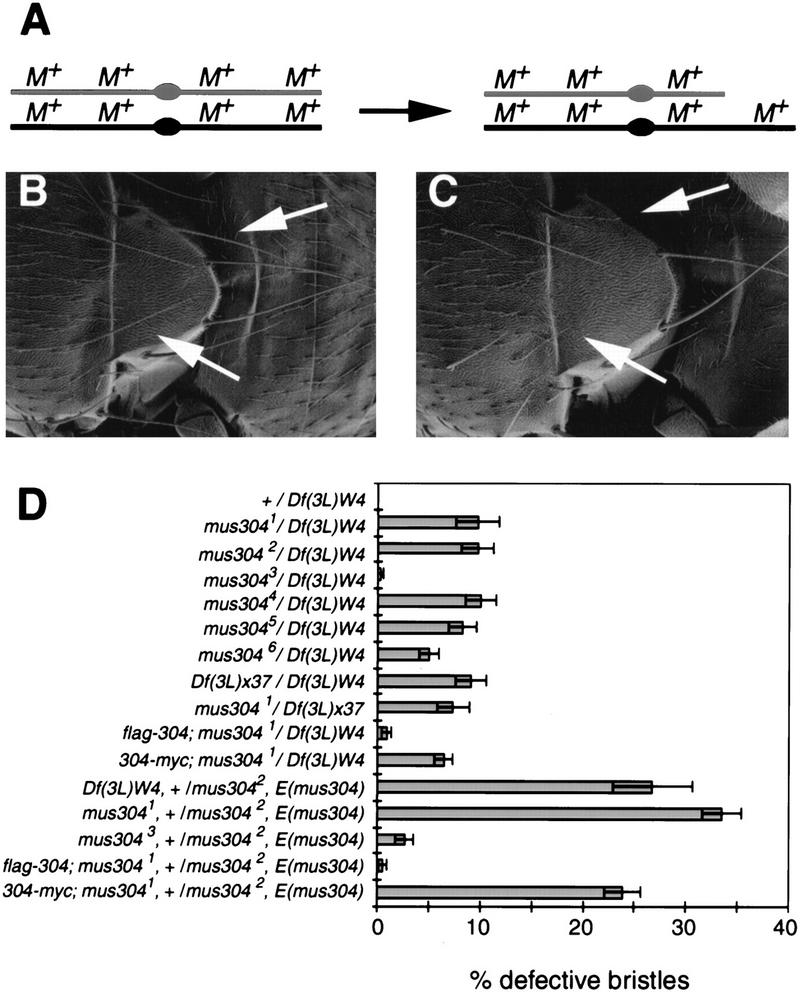
Macrochaete defects in mus304 adults. (A) The genetic basis of bristle defects due to genomic instability. Minute loci (M) are present throughout the genome and are haploinsufficient for macrochaete (bristle) development. If one of the two wild-type copies of a Minute locus is lost in a macrochaete precursor, the resulting macrochaete will exhibit a Minute phenotype. (B) A wild-type notum. Two specific macrochaetae are indicated (arrows). (C) A mus304 notum. The indicated macrochaetae exhibit a Minute phenotype (i.e., shorter and thinner). (D) The frequency of Minute bristles in different genetic backgrounds. Error bars, s.e.m..
Meiotic defects in mus304 females
Females carrying mus304 mutations display defects in meiotic recombination and chromosome segregation (Green 1982). To characterize the meiotic recombination defect more thoroughly, we measured meiotic crossing over in six intervals extending from net, at the tip of chromosome arm 2L, to cn, at the base of 2R (Table 2). Because strong alleles of mus304 severely decrease female fertility, we used mus3043, a partial loss-of-function allele that does not affect female fertility. The total genetic distance from net to cn was 40.0 map units in wild-type females (n = 1987), but only 20.9 map units in mus3043/Df(3L)WR4 females (n = 2680). A quantitatively similar decrease in recombination was observed in females bearing stronger alleles of mus304 (data not shown). The decrease in recombination was not uniform in the six intervals tested. The decrease was most severe in distal and medial intervals (35% of control between net and dp; 45% of control between dp and b). In contrast, there was actually a slight increase in the interval that spans the centromere (130% of control between b and cn).
Table 2.
Decreased rates and altered distribution of meiotic recombination in mus304 mutant females
| Maternal genotype (total progreny scored)
|
Recombination frequencies within specific genetic intervalsa
|
||||||
|---|---|---|---|---|---|---|---|
|
net–cn (total)
|
I. net–ho
|
II. ho–dp
|
III. dp–Sp
|
IV. Sp–b
|
V. b–pr
|
VI. pr–cn
|
|
| A. mus3043/+ (n = 1333) | 40.0 | 4.3 | 7.7 | 8.8 | 15.9 | 2.2 | 1.1 |
| B. mus3043/Df(3L)WR4 (n = 1378) | 20.9 | 1.5 | 2.7 | 3.6 | 8.8 | 2.9 | 1.4 |
| B/Ab | 0.52 | 0.35 | 0.35 | 0.41 | 0.55 | 1.32 | 1.27 |
Recombination frequencies are expressed as centiMorgans (100 × number of recombination events/number progeny scored). The sum of the recombination frequencies of all six intervals is listed in the first column.
The recombination frequency of mus3043 hemizygotes divided by the recombination frequency of mus3043 heterozygotes.
A meiotic chromosome segregation defect in mus304 females (Table 3) was manifested as a moderate increase in exceptional female progeny (derived from diplo-X ova) and a pronounced increase in exceptional male progeny (derived from nullo-X ova). The most straightforward interpretation of this data is that mus304 females exhibit an ∼10-fold increase in X chromosome nondisjunction and an ∼50-fold increase in X chromosome loss.
Table 3.
Meiotic segregation defects in mus304 mutant females
| Maternal genotype (total progeny scored)
|
Exceptional females (%)a
|
Exceptional males (%)b
|
Progeny/female
|
|---|---|---|---|
| mus3043/+ (n = 1333) | 0.02 | 0.03 | 71 |
| mus3043/mus3043(n = 820) | 0.5 | 2.7 | 68 |
| mus3043/Df(3L)WR4 (n = 1378) | 0.4 | 3.3 | 85 |
| mus3041/Df(3L)WR4 (n = 221) | 0.5 | 3.6 | 3.1 |
Numbers given are percentage of total progeny in each class, corrected for inviability of some genotypes.
