

Abstract
The p53 tumor suppressor activates either cell cycle arrest or apoptosis in response to cellular stress. Mouse embryo fibroblasts (MEFs) provide a powerful primary cell system to study both p53-dependent pathways. Specifically, in response to DNA damage, MEFs undergo p53-dependent G1 arrest, whereas MEFs expressing the adenovirus E1A oncoprotein undergo p53-dependent apoptosis. As the p53-dependent apoptosis pathway is not well understood, we sought to identify apoptosis-specific p53 target genes using a subtractive cloning strategy. Here, we describe the characterization of a gene identified in this screen, PERP, which is expressed in a p53-dependent manner and at high levels in apoptotic cells compared with G1-arrested cells. PERP induction is linked to p53-dependent apoptosis, including in response to E2F-1-driven hyperproliferation. Furthermore, analysis of the PERP promoter suggests that PERP is directly activated by p53. PERP shows sequence similarity to the PMP-22/gas3 tetraspan membrane protein implicated in hereditary human neuropathies such as Charcot–Marie–Tooth. Like PMP-22/gas3, PERP is a plasma membrane protein, and importantly, its expression causes cell death in fibroblasts. Taken together, these data suggest that PERP is a novel effector of p53-dependent apoptosis.
Keywords: p53, apoptosis, PERP, MEFs, PMP-22/gas3
The p53 tumor suppressor gene plays a crucial role in protecting organisms from developing cancer (Levine 1997). p53 is mutated in over half of all human cancers, reflecting a selective pressure to remove this negative regulator of cell proliferation during the course of tumorigenesis (Levine 1997). p53 mutations are found in tumors of a wide variety of cell types, suggesting that p53 normally inhibits tumor formation in many tissues. Moreover, individuals with Li–Fraumeni syndrome, who are heterozygous for a mutant p53 allele, are highly prone to developing a variety of different cancer types (Malkin et al. 1990). In addition, mice carrying targeted mutations in the p53 gene develop tumors at 100% frequency within a few months of birth (for review, see Attardi and Jacks 1999).
Mechanistically, the p53 protein acts as a cellular stress sensor (Giaccia and Kastan 1999). In response to a number of forms of stress, including hyperproliferation, DNA damage, and hypoxia, p53 levels rise, causing the cell to undergo one of two fates: arrest in the G1 phase of the cell cycle or genetically programmed cell death, known as apoptosis (Levine 1997). The G1 arrest is part of a checkpoint response whereby cells that have sustained DNA damage pause in G1 to allow for DNA repair before progression through the cell cycle, thereby limiting the propagation of potentially oncogenic mutations. The p53-dependent apoptotic pathway is also induced by DNA damage in certain cell types, as well as in cells undergoing inappropriate proliferation. Importantly, however, the mechanism by which p53 dictates the choice between the G1 arrest and the apoptotic pathways is presently not well understood.
Mouse embryo fibroblasts (MEFs) represent an ideal cell system in which to study both the G1 arrest and apoptotic activities of p53. When treated with DNA-damaging agents, wild-type MEFs activate the cell cycle checkpoint response by arresting in G1 (Kastan et al. 1992). This response is clearly p53 dependent as p53-null MEFs fail to undergo G1 arrest upon DNA damage treatment. In contrast, hyperproliferative MEFs, such as those expressing the adenovirus E1A oncoprotein, respond differently to DNA damage treatment. By mechanisms that are not well understood, these growth-deregulated fibroblasts are reprogrammed to undergo p53-dependent apoptosis (Debbas and White 1993; Lowe et al. 1993). Similarly, expression of c-Myc or E2F-1 renders cells susceptible to p53-dependent apoptosis, which is greatly enhanced by exposure to stresses such as serum starvation or DNA-damaging agent treatment (Hermeking and Eick 1994; Qin et al. 1994; Wagner et al. 1994; Wu and Levine 1994; Soengas et al. 1999).
These observations and data from in vivo systems support the concept that activation of apoptosis by p53 in incipient tumor cells plays an important role in tumor suppression. For example, E1A and ras-expressing p53−/− MEFs transplanted into nude mice form rapid tumors compared with E1A/ras–p53+/+ MEFs, as do E1A/ras-expressing MEFs lacking the apoptotic effector proteins caspase 9 or Apaf-1 (Lowe et al. 1994; Soengas et al. 1999). These data indicate that defects in the p53-dependent apoptotic pathway contribute to tumor development. Another example comes from transgenic mice that express SV40 large T antigen in the choroid plexus epithelium and, consequently, rapidly develop brain tumors (Symonds et al. 1994). If a mutant T-antigen protein incapable of binding and sequestering p53 is expressed, tumor development is slowed dramatically, because of the activation of p53-dependent apoptosis. Additional support for the role of p53-induced apoptosis in tumor suppression comes from the demonstration that some human tumor-derived p53 mutants are defective only in inducing the apoptotic pathway but not in activating the G1 arrest pathway (Ryan and Vousden 1998).
Although the mechanism by which p53 activates G1 arrest is well characterized and involves primarily transcriptional activation of the cyclin-dependent kinase inhibitor p21 (El-Deiry et al. 1993; Brugarolas 1995; Deng et al. 1995), p53-dependent apoptosis is not well understood. A wealth of evidence indicates that p53 is a transcriptional activator protein (Levine 1997). However, both transcriptional activation-dependent and -independent functions of p53 have been shown to be important for its ability to induce apoptosis, possibly reflecting distinct mechanisms of p53 action in different cell types (Caelles et al. 1994; Wagner et al. 1994; Haupt et al. 1995; Sabbatini et al. 1995; Attardi et al. 1996; Yonish-Rouach 1996). Previously, by testing a collection of p53 mutants, we established that transcriptional activation by p53 is critical for the induction of both G1 arrest in MEFs and apoptosis in E1A-expressing MEFs (Attardi et al. 1996). However, the mode of activation of these pathways by p53 could be distinguished by examination of p53 transcriptional target gene requirements. In particular, although p21 was critical for mediating the p53-activated G1 arrest response to DNA damage, it was not required for the apoptotic response in E1A MEFs. This finding suggested that p53 acts via distinct mechanisms to activate the two pathways, possibly through the activation of different sets of target genes.
Several previously identified p53 target genes have been proposed to play a role in cell death. Bax, a proapoptotic member of the Bcl-2 family, has p53 binding sites in its promoter, and direct activation of its promoter could constitute the link between p53 and the apoptotic machinery (Miyashita and Reed 1995). However, the requirement for Bax in p53-dependent cell death is, at best, partial. For example, in E1A MEFs or in the T antigen-induced brain tumor mouse model, levels of apoptosis are reduced in a bax null background, in contrast to being totally eliminated in the absence of p53 (McCurrach et al. 1997; Yin et al. 1997). In addition, Bax is fully dispensable for p53-dependent cell death of thymocytes in response to γ-irradiation, indicating that it may be more relevant in some cellular contexts than others (Knudson et al. 1995). Other potential apoptosis target genes have been discovered, including KILLER/DR5 and PIGs (p53 inducible genes), but it remains to be seen whether they play a role in p53-dependent apoptosis (Polyak et al. 1997; Wu et al. 1997). As Bax is the only p53 target gene for which loss-of-function experiments suggest a function in the p53 cell death pathway and as it is only a partial role, it is likely that other p53 target genes in this pathway remain to be identified.
To further dissect the p53-dependent apoptotic pathway activated in incipient tumor cells, we sought to identify p53 target genes specifically induced during apoptosis. Toward this end, we performed a differential screen in which G1-arrested MEF RNA populations were subtracted from apoptotic E1A MEF RNA populations. The rationale for this strategy was to select against genes induced by p53 in nonapoptotic cells, allowing for the isolation of genes specifically up-regulated by p53 during apoptosis. Although subtractive hybridization strategies have been used previously to identify p53-responsive genes such as p21, they have generally failed to identify genes clearly involved only in apoptosis and not in G1 arrest (El-Deiry et al. 1993; Okamoto and Beach 1994; Buckbinder et al. 1995; Polyak et al. 1997). These approaches have typically entailed comparing gene expression profiles of cell lines lacking p53 with cell lines overexpressing p53, without distinguishing between the G1 arrest and apoptotic responses. Another advantage of the strategy used here is that it relies on the response of endogenous cellular p53 to DNA damage and is performed using primary cells, making it very physiological. Using this novel subtractive cloning approach, we have isolated a new p53 target gene, PERP, a candidate effector in the p53-dependent apoptotic pathway.