Exceptional (or matroclinous) females received both X chromosomes from their mother (i.e., are derived from diplo-X ova).
Exceptional (or patroclinous) males received their X chromosome from their father (i.e., are derived from nullo-X ova).
Embryonic defects in mus304 embryos
It has been suggested that checkpoint genes regulate cell cycle progression and zygotic gene transcription in early embryos (Fogarty et al. 1997; Sibon et al. 1997, 1999). We examined these phenotypes in embryos laid by mus304 females. Because early embryonic development is entirely regulated by maternal gene products, only the maternal genotype is described. During the first 2 hr of wild-type embryogenesis, 13 synchronized nuclear divisions occur within a syncytial environment (Foe et al. 1993). Interphase lengthens during cycles 10–13. Following mitosis 13, the embryo enters a prolonged interphase 14 during which cellularization and high levels of zygotic transcription begin. At this stage, most nuclei have a uniform morphology and are evenly distributed near the surface of the embryo. DAPI staining of fixed mus304 embryos revealed three classes: A total of 24% (107/453) exhibited the uniform nuclear morphology and spacing seen in wild-type embryos (Figs. 5A and 6F); 16% (73/453) were arrested at an early stage of embryogenesis, apparently between cycles 3 and 5, and had highly fragmented nuclei (not shown); 60% (273/453) showed a complete or partial arrest at about cycle 14 with severe disruptions in nuclear morphology and spacing (Figs. 5 and 6G,H). Previous studies report that all grp and mei-41 embryos exhibit this last phenotype (Fogarty et al. 1994; Sibon et al. 1997, 1999).
Figure 5.

Cell cycle defects in mus304 embryos. (A) Nuclear morphologies are visualized in wild-type and mus304 embryos using a maternally contributed Histone–GFP fusion protein. Following interphase 14, the mus304 embryo enters an additional, asynchronous mitosis. (B) The length of interphase (white bars) and M phase (black bars) is indicated for the wild-type and mus304 embryos shown in A.
Figure 6.
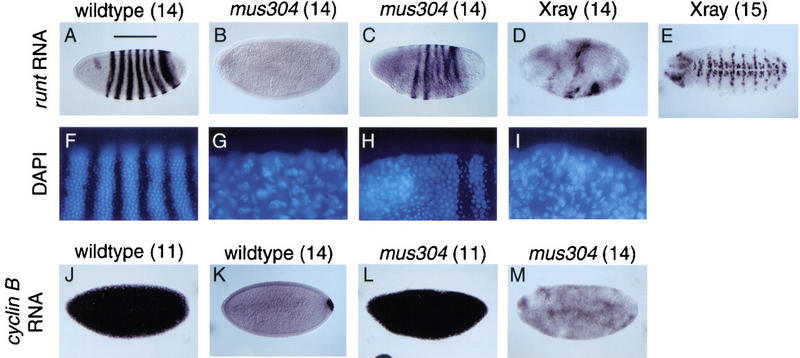
Disrupted zygotic gene expression in irradiated or mus304 embryos. Embryos from wild-type mothers (A,D,E,F,I,J,K) or mus304 mothers (B,C,G,H,L,M) were processed for either runt RNA expression (A–E) and DAPI staining (F–I), or cyclin B RNA expression (J–M) and DAPI staining (not shown). The nuclear division cycle for each embryo, as determined by timed collection and DAPI staining, is indicated in parentheses. X-ray indicates wild-type embryos irradiated 1 hr prior to fixation (D,E,I). Note that the stripes of staining by the RNA in situ procedure interferes with the DAPI signal, causing the apparent stripes in F.
We used a histone–GFP fusion (Clarkson and Saint 1999) to examine nuclear divisions 11–14 in live embryos (Fig. 5). Of seven wild-type embryos examined, all exhibited synchronous nuclear divisions followed by a prolonged interphase 14. Of six mus304 embryos examined, two resembled wild-type and four showed defective nuclear divisions. Defects were visible during anaphase 12 and became more severe during cycles 13 and 14. Defects included asynchronous and incomplete chromosome segregation during anaphase as well as fused and irregularly spaced nuclei during interphase. To determine cell cycle timing in these embryos, a small group of nuclei in the center of each image was followed after exit from telophase 10 (Fig. 5B). In three of six mus304 embryos examined, interphase failed to lengthen until cycle 14 and interphase 14 was interrupted by a premature mitosis 14. Although the disruption of nuclear morphology may affect control of cycles 13 and 14, the alteration of interphase 12 occurs prior to the most severe defects. These observations are consistent with a model (Sibon et al. 1997) in which a DNA replication checkpoint is required to slow nuclear division cycles 11–14 during normal embryonic development.
We examined how these defects in cell cycle progression affect gene expression during interphase 14. In wild-type embryos, strong expression of zygotic genes begins in cycle 14. We used RNA in situ hybridization to the runt (Fig. 6A–E) and tailless (not shown) genes to detect zygotic gene transcription. The variable nuclear morphology phenotype was mirrored as a variable defect in zygotic transcription. In some mus304 embryos, runt and tailless gene expression was completely blocked (Fig. 6B). Among the embryos with some runt expression, many exhibited an abnormal pattern (Fig. 6C). Embryos with partial defects in runt transcription demonstrate that the block in transcription was not simply due to a global delay in embryonic development. Double staining of mus304 embryos with antisense RNA probes and DAPI revealed a spatial correlation between defects in nuclear morphology and defects in zygotic transcription (Fig. 6F–I).
In grp embryos, nuclei accumulate chromosome damage (Fogarty et al. 1997) possibly due to entering mitosis with incompletely replicated DNA. To address whether chromosome damage during nuclear division is sufficient to explain subsequent developmental defects, we examined the phenotypes of irradiated embryos. Irradiation of wild-type embryos produces a phenocopy of the nuclear defects observed in grp and mus304 embryos (Fogarty et al. 1997; Fig. 6I). We examined runt transcription in irradiated wild-type embryos. Irradiation of embryos with actively dividing nuclei (i.e., before interphase 14) with 300 rads, significantly disrupted runt transcription (Fig. 6D). Irradiation with 600 rads completely blocked transcription in all embryos (not shown). Cellularization and gastrulation were also blocked in these embryos. To determine whether the effect of irradiation on transcription required cell cycle progression, we also examined embryos irradiated during the prolonged interphase 14. This treatment did not affect later runt expression (Fig. 6E). These results suggest that the block in zygotic transcription in mus304 embryos may be due to disrupted nuclear structure.
During cycle 14, several maternal transcripts, including the cell cycle regulator cyclin B, are degraded (Foe et al. 1993). In mus304 embryos, the degradation of cyclin B RNA occurred at approximately the correct stage of development (Fig. 6J–M); however, the patchy appearance of the cyclin B transcript may indicate that the degradation is less synchronous than in wild type. Thus, whereas defects in nuclear structure appear to block activation of transcription, other aspects of gene regulation, such as RNA degradation during cycle 14, may be unaffected.
Molecular analysis of mus304
We used deletion mapping to identify the mus304 coding sequence (Materials and Methods). Characterization of deletions that include mus304 revealed that the overlap between deletions Df(3L)x37 and Df(3L)WR4 includes mus304, but no genes required for viability. On the basis of previous mapping of these deletions (Segraves 1988), this overlap is ∼2 kb.
To identify the mus304 transcripts, we screened an embryonic cDNA library with a 10-kb genomic fragment surrounding the Df(3L)x37 and Df(3L)WR4 overlap. We identified seven cDNAs that spanned the deficiency overlap and contained a single ORF (Fig. 7A). Sequence analysis of the six point mutant alleles of mus304 revealed that each contains either an amino acid change or a premature stop codon within this ORF. Transgenes (Materials and Methods) containing either the cDNA fused to 300 bases of upstream genomic sequence or the cDNA with a Glass-responsive promoter were able to rescue a mus304-dependent rough eye phenotype (data not shown). The transgene with the genomic promoter also rescued female infertility and genomic instability. Thus, these cDNAs correspond to the mus304 gene.
Figure 7.
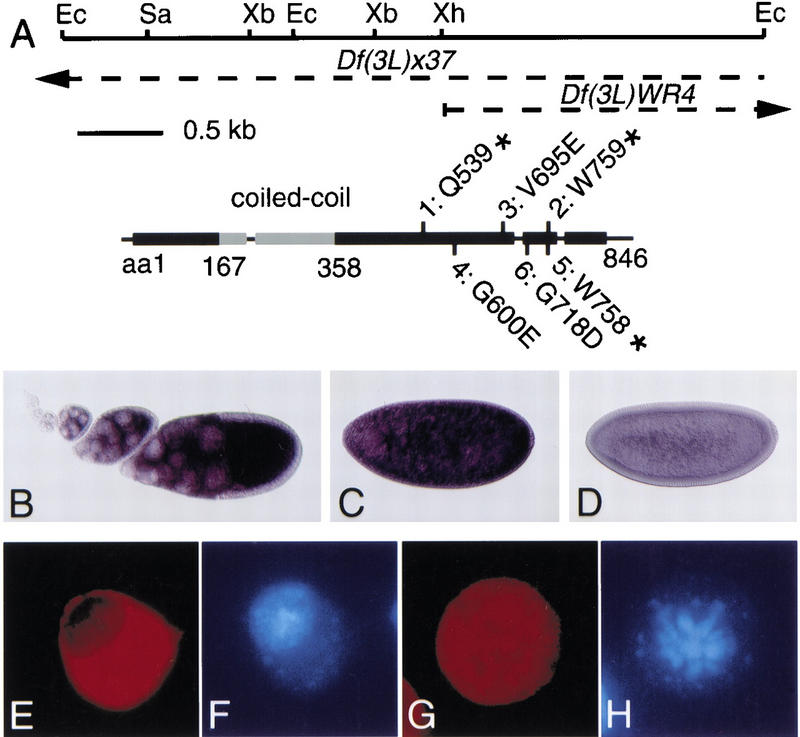
mus304 encodes a novel protein targeted to the cytoplasm. (A) mus304 gene structure. (Top) A map of the mus304 genomic sequence with the following restriction enzyme sites indicated: EcoRI (Ec); SacII (Sa); XbaI (Xb); XhoI (Xh). The broken lines indicate genomic DNA deleted in two deficiencies that contain mus304. The mus304 cDNA (GenBank accession no. AF224715) is shown to scale with the restriction map. Untranslated regions and introns are indicated as a thin line and coding regions are indicated as boxes. The position of each mutant allele is shown; the allele number is followed by the wild-type amino acid, the amino acid number, and the mutant amino acid or stop codon. The position of a potential dimeric coiled-coil region within the coding region is highlighted in gray. (B) mus304 RNA expression in a wild-type ovary. (C) mus304 RNA expression in a wild-type interphase 13 embryo. (D) mus304 RNA expression in a wild-type interphase 14 embryo. (E) Anti-Flag staining (red) of an interphase S2 cell transfected with a Flag–MUS304 transgene. (F) DAPI staining (blue) of the cell in E. (G) Anti-Flag staining (red) of a mitotic S2 cell transfected with a Flag–MUS304 transgene. (H) DAPI staining (blue) of the cell in G.
We used this cDNA to examine the distribution of the mus304 transcript in ovaries and embryos. Consistent with a maternal requirement, high levels of mus304 RNA were seen in the oocyte and nurse cells from stage 5 onward (Fig. 7B) and in embryos prior to during nuclear division 14 (Fig. 7C). Transcript levels decrease to background levels during interphase 14 (Fig. 7D). In later stage embryos and imaginal discs, no signal was detected above background (data not shown). Previous analysis of mei-41 and grp expression indicated that these genes are also highly expressed in ovaries and early embryos (Hari et al. 1995; Fogarty et al. 1997).