Results
PERP, a novel p53 target gene, is expressed preferentially during p53-mediated apoptosis
In an effort to isolate genes up-regulated by p53 specifically during apoptosis, we undertook a subtractive hybridization strategy in which G1-arrested cDNA populations were subtracted from apoptotic cDNA populations (Fig. 1A). In our screen, the tester population, from which the target genes were sought, was comprised of cDNA from DNA damage-treated E1A–p53+/+ MEFs (apoptotic cells), whereas the driver consisted of cDNA from DNA damage-treated p53+/+ MEFs (G1-arrested cells). In addition, DNA damage-treated E1A–p53−/− populations were included in the driver population to remove messages induced by E1A in a p53-independent manner. We chose to use the DNA-damaging agent doxorubicin for this screen as it effectively induced p53-dependent G1 arrest in MEFs and p53-dependent apoptosis in E1A MEFs at the same dose and with similar kinetics (L.D. Attardi and T. Jacks, in prep.). To prepare the G1-arrested RNA population for the screen, wild-type MEFs were arrested with 0.2 μg/ml doxorubicin. As doxorubicin was shown previously to induce arrest responses in multiple phases of the cell cycle, some of which are p53 independent (L.D. Attardi and T. Jacks, in prep.), we obtained a G1-arrested population by first synchronizing the cells. Wild-type MEFs were synchronized in G0 by serum starvation, stimulated to re-enter the cell cycle by the addition of serum, and shortly thereafter, treated with 0.2 μg/ml doxorubicin for 12 hr. This time point was chosen because we had previously shown it to be a point of peak expression of known p53 target genes (L.D. Attardi and T. Jacks, in prep.). That G1 arrest had occurred was confirmed by FACS analysis, which showed a G1 arrest in the wild-type but not in p53−/− MEFs (Fig. 1B). To prepare apoptotic RNA for the screen, E1A–p53+/+ MEFs were treated with 0.2 μg/ml doxorubicin for 17–18 hr, a time at which apoptosis had clearly been initiated (Fig. 1C) and when p53 target gene expression was maximal (L.D. Attardi, in prep.). At this time point, between 23% and 31% of the cells were undergoing apoptosis (two independent experiments), as assessed by both trypan blue exclusion, which measures loss of membrane integrity, and DAPI staining, which allows analysis of nuclear integrity. When E1A–p53−/− MEFs were treated with 0.2 μg/ml doxorubicin for ∼17–18 hr, they failed to undergo apoptosis, as expected (Fig. 1C).
Figure 1.
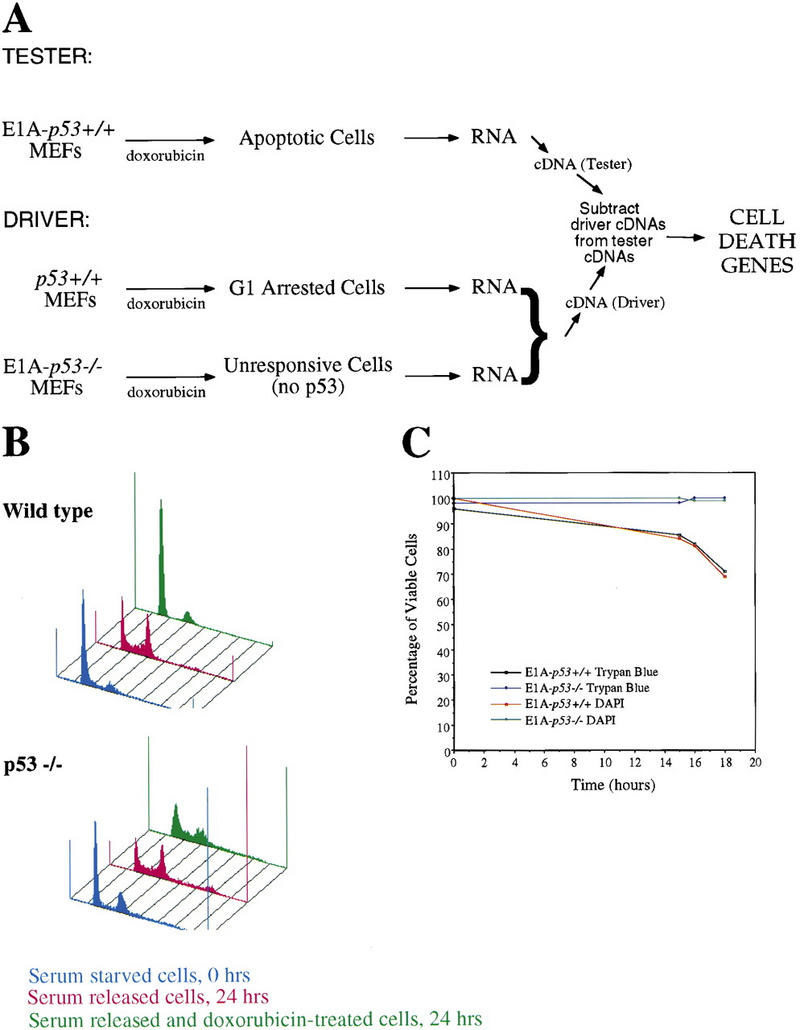
Subtractive hybridization strategy. (A) Scheme for differential screen. cDNA from G1-arrested MEFs (doxorubicin-treated MEFs) and doxorubicin-treated E1A–p53−/− MEFs was pooled and subtracted from cDNA from apoptotic MEFs (doxorubicin-treated E1A–p53+/+ MEFs). (B) Doxorubicin induces a p53-dependent G1 arrest. For the G1-arrested population in the screen, wild-type MEFs were synchronized by serum starvation, released into the cell cycle, and then treated with doxorubicin to induce G1 arrest. FACS profiles of both wild-type and p53-null MEFs are shown and include synchronized cells, cells stimulated to re-enter the cell cycle for 24 hr, and cells G1-arrested with doxorubicin. (C) Doxorubicin induces a p53-dependent apoptotic response in E1A MEFs. Shown here are data from one experiment of two performed to prepare RNA for the subtraction experiment. The percentage of viable E1A–p53+/+ and E1A–p53−/− cells after doxorubicin treatment is plotted as a function of time. Cell death was measured both by trypan blue exclusion, to measure loss of membrane integrity, and DAPI staining, to examine nuclear morphology, and the techniques gave equivalent results.
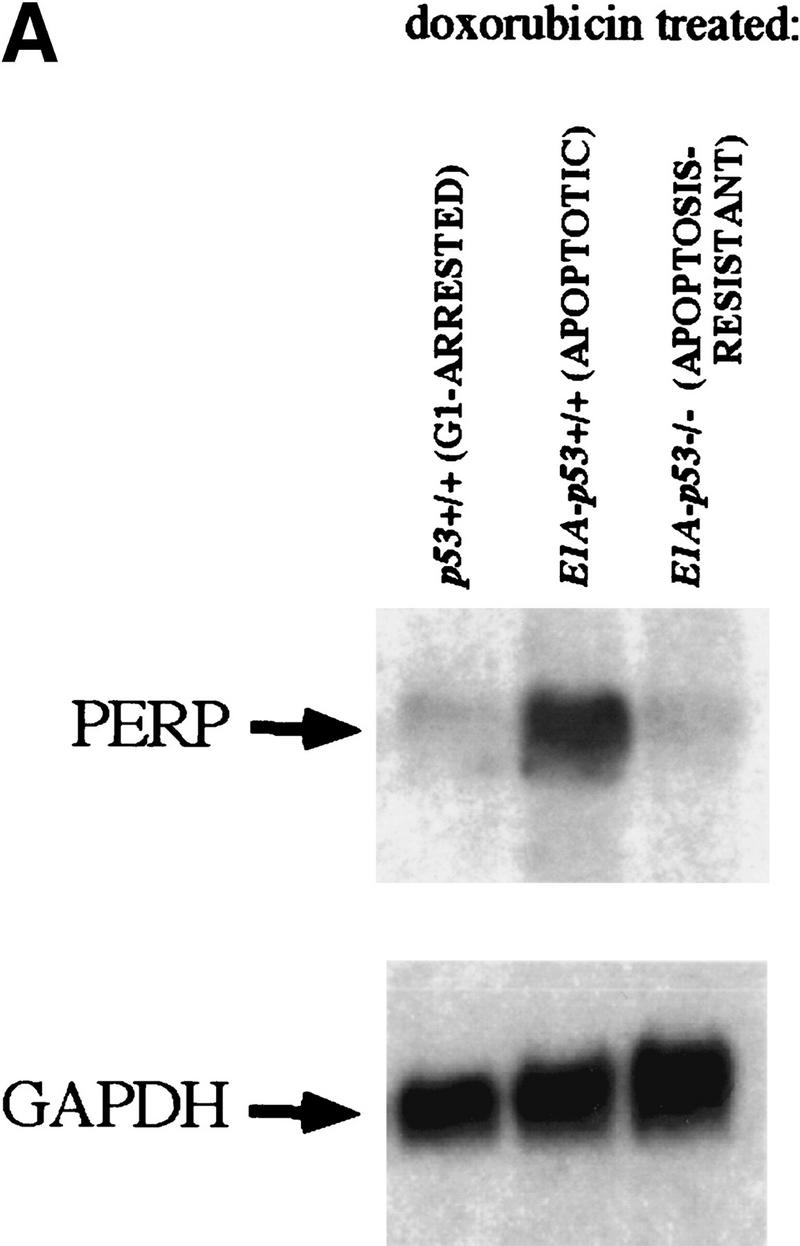
cDNA was made from poly(A)+ RNA derived from each population, and the driver population was subtracted from the tester population using a variant of representational difference analysis (RDA), termed PCR-Select (Clontech; see Materials and Methods). Cloned cDNAs were sequenced to determine their identities and were validated by probing Northern blots to confirm apoptosis-specific expression. Initially, cDNAs were used to probe blots containing RNA from G1-arresting cells (doxorubicin-treated wild-type MEFs), apoptotic cells (doxorubicin-treated E1A–p53+/+ MEFs), and doxorubicin-treated E1A–p53−/− MEFs. One clone was particularly notable because it was isolated multiple times and because it showed a dramatic difference in levels between apoptotic and G1-arrested populations. This gene, PERP (p53 apoptosis effector related to PMP-22), was expressed at high levels in apoptotic cells compared with G1-arrested or doxorubicin-treated E1A–p53−/− cells (Fig. 2A) and thus represented a good candidate for a downstream target of p53 in the apoptotic pathway.
Figure 2.


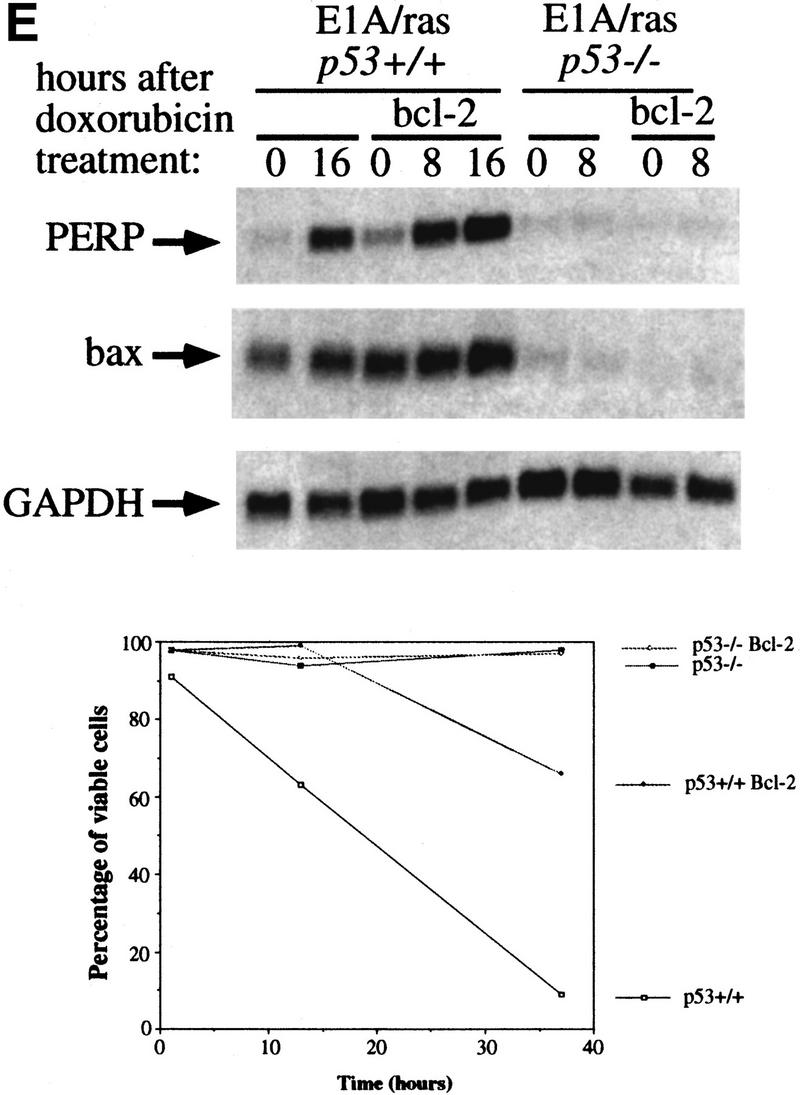
PERP induction is correlated with activation of the p53-dependent apoptotic pathway. (A) Northern blot analysis shows a 1.9-kb message, PERP, that is up-regulated in apoptotic E1A MEFs (middle lane) compared with G1-arrested MEFs (left lane) and E1A–p53−/− MEFs (right lane). The blot was reprobed with GAPDH as a loading control. (B) Time course analysis of PERP and p53 target gene message levels in MEFs undergoing G1 arrest, E1A MEFs undergoing apoptosis, and doxorubicin-treated E1A–p53−/− MEFs. PERP message accumulates to significantly higher levels in apoptotic cells than in G1-arrested cells. The blot was probed with GAPDH as a loading control. (C) PERP is induced in another context of p53-dependent apoptosis. Northern blot analysis shows that PERP is induced as wild-type MEFs undergo apoptosis in response to UV light. It is not induced in the p53−/− MEFs, which are not undergoing apoptosis. The percentages of cells undergoing apoptosis are indicated (bottom). The blot was probed with GAPDH as a loading control. (D) PERP is not induced during p53-independent apoptosis. Northern blot analysis shows RNA derived from TNF-α-treated E1A/ras–p53−/− (transformed) fibroblasts. The percentages of cells undergoing apoptosis are indicated (bottom). For comparison, lane 5 includes RNA from E1A MEFs undergoing apoptosis, in which PERP mRNA levels are much higher than in TNF-α-treated samples. The blot was probed with GAPDH as a loading control. (E) PERP Induction is not inhibited by Bcl-2. Northern blot analysis shows that PERP is still induced in response to doxorubicin in cells that express Bcl-2. Bax levels, in contrast, remain stable in response to doxorubicin. The blot was probed with GAPDH as a loading control. The graph (bottom) shows the cell death profiles in cells of various genotypes, with only p53+/+, non-Bcl-2-expressing cells undergoing apoptosis.
PERP induction is linked specifically to the p53-dependent apoptotic pathway
To characterize PERP more fully, we analyzed its expression in a variety of contexts. We first examined the kinetics of PERP expression during apoptosis by performing a time course analysis. p53+/+, E1A–p53+/+, and E1A–p53−/− MEFs were treated with 0.2 μg/ml doxorubicin, and cells were collected for RNA preparation at various time points after the initiation of DNA damage treatment (Fig. 2B). Northern blot analysis revealed some induction of PERP during G1 arrest, but to very moderate levels. Induction was more clearly observed upon doxorubicin treatment in apoptotic E1A MEFs, with accumulation of PERP message to high levels compared with the G1-arresting population. High-level PERP expression correlated with the induction of cell death, with 21% of cells undergoing apoptosis by 16 hr. PERP was not expressed significantly in E1A–p53−/− MEFs, indicating that both basal expression and induction of this gene by DNA damage are p53 dependent. Accumulation of high levels of PERP gene expression are thus dependent on p53, E1A, and DNA damage, all of which are essential for E1A-induced apoptosis in MEFs.