Sequence analysis of mus304 cDNAs predicted a novel 846 amino acid protein. Motif searching programs predicted a coiled-coil domain in the amino-terminal region of the protein (Fig. 7A). Database searching with this ORF revealed many proteins with homology to the coiled-coil region only. Searches excluding the coiled-coil domain did not reveal any specific sequence similarities. Despite the absence of sequence motifs or similarities in the carboxyl terminus of MUS304, analysis of mus304 mutations indicated that this region is critical for MUS304 function (Fig. 7A). All six mutant alleles have point mutations in the carboxy-terminal half of the coding sequence. In two strong alleles, mus3042 and mus3045, stop codons occur 87 and 88 residues from the end of the coding region. The weakest allele, mus3043, has an amino acid change at position 695.
To examine MUS304 subcellular localization, we constructed a Flag-tagged version of MUS304. A single transgene containing the mus304 promoter driving Flag–MUS304 rescued multiple mus304 phenotypes including female infertility, genomic instability, and the rough eye phenotype seen in combination with E(mus304) (Figs. 3 and 4; data not shown). The mus304 promoter did not drive sufficient expression to detect the Flag epitope in embryos or discs. However, when this construct was transfected into Drosophila tissue culture cells, Flag–MUS304 was largely or entirely localized to the cytoplasm of interphase cells (Fig. 7E,F) and was not associated with the condensed chromosomes of metaphase cells (Fig. 7G,H). Similar results were obtained following overexpression of Flag–MUS304 in eye discs using a Glass-dependent promoter. Irradiation of S2 cells or imaginal discs expressing Flag–MUS304 did not alter the cytoplasmic localization of the protein (data not shown).
Discussion
In this study, we have characterized a novel checkpoint gene, mus304. To better understand how checkpoint pathways regulate Drosophila DNA repair and development, we have compared the phenotypes of mus304 animals with phenotypes described previously for two other DNA damage checkpoint genes, mei-41 and grp. We find that mus304 animals exhibit reduced chromosome break repair and remarkably high levels of genetic instability. In addition, mus304 animals show defects in meiotic recombination, bristle development, and early embryogenesis. We discuss how these specific phenotypes may be a direct or indirect consequence of the cell cycle checkpoint defect.
mus304 is a novel checkpoint regulator in Drosophila
To characterize previously identified checkpoint homologs, we assayed a DNA damage checkpoint in third instar larvae. mei-41 is absolutely required for this checkpoint in imaginal discs (this study) and in the larval brain (Hari et al. 1995). Because these tissues include cells in logarithmic growth phase as well as cells undergoing their final division, mei-41 may be required for damage-induced cell cycle arrest in all dividing larval cells. Larvae with null mutations in grp and 14-3-3ε have a partial checkpoint defect, suggesting that other genes act in parallel to these during cell cycle arrest. Because the S. pombe mitotic checkpoint requires the combined action of Chk1 and Cds1 (Rhind and Russell 1998; Russell 1998), the Drosophila homolog of Cds1 (Oishi et al. 1998) may act in parallel to grp, the Chk1 homolog (Fogarty et al. 1997). In 14-3-3ε animals, another 14-3-3 isoform, such as 14-3-3ζ (Kockel et al. 1997), may help regulate the G2/M checkpoint. Our genetic results are consistent with conclusions reached in S. pombe: The ATM kinase homolog is absolutely required for the G2/M checkpoint, whereas the 14-3-3 and Chk1 homologs cooperate with other genes.
We also show that the gene mus304 is also required for the DNA damage checkpoint. For this phenotype and others, mus304 strongly resembles mei-41. Combined with double mutant analysis, these results suggest that mus304 and mei-41 act in the same genetic pathway during DNA repair and development. However, a comparison of mus304 and grp mutant phenotypes indicates that the relative importance of individual checkpoint genes can vary in different developmental contexts. The DNA damage checkpoint is completely absent in mus304 imaginal discs, whereas grp discs retain a partial checkpoint. In contrast, 100% of grp embryos fail to undergo cellularization, whereas many mus304 embryos show at least some cellularization. mei-41 animals show severe defects in both the damage checkpoint and during embryogenesis. One model that could explain these results is that grp and mus304 act in parallel pathways downstream of mei-41 and that these branches contribute in varying degrees depending on the cell type or the activating signal.
To help determine how mus304 regulates DNA damage responses, we have begun to characterize the gene product. The amino-terminal region of MUS304 contains the only recognizable motif, a coiled-coil domain that might mediate protein–protein interactions. Analysis of mutations in mus304 indicates that the carboxy-terminal region of MUS304 is critical for its function. The lack of an obvious mus304 homolog in yeast may reflect either a biochemical mechanism that is not conserved in yeast, or a protein whose primary sequence can rapidly diverge. When expressed in tissue culture, epitope-tagged MUS304 is cytoplasmic, although we cannot rule out the possibility that a small amount of this protein is active in the nucleus. Potential cytoplasmic targets of DNA damage checkpoints in animal cells include Cyclin B (Jin et al. 1998; Toyoshima et al. 1998) and Myt1, a kinase that phosphorylates Cdc2 (Mueller et al. 1995; Liu et al. 1997).
DNA repair defects in mus304 mutants
In previous studies, X-linked DNA repair loci were examined for defects in DNA repair (Baker et al. 1978; Baker and Smith 1979; Gatti 1979). Among these loci, mei-41 mutations result in the strongest disruption of chromosome break repair and genomic stability. The mutagen-sensitivity phenotype of mus304 suggests that it also mediates DNA repair. Compared with wild type, mus304 neuroblasts show a threefold increase relative in irradiation-induced chromosome breaks (this study), whereas mei-41 neuroblasts show a sixfold increase (Gatti 1979). These results confirm that G2/M checkpoint genes play a role in chromosome break repair. The difference between mus304 and mei-41 may indicate that mei-41 has roles in DNA damage repair not shared by mus304.