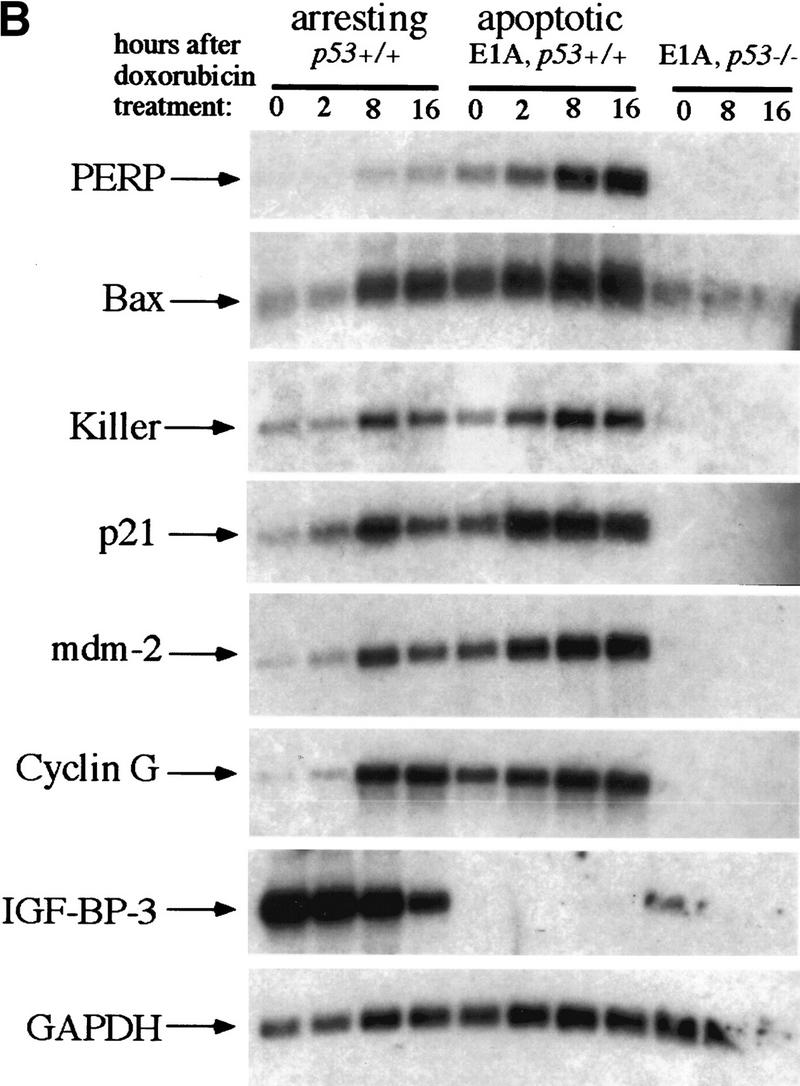
Sequential probing of the time course Northern blot revealed that other p53 target gene mRNAs generally accumulate to similar levels in G1-arresting and apoptotic cells (Fig. 2B). Genes tested included p21, cyclin G, mdm-2, IGF–BP-3, KILLER/DR5, and most interestingly, the proapoptotic regulator bax (for review, see El-Deiry 1998). As Bax is a Bcl-2 family member that is sufficient to induce apoptosis in certain contexts, its activation by p53 has been proposed to lead to apoptosis (Oltvai et al. 1993). In addition, E1A–bax−/− MEFs are partially compromised for apoptosis induced by DNA damage (McCurrach et al. 1997). However, we find that bax is induced to similar levels in arrested and apoptotic cells (Fig. 2B,E), indicating that p53 activation of bax transcription does not dictate the specific fate that MEFs undergo. Curiously, IGF–BP-3, a p53 target gene proposed to be involved in apoptosis by binding and neutralizing the IGF-1 survival factor (Buckbinder et al. 1995), is expressed in arresting but not apoptotic cells and is inhibited by DNA damage treatment. Moreover, although it is not expressed in apoptotic E1A–p53+/+ MEFs, it is expressed in E1A–p53−/− MEFs. KILLER/DR5 is another p53 target gene that has been shown in some systems to be sufficient to induce apoptosis (Wu et al. 1997). Here, we observed that the levels of KILLER/DR5 are similar in arresting and apoptotic cells. In contrast, PERP represents a good candidate to specify the cell death decision, as its levels in apoptotic and arresting cells differ significantly. In addition, the induction kinetics of PERP in response to DNA damage and its p53 dependence are similar to those of other p53 target genes, suggesting that PERP is a direct target of p53 (see below). Also, as with other p53 target genes, basal levels of PERP are higher in E1A MEFs than in wild-type MEFs, because of stabilization of p53 in these cells (Lowe and Ruley 1993).
To determine whether PERP is a component specific to the p53-activated apoptotic pathway or if it is generally induced during apoptosis, we examined the expression of PERP in other apoptotic contexts. To examine the expression of PERP in another situation of p53-dependent cell death, we examined wild-type MEFs treated with UVC light, which undergo p53-dependent apoptosis at early time points (A. de Vries and L.D. Attardi, unpubl.). After 20 J/m2 of UVC treatment, PERP levels increased in wild-type MEFs, concomitant with the induction of apoptosis (Fig. 2C). In contrast, PERP was not induced in p53−/− cells, which also do not undergo apoptosis at these time points. Even as the p53−/− cells initiated apoptosis, however, there was no obvious induction of PERP (Fig. 2C, last lane). These data suggest that induction of PERP is associated specifically with p53-dependent apoptosis. Furthermore, as in the previous analysis, the similar kinetics of induction of PERP and the well-characterized p53 target gene mdm-2 by UVC suggests that PERP is a direct target of p53.
The analysis of transformed p53-deficient MEFs confirmed the specificity of PERP induction for the p53 apoptotic pathway. E1A/ras-expressing-p53−/− MEFs, when treated with TNFα, undergo p53-independent apoptosis (Lanni et al. 1997). Upon induction of cell death by 20 ng/ml TNFα treatment, there was no increase in PERP RNA levels, despite the fact that cell death had been induced (Fig. 2D). Moreover, the levels of expression were much lower than those seen in apoptotic E1A MEFs (Fig. 2D, lane 5). These data indicate that PERP is not generally induced in all contexts of apoptosis in fibroblasts but, rather, that it is linked to the p53-dependent apoptotic response.
To determine whether PERP activation is an early event in the pathway thus far defined for p53-mediated apoptosis, we examined PERP induction in cells blocked in the apoptotic pathway by expression of Bcl-2. Bcl-2 has been shown previously to inhibit p53-induced apoptosis in MEFs (McCurrach et al. 1997), presumably by blocking caspase 9 activation, a late event in the pathway. E1A/ras-expressing cells, which, like E1A-expressing cells, are sensitive to p53-dependent apoptosis, were tested for induction of PERP, either in the absence or presence of Bcl-2 expression. As expected, in the absence of p53, doxorubicin treatment did not induce PERP expression, irrespective of bcl-2 status (Fig. 2E). In the presence of p53, and in the absence of Bcl-2, induction of PERP was observed upon doxorubicin treatment, as seen previously. Interestingly, in the presence of Bcl-2, which successfully blocked apoptosis (see Fig. 2E), PERP was still induced by doxorubicin. Bax, in contrast, was only marginally induced under these conditions. These data indicate that induction of PERP is upstream of the block imposed by Bcl-2 and suggest that its activation is an early event during apoptosis rather than a consequence of cell death. Taken together, these findings support the candidacy of PERP as a key mediator between p53 and the apoptotic machinery.
The PERP promoter is p53 responsive
The p53-dependence and kinetics of PERP expression in response to DNA damage resemble that of other well-defined p53 target genes. To begin to establish whether PERP is a direct transcriptional target of p53, we isolated 4 kb of PERP genomic sequences upstream of the putative transcriptional start site and tested them for responsiveness to p53. Upon visual inspection of this genomic sequence, we found two putative p53 binding sites with significant similarity to the p53 consensus binding site (Fig. 3A). These sites are located at ∼200 bp and ∼2.1 kb upstream of the proposed start site of transcription, which we assigned based on primer extension analysis, cDNA cloning, and EST analysis (data not shown). We also noted additional potential p53 binding sites in the first intron (data not shown). To examine the p53 responsiveness of the PERP promoter, we fused this upstream region to a luciferase reporter construct to create pPERPluc1 and introduced it into MEFs (Fig. 3B). As a negative control, we tested the pGL3Basic vector that serves as the backbone for the PERP reporter construct. As a positive control for p53 responsiveness, we tested the PG13–Luc vector that contains 13 multimerized p53 binding sites (El-Deiry et al. 1993). Upon introduction of the PERP reporter plasmid into wild-type MEFs, there was slightly enhanced activity (2.5-fold) relative to the p53-null MEFs. Upon cointroduction of the PERP reporter and a p53 expression vector into p53-null MEFs, however, a dramatic increase in luciferase activity was observed (29-fold relative to p53-null MEFs without exogenous p53), indicating that the promoter is p53 responsive. The pGL3Basic reporter showed a marginal change in activity upon introduction of p53. The PG13–Luc positive control, like the PERP reporter, displayed increased activity in wild-type relative to p53-null MEFs and even greater activity in p53-null MEFs upon introduction of exogenous p53. This more marked response seen with PG13–Luc than with pPERPluc1 presumably reflects the larger number of p53 binding sites. The MG15–Luc reporter containing mutant p53 sites did not respond to endogenous or exogenous p53. These data support the view that PERP is a direct target of p53, involved in p53-dependent apoptosis.
Figure 3.
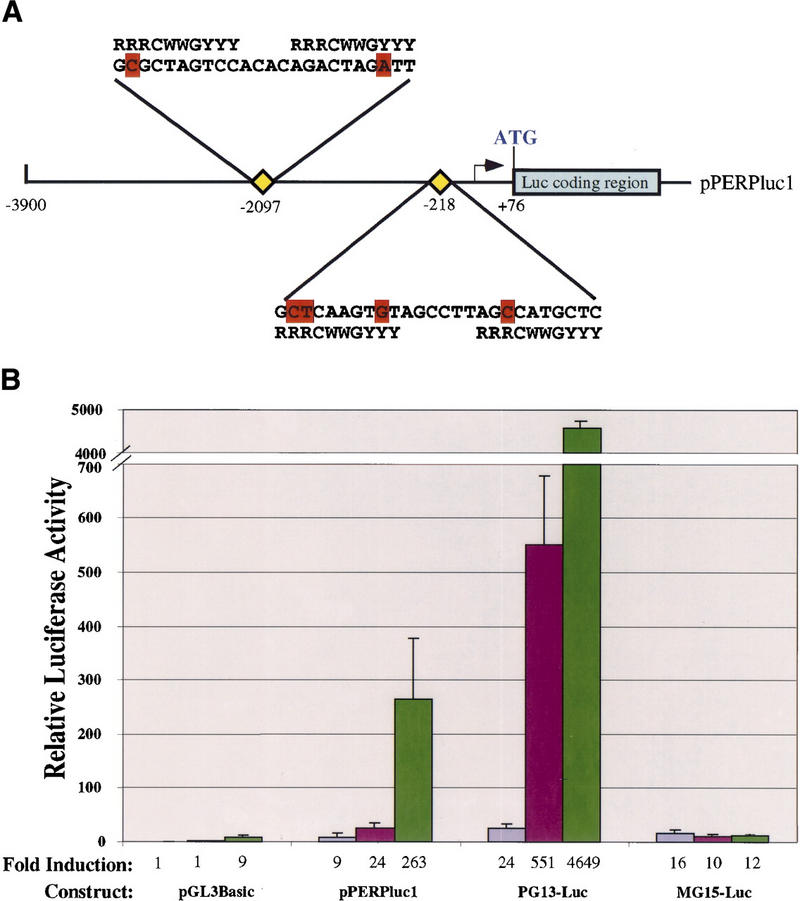
The PERP promoter is p53 responsive. (A) Schematic of the pPERPluc1 luciferase reporter containing the PERP promoter region. The arrow represents the putative start site of transcription. The position of two potential p53 binding sites (at −2097 and −218) with significant homology to the published consensus sequence (18 out of 20 and 16 out of 20 matches, respectively) are indicated by yellow diamonds and their sequences are shown. Mismatches are indicated in red. (B) The 4-kb region containing the PERP promoter is activated by p53. The pPERPluc1 reporter was transfected into p53−/− MEFs (lavender bars), wild-type MEFs (fuchsia bars), or p53−/− MEFs cotransfected with human p53 (green bars). Values shown are given relative to the pGL3Basic backbone plasmid in p53−/− MEFs and represent the average of three separate experiments. Reporters include pPERPluc1 (diagramed above); pGL3Basic luciferase reporter with no promoter/enhancer (Promega); PG13–Luc1 (13 copies of a p53 binding site placed upstream of the polyoma promoter and luciferase); and MG15–Luc (15 copies of a mutant p53 binding site upstream of the polyoma promoter and luciferase).
PERP is induced during E2F-1 activation of the p53-dependent cell death pathway
If PERP is an important player in the p53-dependent cell death pathway, it would be predicted that proliferative signals other than E1A that activate this pathway would induce PERP expression. Thus, we tested whether PERP could be induced by E2F-1, which also causes both inappropriate cell proliferation and p53-dependent cell death (Qin et al. 1994; Wu and Levine 1994). Wild-type E2F-1 or a carboxy-terminal truncation mutant lacking the transactivation domain (E2F-1ΔC) were transduced into wild-type or p53−/− MEFs by retroviral infection. Forty-eight hours after the infection, when wild-type cells expressing full-length E2F-1 were clearly undergoing apoptosis, PERP levels were induced (Fig. 4). This high-level induction was not observed in the cells expressing the transcriptionally compromised form of E2F-1 (E2F-1ΔC) or in p53−/− cells expressing full-length E2F-1, indicating that both transcriptionally active E2F-1 and p53 are required to up-regulate PERP expression to high levels. Examination of the previously characterized E2F-1 target gene cyclin A (Hurford et al. 1997) shows comparable induction in full-length E2F-1-infected wild-type and p53−/− cells (whereas the transactivation mutant E2F-1 failed to induce cyclin A, as expected), indicating that the absence of PERP induction in the p53-null cells was not due to inefficient retroviral infection. Rather, these data suggest a specific link between PERP induction and E2F-1-induced apoptosis.
Figure 4.

PERP is up-regulated during E2F-1-induced cell death. Wild-type and p53-null MEFs were infected with retroviruses carrying either a full-length or a mutant form of E2F-1 lacking the transactivation domain (E2F-1ΔC, comprising residues 1–409 of E2F-1). Forty-eight hours after infection, PERP expression was observed only in the wild-type cells infected with full-length E2F-1, correlating with a visible induction of apoptosis. The E2F-1 target gene cyclin A, in contrast, is induced in full-length E2F-1-infected wild-type and p53−/− MEFs, relative to background endogenous cyclin A levels (seen in E2F-1ΔC-infected cells). The blot was probed with GAPDH as a loading control.
PERP encodes a new member of the PMP-22/gas3 family
The full-length PERP cDNA was cloned by RACE (see Materials and Methods). The size of the isolated cDNA matched the size of the mRNA detected by Northern blot, suggesting that a complete cDNA was obtained. Furthermore, the cloned cDNA matches several entries in the EST database, which included cDNAs with 5′ ends only a few nucleotides longer than the PERP cDNA we isolated. Analysis of the cDNA revealed a 193-amino-acid ORF, encoding a protein of predicted molecular mass of 21 kD (Fig. 5A). Examination of the amino acid sequence by Pfam and BLOCKS analysis identified a similarity to PMP-22 (Fig. 5B), a member of an expanding family of tetraspan membrane proteins associated with growth regulation. This family also includes EMP-1 (epithelial membrane protein 1), EMP-2, EMP-3, and MP20 (lens membrane protein) (for review, see Naef and Suter 1999). Importantly, the PMP-22 gene is implicated in several common human demyelinating peripheral neuropathies, including Charcot–Marie–Tooth Type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP), both characterized by Schwann cell growth deregulation and myelin defects (Naef and Suter 1999). Moreover, PMP-22 was cloned previously and characterized as gas3 (growth arrest specific 3) a gene up-regulated in response to serum starvation of NIH-3T3 cells (Schneider et al. 1988). Similar to PERP, the PMP-22/gas3 message is ∼1.8 kb, with a small open reading frame (ORF; 160 amino acids) (Schneider et al. 1988; Spreyer et al. 1991; Welcher et al. 1991). Additionally, consistent with the sequence similarity of PERP to PMP-22/gas3, the Kyte–Doolittle hydropathy profile prediction for PERP indicates that it also has four membrane-spanning regions (modeled in Fig. 5C). Significantly, however, PMP-22/gas3 regulation is clearly distinct from that of PERP, as demonstrated by analysis of its expression during p53-mediated G1 arrest and apoptosis in the MEF system (Fig. 5D). PMP-22/gas3 levels of expression were highest in the E1A–p53−/− cells, and levels were higher in the G1-arresting population than in the apoptotic population. Taken together, these data indicate that PMP-22/gas3 and PERP, although potentially functionally similar, are expressed in very different patterns.
Figure 5.