In both mei-41 (Gatti 1979) and mus304 neuroblasts, the frequency of spontaneous chromosome breaks is at least 10-fold higher than in wild type. As expected from the high frequency of breaks, elevated levels of spontaneous cell death are observed in mus304 or mei-41 discs (M.H. Brodsky, unpubl.) that may help prevent cells with damaged genomes from contributing to adult tissues. The LOH assay measures the number of adult cells with compromised genetic material. In this assay, mus304 mutants have a severe defect, only slightly less than reported for mei-41 (Baker et al. 1978). The similar effect on genetic instability due to mus304 and mei-41 mutations suggests that the G2/M checkpoint transition plays a critical role in preventing genetic loss during normal animal development.
Developmental roles of checkpoint genes
mus304 function is required for meiotic recombination, embryonic development, and bristle formation. mei-41 is also required for these processes (Baker and Carpenter 1972; Sibon et al. 1999; M.H. Brodsky and G.M. Rubin, unpubl.). Below, we argue that the developmental phenotypes shared by mei-41 and mus304 arise in cell types with particular sensitivity to checkpoint defects. These cell types have either developmentally programmed DNA breaks, abbreviated cell cycle control, or unusual susceptibility to aneuploidy.
Consistent with the notion that cells undergoing developmentally programmed DNA recombination are especially susceptible to defects in checkpoint signaling, mus304 is required for normal meiosis. Meiotic recombination is initiated by double-strand break formation and is often disrupted by DNA repair mutations (Baker et al. 1976; Paques and Haber 1999). mei-41 mutations cause an altered frequency and distribution of meiotic recombination events in Drosophila (Baker and Carpenter 1972). During meiosis in Saccharomyces cerevisiae, MEC1, a homolog of mei-41, has been proposed to prevent the cell from entering anaphase in the presence of incomplete recombination events (Lydall et al. 1996). In Drosophila ovaries with defects in double-strand break repair, a mei-41-dependent checkpoint regulates dorsoventral patterning (Ghabrial and Schüpbach 1999). In ovaries with normal double-strand break repair, mei-41 is not required for patterning, but might regulate the timing of cell cycle progression in response to meiotic recombination intermediates (Sekelsky et al. 1998). The mus304 meiotic recombination and segregation phenotypes are similar, both qualitatively and quantitatively, to that reported for mei-41. The shared meiotic defects in these DNA damage checkpoint mutants suggest that they act in a common pathway that couples progress of recombination to the meiotic cell cycle.
Among the phenotypes we have examined, Drosophila meiosis is particularly sensitive to a partial loss of mus304 function. The mus3043 allele shows a partial defect in the DNA damage checkpoint, acts as a weak allele in mutagen sensitivity and LOH assays, and has no effect on embryo development. However, its effects on recombination and nondisjunction rates are similar to those seen in genetic null alleles. Thus, meiosis, the one developmental event in Drosophila utilizing DNA strand breaks, is exceptionally sensitive to reduced checkpoint function.
Another developmental context with a specific requirement for checkpoint function is during the rapid nuclear division cycles in the Drosophila syncytial blastoderm. These cycles are ∼10 min and lack the G1 and G2 phases present in most dividing cells (Foe et al. 1993). In contrast, the cell cycle of imaginal disc cells typically requires 10 hr. In the absence of gap phases, it is critical to prevent mitosis until DNA replication is complete. In grp (Fogarty et al. 1994, 1997; Sibon et al. 1997), mei-41 (Sibon et al. 1999), or mus304 embryos (this study), mitotic delay fails, possibly leading to aberrant mitoses and disruptions in nuclear structure. Alternatively, a defect in cyclin proteolysis may be responsible for the nuclear division phenotypes (Su et al. 1999). Regardless of the initial defect, the similar phenotypes seen in checkpoint deficient embryos and in irradiated embryos (this study; Fogarty et al. 1994, 1997) suggest that a defect in chromosome structure leads to subsequent defects in zygotic transcription and cellularization. In general, cell types with abbreviated cell cycle controls may have a greater dependency on checkpoint signaling to block cell cycle progression in the presence of unreplicated or unrepaired DNA.
We propose that a third developmental phenotype of mus304 mutants is due to a combination of loci haploinsufficient for bristle development (see below) and a high level of genetic instability. In a mus304 wing, ∼0.5% (106/20,000) of cells show LOH at the mwh locus. The high frequency of chromosome breaks in dividing cells (Table 1) and the clone size distribution in the adult wing (M.H. Brodsky and G.M. Rubin, unpubl.; see Baker et al. 1978) suggest that most LOH in mus304 animals is due to chromosome breaks. A similar conclusion was reached in a genetic analysis of LOH in mei-41 animals (Baker et al. 1978). Thus, ∼0.5% of mus304 wing cells lost part of the chromosome arm carrying the wild-type mwh. This assay only measures genetic loss at a single point on one chromosome arm. Assuming that the 10 major chromosome arms are equally affected, the frequency of wing hair cells with genetic loss would be at least 10-fold higher, or 5%.
If the wing is typical of other tissues in the adult, the LOH measurements suggest that on the order of 5% of cells in a mus304 adult have lost some genetic material. Most cell types appear unaffected by this high level of aneuploidy, consistent with the observation that only a few Drosophila genes have visible haploinsufficient phenotypes (Ashburner 1989). Minute loci encode ribosomal proteins and have several haploinsufficient phenotypes including smaller or missing macrochaetae. Because the ∼50 Minute loci are distributed throughout the genome, Minute macrochaetae should appear at high frequencies in animals with large numbers of aneuploid cells. A previous analysis of mei-41 demonstrated that LOH in abdominal bristles is often accompanied by a Minute phenotype (Baker et al. 1978). In mus304 adults, nearly 10% of examined macrochaetae exhibit a Minute-like phenotype. This result is roughly consistent with the levels of LOH measured in the wing. We suggest that the mus304 macrochaetae phenotype may be explained by the sensitivity of this cell type to haploinsufficiency at Minute loci.