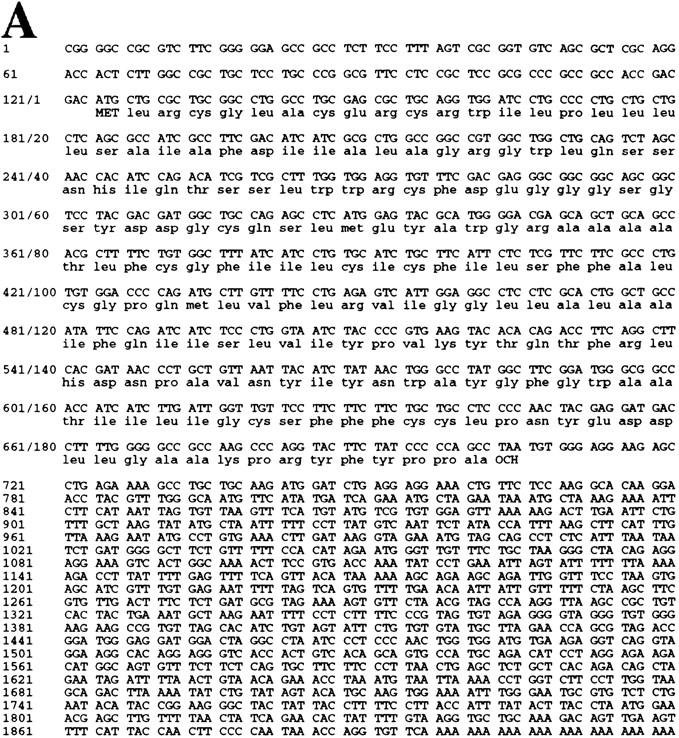
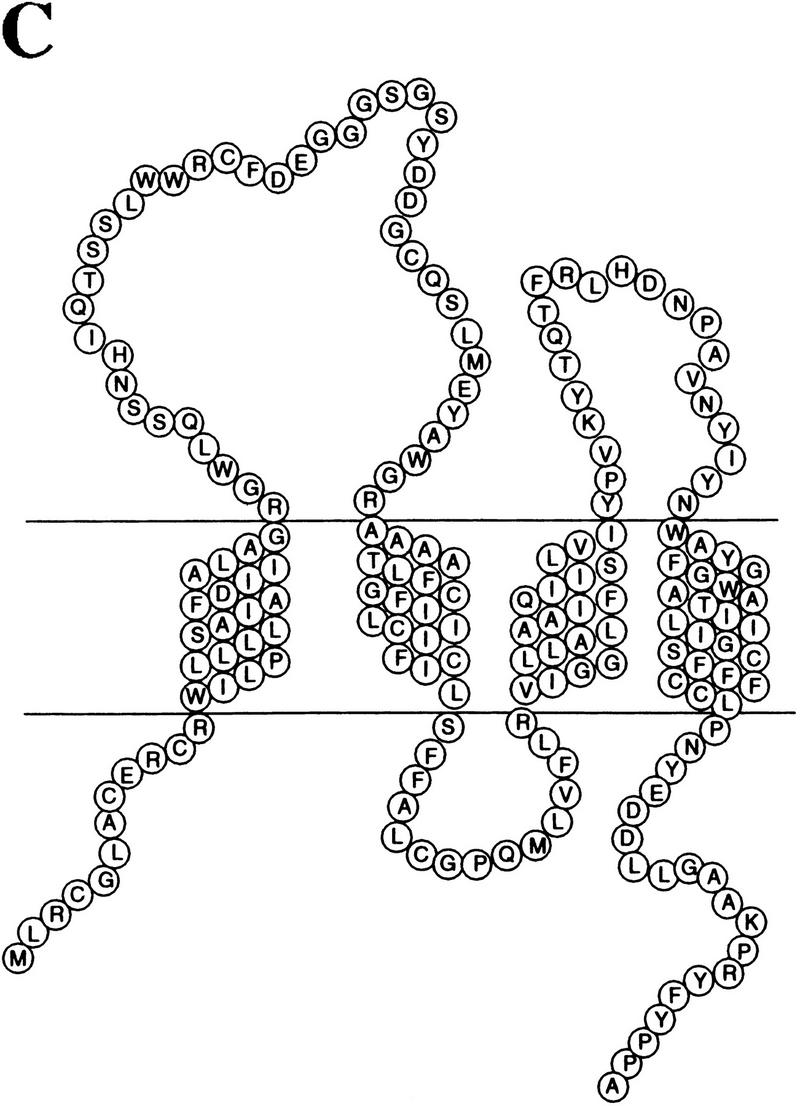

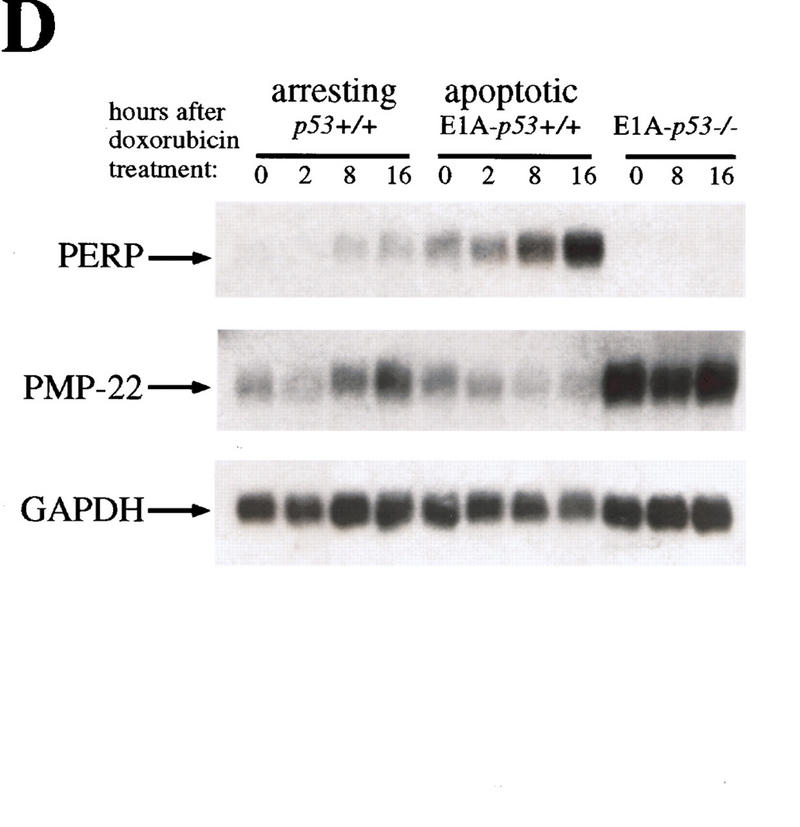
PERP is a new member of the PMP-22/gas3 family. (A) cDNA and amino acid sequence of PERP. (B) Alignment of PERP and PMP-22/gas3 from several species (human, mouse, rat) is shown. Identical amino acids are highlighted in red, and similar amino acids are highlighted in yellow. (C) Molecular model of PERP in the plasma membrane, as determined by transmembrane prediction programs. (D) Northern blot analysis shows that PMP-22/gas3 is expressed in a pattern distinct from PERP during p53-dependent G1 arrest in MEFs and apoptosis in E1A MEFs. The blot was probed with GAPDH as a loading control.
In addition, human EST homologs of mouse PERP were present in the database. According to the Sanger Centre human genome project, PERP has been assigned to human chromosome 6q24. This region is mutated in a variety of cancers, including melanoma, cervical cancer, ovarian cancer, and breast cancer (Millikin et al. 1991; Foulkes et al. 1993; Noviello et al. 1996; Mazurenko et al. 1999), raising the possibility that PERP might be the target of mutation during human tumorigenesis.
PERP localizes to the plasma membrane
The similarity of PERP protein to the PMP-22/gas3 protein family and its hydropathy profile suggested that it is a membrane protein. PMP-22/gas3 is present in the ER, Golgi, and on the plasma membrane (for review, see Naef and Suter 1999). To determine the subcellular localization of PERP, a carboxy terminally HA-tagged variant was expressed in NIH-3T3 or E1A–p53−/− cells. Upon immunostaining with anti-HA antibodies, PERP protein was observed in discrete cytoplasmic vesicular structures and on the plasma membrane (Fig. 6A,C,E). Double immunofluorescence analysis was performed to identify the subcellular structures showing PERP staining. Costaining with MitoTracker to detect mitochondria showed some potential overlap, but many distinct regions as well, suggesting that PERP is not exclusively localized to mitochondria (Fig. 6A,B). Costaining with an antibody directed against β-COP (a Golgi apparatus marker), however, revealed colocalization of PERP and the Golgi apparatus (Fig. 6C–F). This pattern of localization to the secretory pathway and the plasma membrane is reminiscent of that of PMP-22/gas3, supporting the idea that PERP is a bona fide member of the PMP-22/gas3 family. The localization of PERP to the Golgi apparatus is likely to reflect protein passing through the secretory pathway en route to the plasma membrane, although it remains possible that PERP has an intracellular function as well.
Figure 6.