Some of the symptoms associated with the human cancer predisposition syndrome A-T may arise from mechanisms similar to those we have described for mus304 and other Drosophila checkpoint genes. Consistent with the homology between ATM and checkpoint genes from yeast and flies, ATM cells have checkpoint defects and chromosome instability. A cellular defect in DNA repair is likely to lead to increased levels of genetic instability and provides a straightforward explanation for the occurrence of lymphomas in A-T patients. Similarly, the immune and reproductive system symptoms are likely to arise from the combination of developmentally programmed DNA breaks (occurring during meiotic and V(D)J recombination) and defective checkpoints. However, other features such as neurodegeneration, dilated blood vessels, premature aging, and diabetes are less easily attributed a checkpoint defect. One possible explanation is that ATM acts in multiple signal transduction pathways. Alternatively, some symptoms may reflect cell types that have an unrecognized sensitivity to checkpoint defects due to unusual cell cycle control or sensitivity to haploinsufficiency, such as found in Drosophila embryos and bristles, respectively. In future studies, the genetic and developmental tools available in Drosophila should provide further clues to the roles of checkpoint genes animal development and disease.
Materials and methods
Checkpoint assay
All animals were kept at 25°C. Climbing third instar larvae were irradiated in a Torrex 120D X-ray machine at 4000 rad. After 1 hr, imaginal discs were dissected and fixed in 4% paraformaldehyde in PBS for 1 hr. Discs were washed in PBS and 0.3% Triton (PBT), and blocked in PBT and 2% goat serum (PBTN). Discs were incubated in a 1:500 dilution of a rabbit anti-phospho-histone H3 antibody (Upstate Biotechnology) in PBTN overnight at 4°C. Discs were incubated with a secondary antibody and stained for HRP activity.
Chromosome instability assays
Chromosome breaks were assayed as described previously (Gatti et al. 1994). Brains were irradiated at 220 rads, dissected, and incubated for 2.5 hr in saline followed by 1.5 hr in colchicine. Mitotic figures were scored using 0.5 mg/ml DAPI to stain chromosomes.
To assay mwh loss, wings were dehydrated in isopropanol and mounted in 1:1 methylsalicilate: Canada balsam (Sigma). All intervein wing hair cells were examined for the mwh phenotype. Five to ten wings were examined for each genotype.
Bristle defects were examined in 1- to 4-day-old adults. A total of 34 bristles were examined per individual, orbitals (6), ocellar (2), verticals (4), postvertical (2), presutural (2), notopleurals (4), supra-alars (4), dorsocentrals (4), anterior post-alars (2), and scutellars (4). Bristles were scored as mutant if at least 50% shorter than the corresponding wild-type bristle. Ten adult females were scored for each genotype.
Analysis of embryonic phenotypes
Video microscopy was performed on embryos laid by mothers heterozygous for a transgene containing a histone–GFP fusion (Clarkson and Saint 1999). Images were collected every 30 sec using the DeltaVision microscope and software (Applied Precision Instruments). Analysis of gene expression in fixed wild-type and mutant embryos was performed by RNA in situ analysis (Tautz and Pfeifle 1989) using antisense RNA probes. Embryos at 1–2 and 2–3 hr after egg laying were used in these experiments. To determine the division cycle and nuclear structure, 0.5 mg/ml DAPI was added to the anti-digoxygenin antibody incubation step. In irradiation experiments, embryos at 1–2 and 2–3 hr after egg laying were irradiated, aged 1 hr, and processed for RNA expression and DAPI staining.
Genetics
All crosses were performed at 25°C. mei-41RT1, mei-41RT2, grpfs(A)4, 14-3-3εj2b10, and mus309d2/mus309d3 were tested for defects in checkpoint assay. w1118 was used as the wild-type control. mus3041, mus3042, and mus3043 were originally described as mus304D1, mus304D2, and mus304D3 (Boyd et al. 1981). E(mus304) was identified as a spontaneous recessive lethal in a mus3042 strain. Recombination and deficiency mapping places E(mus304) in cytological region 93D–F.
Additional alleles of mus304 were identified in two noncomplementation screens. In the first screen, st males were mutagenized with EMS and crossed to mus3042, E(mus304)/TM3Sb females. Male progeny were screened for the rough eye phenotype associated with animals that are homozygous for mus304 and heterozygous for E(mus304) (see Fig. 2). Because mus3042, E(mus304) is viable over deficiencies for the mus304 locus, this screen is not biased against lethal alleles. mus3044, mus3045, and mus3046 were recovered from this screen. In addition, l(3)j6A11, a P element near the mus304 gene, was mobilized using a genomic source of transposase, delta2-3, to create two new deficiencies Df(3L)mus304-1 and Df(3L)mus304-2 in this region (M.H. Brodsky, unpubl.). Cytological and Southern blot analysis indicate that the left breakpoint of the deletions begin at the original P insertion site, whereas the right breakpoint extends into cytological region 75C1-2.
Previous results placed mus304 at meiotic map position 3-46 (Boyd et al. 1981). mus304 was localized by deficiency mapping (Segraves 1988; Grether et al. 1995) to 75B8–75C1 between E75 and hid. mus304 failed to complement both Df(3L)W4 and Df(3L)x37 which overlap in a single genomic clone from this region, λ3510 (Segraves 1988; Segraves and Hogness 1990).
Molecular analysis of mus304
Subclones from λ3510 were used to screen the LD embryonic cDNA library. Standard procedures were used for library screening, subcloning, and sequencing of mus304 cDNAs. Intron-exon structure was determined by sequencing of subclones from λ clone λ3510. PCR products from mutant alleles were directly sequenced. The breakpoint of Df(3L)WR4 was determined by sequencing of λWR4Xho (Segraves 1988), which contains the breakpoint. This deficiency interrupts the predicted protein after amino acid number 586 and adds 22 residues before the next stop codon.