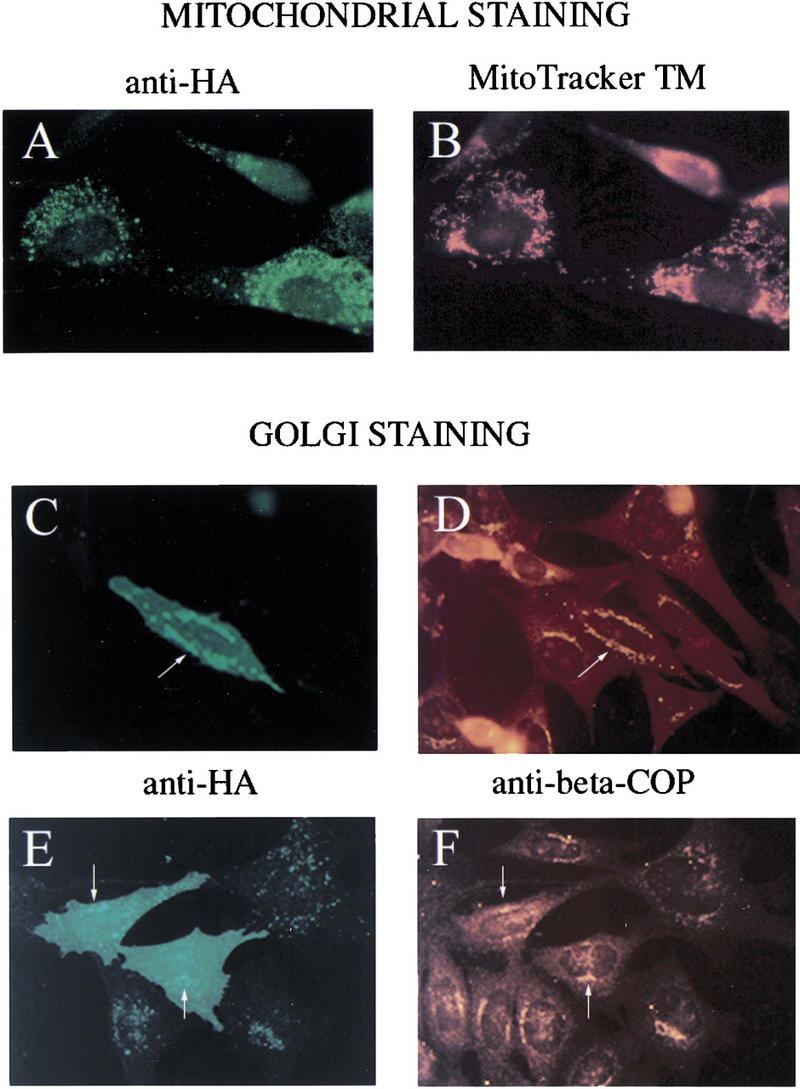
PERP localizes to the Golgi apparatus and the plasma membrane. (A,B) Fibroblasts were transfected with PERP–HA and immunostained with anti-HA antibodies (A) and MitoTracker (B). Many regions of staining are nonoverlapping, suggesting that PERP is not exclusively localized to the mitochondria. (C–F) E1A–p53−/− MEFs were transfected with PERP–HA and immunostained with anti-HA (C,E) and anti-β-COP (D,F). Regions of vesicular HA staining coincide with β-COP staining, suggesting that PERP is localizing to the Golgi apparatus (arrows). In addition, some cells show plasma membrane staining, seen as uniform cell staining (E).
PERP is sufficient to induce cell death
To determine whether PERP might be a downstream mediator of p53-dependent apoptosis, capable of substituting for p53 itself, PERP was introduced into E1A–p53−/− cells. We have shown previously that reintroduction of p53 into these cells resensitizes them to undergo apoptosis (Attardi et al. 1996). E1A–p53−/− cells were transfected with PERP, with p53 as a positive control, or with β-galactosidase as a negative control. After 48 hr, cells were fixed, immunostained for each expressed protein, and examined morphologically. Many cells expressing either p53 or PERP showed characteristic cell rounding and cytoplasmic shrinkage, with blebbing nuclei, typical of apoptotic cells (Fig. 7A). Control cells expressing β-galactosidase, in contrast, exhibited only background levels of cell death. Levels of PERP-induced cell death were intermediate between the background levels and those induced by p53 (Fig. 7B). The difference in potency between p53 and PERP in this assay could be explained by PERP being only one of two or more components of the p53-mediated cell death pathway and, thus, only partially reconstituting the p53 response. Alternatively, this difference could reflect the increased sensitivity of the HA antibody (relative to the p53 antiserum) in detecting low level-expressing cells, which are not as prone to undergo apoptosis. Although such an overexpression assay cannot definitively prove that inducing cell death is an important physiological function of PERP, it is consistent with PERP acting as a p53 cell death target gene.
Figure 7.
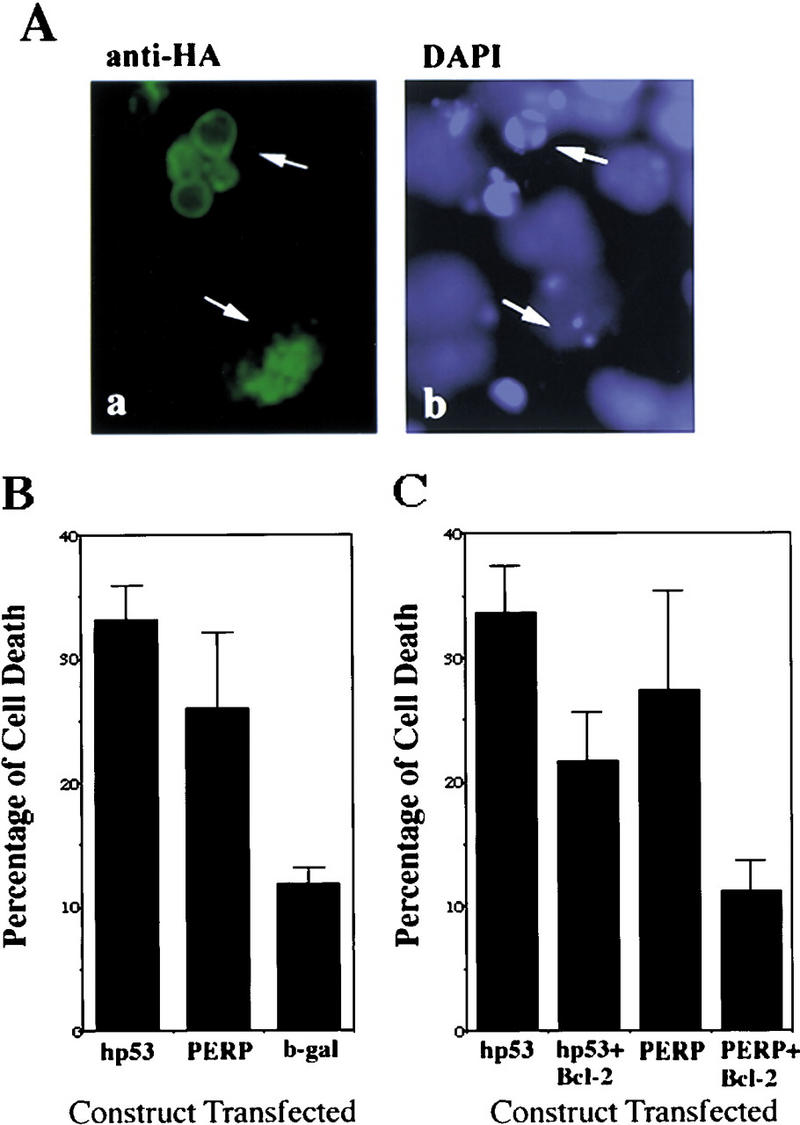
PERP is sufficient to induce cell death. (A) Expression of PERP induces apoptotic morphology. Cells transfected with PERP–HA and immunostained with anti-HA and DAPI show apoptotic morphology (shrunken cell bodies and blebbing nuclei) when examined by immunofluorescence. (B) Expression of PERP induces cell death. PERP induces cell death in E1A–p53−/− MEFs at a level intermediate between p53 and the background level of the negative control (β-galactosidase). Data represent the average of four experiments. (C) Bcl-2 expression inhibits PERP-induced cell death. Coexpression of Bcl-2 lowers the percentage of cells undergoing apoptosis in both p53- and PERP-expressing samples. Data represent the average of three experiments.
To characterize the nature of PERP-induced cell death more fully, we determined the effect of PERP expression in the presence of the general apoptosis inhibitor Bcl-2. Coexpression of Bcl-2 with either p53 or PERP resulted in an inhibition of cell death (Fig. 7C), suggesting that the cell death induced by PERP occurs via apoptosis. Furthermore, these data are consistent with PERP functioning through the same pathway as p53 (McCurrach et al. 1997) and acting as an important mediator of p53-induced apoptosis.