Rescue constructs were created by placing a mus304 cDNA in the pCaSpeR2 vector with the mus304 promoter region (300 bp from the beginning of the longest cDNA). Epitope-tagged versions were created by PCR. A Flag tag was added immediately before the start codon with the following oligonucleotides: mus304.f1 ATGGACTACAAGGACGACGACGACAAGGGCGGCATGGCCAAGCGCTTTTCTGCC; mus304.f2 GTCGTCGTCGTCCTTGTAGTCCATGTTTTGCAGTTTCCGCTATATTTCGTTAC. A MYC tag was added following the final amino acid with the following oligonucleotides: mus304.m1 CAGAAGCTGATCTCCGAGGAGGACCTGTAGTTATAGTTAGCTTAAACGTT; mus304.m2 CTCCTCGGAGATCAGCTTCTGCTCGCCGCCCTCAAAAGTGTTCTCCTCCCGT.The tagged cDNA derivatives were also cloned into the pGUS vector, which contains upstream binding sites for both the Glass and Gal4 transcription factors (M.H. Brodsky and G.M. Rubin, unpubl.). The carboxy-terminal Myc-tagged constructs showed the same localization as the amino-terminal Flag-tagged constructs, but only provided partial rescue of mus304 phenotypes (data not shown).
For immunostaining, the Flag–mus304 fusion under control of the mus304 promoter was transfected into S2 cells using the SuperFect reagent (Qiagen). A 1:200 dilution of the M5 anti-flag antibody (Sigma) and a 1:2000 dilution of a Cy3 secondary antibody (Jackson labs) were used to detect the expressed protein.
Acknowledgments
For providing Drosophila stocks, DNA clones, and other reagents, we thank Bill Segraves, Ed Garza, Julie Agapite, Rob Saint, Ling Hong, and the Bowling Green and Bloomington Stock Centers. We also thank Susan Mullaney, Elaine Kwan, Chris Suh, and Todd Laverty for technical assistance, Peter Carlton, Carrie Cowan, and Satoru Uzawa for video microscopy advice and help, and Catherine Nelson, Bobby Williams, Andrea Page-McCaw, and Audrey Huang for helpful comments. Support was provided by the Howard Hughes Medical Institute, the California Division of the American Cancer Society (M.H.B.), and the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation (J.J.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL brodsky@uclink4.berkeley.edu; FAX (510) 643-9947.
References
- Abbott MK, Lengyel JA. Embryonic head involution and rotation of male terminalia require the Drosophila locus head involution defective. Genetics. 1991;129:783–789. doi: 10.1093/genetics/129.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. Drosophila: A laboratory handbook. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Baker BS, Boyd JB, Carpenter AT, Green MM, Nguyen TD, Ripoll P, Smith PD. Genetic controls of meiotic recombination and somatic DNA metabolism in Drosophila melanogaster. Proc Natl Acad Sci. 1976;73:4140–4144. doi: 10.1073/pnas.73.11.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BS, Carpenter AT. Genetic analysis of sex chromosomal meiotic mutants in Drosophila melanogaster. Genetics. 1972;71:255–286. doi: 10.1093/genetics/71.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BS, Smith DA. The effects of mutagen-sensitive mutants of Drosophila melanogaster in nonmutagenized cells. Genetics. 1979;92:833–847. doi: 10.1093/genetics/92.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BS, Carpenter ATC, Ripoll P. The utilization during mitotic cell division of loci controlling meiotic recombination and disjunction in Drosophila melanogaster. Genetics. 1978;90:531–578. doi: 10.1093/genetics/90.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall EL, Rio DC. Drosophila IRBP/Ku p70 corresponds to the mutagen-sensitive mus309 gene and is involved in P-element excision in vivo. Genes & Dev. 1996;10:921–933. doi: 10.1101/gad.10.8.921. [DOI] [PubMed] [Google Scholar]
- Bonini NM, Choi KW. Early decisions in Drosophila eye morphogenesis. Curr Opin Genet Dev. 1995;5:507–515. doi: 10.1016/0959-437x(95)90056-m. [DOI] [PubMed] [Google Scholar]
- Boyd JB, Golino MD, Nguyen TD, Green MM. Isolation and characterization of X-linked mutants of Drosophila melanogaster which are sensitive to mutagens. Genetics. 1976;84:485–506. doi: 10.1093/genetics/84.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JB, Golino MD, Shaw KE, Osgood CJ, Green MM. Third-chromosome mutagen-sensitive mutants of Drosophila melanogaster. Genetics. 1981;97:607–623. doi: 10.1093/genetics/97.3-4.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JB, Mason JM, Yamamoto AH, Brodberg RK, Banga SS, Sakaguchi K. A genetic and molecular analysis of DNA repair in Drosophila. J Cell Sci Suppl. 1987;6:39–60. doi: 10.1242/jcs.1984.supplement_6.3. [DOI] [PubMed] [Google Scholar]
- Brown KD, Chakravarti A. Multiple ATM-dependent pathways: An explanation for pleiotropy. Am J Hum Genet. 1999;64:46–50. doi: 10.1086/302223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Rubin GM. 14-3-3ε positively regulates Ras-mediated signaling in Drosophila. Genes & Dev. 1997;11:1132–1139. doi: 10.1101/gad.11.9.1132. [DOI] [PubMed] [Google Scholar]
- Clarkson M, Saint R. A His2AvDGFP fusion gene complements a lethal His2AvD mutant allele and provides an in vivo marker for Drosophila chromosome behavior. DNA Cell Biol. 1999;18:457–462. doi: 10.1089/104454999315178. [DOI] [PubMed] [Google Scholar]
- Foe VE, Odell GM, Edgar BA. Mitosis and morphogenesis in the Drosophila embryo: Point and counterpoint. In: Bate M, Arias AM, editors. The development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 149–300. [Google Scholar]
- Fogarty P, Kalpin RF, Sullivan W. The Drosophila maternal-effect mutation grapes causes a metaphase arrest at nuclear cycle 13. Development. 1994;120:2131–2142. doi: 10.1242/dev.120.8.2131. [DOI] [PubMed] [Google Scholar]
- Fogarty P, Campbell SD, Abu-Shumays R, Phalle BS, Yu KR, Uy GL, Goldberg ML, Sullivan W. The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr Biol. 1997;7:418–426. doi: 10.1016/s0960-9822(06)00189-8. [DOI] [PubMed] [Google Scholar]
- Gatti M. Genetic control of chromosome breakage and rejoining in Drosophila melanogaster: Spontaneous chromosome aberrations in X-linked mutants defective in DNA metabolism. Proc Natl Acad Sci. 1979;76:1377–1381. doi: 10.1073/pnas.76.3.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti M, Bonaccorsi S, Pimpinelli S. Looking at Drosophila mitotic chromosomes. Methods Cell Biol. 1994;44:371–391. doi: 10.1016/s0091-679x(08)60924-3. [DOI] [PubMed] [Google Scholar]
- Ghabrial A, Schüpbach T. Activation of a meiotic checkpoint regulates translation of Gurken during Drosophila oogenesis. Nat Cell Biol. 1999;1:354–357. doi: 10.1038/14046. [DOI] [PubMed] [Google Scholar]
- Green MM. On the meiotic effect of mutants at the mutagen-sensitive locus, mus(3)304, in Drosophila melanogaster. Biol Zentbl. 1982;101(2):223–226. [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes & Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Hari KL, Santerre A, Sekelsky JJ, McKim KS, Boyd JB, Hawley RS. The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell. 1995;82:815–821. doi: 10.1016/0092-8674(95)90478-6. [DOI] [PubMed] [Google Scholar]
- Hawley RS, Friend SH. Strange bedfellows in even stranger places: The role of ATM in meiotic cells, lymphocytes, tumors, and its functional links to p53. Genes & Dev. 1996;10:2383–2388. doi: 10.1101/gad.10.19.2383. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol. 1998;141:875–885. doi: 10.1083/jcb.141.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazono A, Matsumoto T. “Isogaba Maware”: Quality control of genome DNA by checkpoints. BioEssays. 1998;20:391–399. doi: 10.1002/(SICI)1521-1878(199805)20:5<391::AID-BIES6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Kockel L, Vorbruggen G, Jackle H, Mlodzik M, Bohmann D. Requirement for Drosophila 14-3-3ζ in Raf-dependent photoreceptor development. Genes & Dev. 1997;11:1140–1147. doi: 10.1101/gad.11.9.1140. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- Liu F, Stanton JJ, Wu Z, Piwnica-Worms H. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol. 1997;17:571–583. doi: 10.1128/mcb.17.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A, Furnari B, Mondesert O, Russell P. Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature. 1999;397:172–175. doi: 10.1038/16488. [DOI] [PubMed] [Google Scholar]
- Lydall D, Nikolsky Y, Bishop DK, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383:840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Morgan SE, Kastan MB. p53 and ATM: Cell cycle, cell death, and cancer. Adv Cancer Res. 1997;71:1–25. doi: 10.1016/s0065-230x(08)60095-0. [DOI] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: A membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- Oishi I, Sugiyama S, Otani H, Yamamura H, Nishida Y, Minami Y. A novel Drosophila nuclear protein serine/threonine kinase expressed in the germline during its establishment. Mech Dev. 1998;71:49–63. doi: 10.1016/s0925-4773(97)00200-1. [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Russell P. Mitotic DNA damage and replication checkpoints in yeast. Curr Opin Cell Biol. 1998;10:749–758. doi: 10.1016/s0955-0674(98)80118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotman G, Shiloh Y. ATM: From gene to function. Hum Mol Genet. 1998;7:1555–1563. doi: 10.1093/hmg/7.10.1555. [DOI] [PubMed] [Google Scholar]
- Russell P. Checkpoints on the road to mitosis. Trends Biochem Sci. 1998;23:399–402. doi: 10.1016/s0968-0004(98)01291-2. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Segraves WA. “Molecular and genetic analysis of the E75 ecdysone-responsive gene of Drosophila melanogaster”, Ph.D. thesis. Stanford, CA: Stanford University; 1988. [Google Scholar]
- Segraves WA, Hogness DS. The E75 ecdysone-inducible gene responsible for the 75B early puff in Drosophila encodes two new members of the steroid receptor superfamily. Genes & Dev. 1990;4:204–219. doi: 10.1101/gad.4.2.204. [DOI] [PubMed] [Google Scholar]
- Sekelsky JJ, Burtis KC, Hawley RS. Damage control: The pleiotropy of DNA repair genes in Drosophila melanogaster. Genetics. 1998;148:1587–1598. doi: 10.1093/genetics/148.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibon OC, Stevenson VA, Theurkauf WE. DNA-replication checkpoint control at the Drosophila midblastula transition. Nature. 1997;388:93–97. doi: 10.1038/40439. [DOI] [PubMed] [Google Scholar]
- Sibon OC, Laurencon A, Hawley R, Theurkauf WE. The Drosophila ATM homologue Mei-41 has an essential checkpoint function at the midblastula transition. Curr Biol. 1999;9:302–312. doi: 10.1016/s0960-9822(99)80138-9. [DOI] [PubMed] [Google Scholar]
- Su TT, Campbell SD, O'Farrell PH. Drosophila grapes/CHK1 mutants are defective in cyclin proteolysis and coordination of mitotic events. Curr Biol. 1999;9:919–922. doi: 10.1016/s0960-9822(99)80399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A nonradioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals a translational control of segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Toyoshima F, Moriguchi T, Wada A, Fukuda M, Nishida E. Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. EMBO J. 1998;17:2728–2735. doi: 10.1093/emboj/17.10.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T. A DNA damage checkpoint meets the cell cycle engine. Science. 1997;277:1450–1451. doi: 10.1126/science.277.5331.1450. [DOI] [PubMed] [Google Scholar]
- ————— DNA damage and checkpoint pathways: Molecular anatomy and interactions with repair. Cell. 1998;94:555–558. doi: 10.1016/s0092-8674(00)81597-4. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- ————— Control of G2 delay by the rad9 gene of Saccharomyces cerevisiae. J Cell Sci Suppl. 1989;12:145–148. doi: 10.1242/jcs.1989.supplement_12.12. [DOI] [PubMed] [Google Scholar]
- Westphal CH. Cell-cycle signaling: Atm displays its many talents. Curr Biol. 1997;7:R789–R792. doi: 10.1016/s0960-9822(06)00406-4. [DOI] [PubMed] [Google Scholar]
- Wolff T, Ready DF. Pattern Formation in the Drosophila Retina. In: Bate M, Arias AM, editors. The development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 1277–1325. [Google Scholar]