Discussion
Activation of a cellular suicide program is a key mechanism by which p53 acts as a tumor suppressor. Hyperproliferative signals, such as those sensed by incipient tumor cells, activate p53, resulting in the induction of the cell death pathway. Mouse or cell culture model systems in which cells express the oncoproteins E1A, SV40 large T antigen, c-Myc, or E2F-1 show increased p53 levels and a consequent propensity to undergo apoptosis (Lowe et al. 1993; Lowe and Ruley 1993; Hermeking and Eick 1994; Qin et al. 1994; Symonds et al. 1994; Wagner et al. 1994; Wu and Levine 1994). Although the events downstream of p53 in this process have been obscure, recently the steps leading to p53 activation have begun to be elucidated. In response to expression of the E1A or c-Myc oncoproteins, induction of the tumor suppressor protein p19ARF and its effects on the Mdm-2 protein cause p53 to be stabilized (Bates et al. 1998; de Stanchina et al. 1998; Sherr 1998; Zindy et al. 1998). The fact that p19ARF is mutated in a large percentage of human tumors (Sherr 1998) further supports the notion that oncogene-induced stabilization of p53 via p19ARF and the subsequent activation of cell death by p53 are important for p53 tumor suppressor function. In terms of more downstream steps in the oncogene-activated p53 cell death pathway, well-characterized components of the apoptotic machinery, Apaf-1 and caspase-9, as well as the release of cytochrome c from the mitochondria have been shown to be involved (Fearnhead et al. 1998; Juin et al. 1999; Soengas et al. 1999). However, what lies between p53 and the proteolytic events stimulated by the Apaf-1/caspase-9/cytochrome c apoptosome is unknown. One potential component is the p53 target gene bax, a proapoptotic member of the Bcl-2 family. The fact that E1A–bax−/− cells are partially defective for apoptosis implicates Bax as a critical component of the apoptosis pathway in this system (McCurrach et al. 1997). However, from our experiments it is clear that transcriptional activation of bax by p53 is not an obvious determinant of apoptosis, as it accumulates to similar levels during the activation of G1 arrest and apoptosis. This finding is supported by previous studies showing that a mutant form of p53, p53ΔPP1, is capable of inducing Bax normally but is defective in activating apoptosis (Sakamuro et al. 1997). Thus, Bax may act as a rheostat, helping to set the threshold levels of signals to induce apoptosis but not specifying the cell death fate directly.
The mechanism by which p53 impinges on the apoptosis machinery has thus remained largely elusive. Generally speaking, both transcriptional activation-dependent and -independent activities of p53 have been proposed to play a role in this process (Caelles et al. 1994; Wagner et al. 1994; Haupt et al. 1995; Sabbatini et al. 1995; Attardi et al. 1996; Yonish-Rouach 1996). For example, transcriptional repression of genes by p53 has been implicated in apoptosis in some systems (Murphy et al. 1996). In this E1A MEF system, however, we showed previously that transcriptional activation by p53 is crucial for its ability to induce cell death and, furthermore, that no other activities of the protein were obviously required (Attardi et al. 1996). As a result, we sought to identify genes activated by p53 and responsible for mediating apoptosis in this setting, as a model for understanding the cell death that occurs in newly forming tumor cells in vivo.
A number of target genes activated by p53 have been described previously, including p21, mdm-2, bax, GADD45, cyclin G, KILLER/DR5, IGF–BP-3, Fas, and PIG3 (for review, see El-Deiry 1998). The roles of these diverse genes in mediating p53 function are understood to variable extents. For example, p21 is a cyclin-dependent kinase inhibitor known to be essential for mediating the G1 arrest checkpoint function of p53 (Brugarolas 1995; Deng 1995). Mdm-2, which binds p53 and consequently causes it to be degraded by the proteasome, is involved in negative feedback regulation of p53 (for review, see Sherr 1998). Several p53 target genes such as bax, KILLER/DR5, Fas, and PIG3 have been proposed to play roles in apoptosis (El-Deiry 1998). As mentioned previously, Bax is clearly important in some contexts of p53-dependent cell death but not others. KILLER/DR5, a death domain-containing receptor for the TRAIL (TNF-related apoptosis inducing ligand) ligand, is also induced by p53, and its expression is sufficient to induce cell death (Wu et al. 1997), but its requirement for p53-dependent cell death is unknown. In addition, transcriptional activation of Fas, another death-domain containing protein, by p53 has been documented (Owen-Scaub et al. 1995), although there is also evidence indicating that Fas is dispensable for p53-mediated apoptosis (O'Connor and Strasser 1999). Finally, the PIGs, which are collectively induced upon overexpression of p53 in a colon cancer cell line, encode proteins that are generally connected to the redox state of the cell. This has led to the hypothesis that p53 acts by affecting the production of reactive oxygen species (ROS), leading to cell death (Polyak et al. 1997). Although this remains an interesting hypothesis, the requirement for the PIGs genes for p53-dependent cell death has not been established. The number of different targets related to p53-dependent apoptosis may suggest that these diverse genes may be relevant in different contexts of p53-dependent apoptosis.
Here, by examining two of these proapoptotic genes, bax and KILLER/DR5, we show that the levels of p53 activation of transcription of these genes does not correlate with the fate that cells undergo, as they show similar levels of accumulation during G1 arrest and apoptosis in this system. We also attempted to examine PIG3 in our MEF system, but we were unable to detect its expression. Previous data have suggested, however, that PIG genes are induced during both G1 arrest and apoptosis in colon cancer cell lines (Polyak et al. 1997). Similar induction of these known p53 target genes in G1 arrest and apoptosis may be a result of having been identified in screens in which tumor cell lines lacking p53 and counterparts overexpressing p53 were compared, without a selection for genes specifically involved in apoptosis (El-Deiry et al. 1993; Okamoto and Beach 1994; Buckbinder et al. 1995; Polyak et al. 1997).
Various lines of evidence suggest that the PERP gene isolated here is a bona fide p53 target gene. The expression profiles of PERP resemble those of other p53 target genes in their kinetics of induction during DNA-damaging agent treatment and in their p53 dependence. Furthermore, evidence for direct regulation of PERP comes from analysis of the PERP promoter, which we have shown contains two putative p53 binding sites and is functionally responsive to p53. PERP is different, however, from other p53 target genes in that it is expressed to significantly higher levels in apoptotic MEFs than in G1-arresting MEFs. This suggests that there may be additional regulatory elements in the promoter that dictate the high level of PERP expression seen during apoptosis. Defining the cis-acting elements involved in PERP expression may help identify trans-acting factors that cooperate with p53 to induce apoptosis.
PERP represents a new member of the PMP-22/gas3 family of tetraspan transmembrane proteins implicated in cell growth regulation (Naef and Suter 1999). PMP-22/gas3 is the best characterized of this family, having been identified in two independent contexts. It was originally identified as a transcript down-regulated during sciatic nerve injury and consequent increased Schwann cell proliferation in culture (Spreyer et al. 1991; Welcher et al. 1991) and then as the gas3 gene up-regulated upon serum starvation of NIH-3T3 cells (Schneider et al. 1988). PMP-22/gas3 was subsequently found to be a commonly altered gene in human demyelinating hereditary neuropathies including CMT1A, HNPP, and Dejerrine Sottas syndrome (DSS) (Naef and Suter 1999). Each of these diseases has a somewhat different phenotype, with the observed spectrum and severity of symptoms in these patients depending on gene dosage (Scherer and Chancer 1995). In addition, the “Trembler” mouse carries point mutations in PMP-22/gas3 and has a series of symptoms consistent with the disease in humans (Naef and Suter 1999).
Several lines of evidence have suggested that PMP-22/gas3 plays dual roles: one as a structural component of myelin and the other as a cell growth regulator (Naef and Suter 1999). The original clue to its growth regulatory function was suggested by the fact that it is induced selectively during serum starvation in NIH-3T3 cells and Schwann cells. Subsequently, overexpression of PMP-22/gas3 was shown to induce G1 arrest in Schwann cells (Zoidl et al. 1995). Furthermore, overexpression of PMP-22/gas3 in NIH-3T3 fibroblasts induces cell death, which is inhibitable by both Bcl-2 and DEVD caspase inhibitors, indicating that it occurs through apoptosis (Fabretti et al. 1995; Zoidl et al. 1997; Brancolini et al. 1999). Disease-associated PMP-22/gas3 mutants were inactive in this apoptosis assay, suggesting that loss of this function may be relevant to pathogenesis. These findings are provocative given the ability of PERP to induce cell death and suggest further that PERP may be functionally similar to PMP-22/gas3.
Activation of PERP, a putative tetraspan transmembrane protein at the plasma membrane, suggests a novel mechanism for p53-induced apoptosis, and various possibilities could be envisioned to explain how PERP activates cell death. The previously characterized p53 targets KILLER/DR5 and Fas are so-called death domain-containing receptors, which bind to adaptor proteins, thereby sending signals from the plasma membrane to directly activate the caspases (El-Deiry 1998). Perhaps analogously to Killer and Fas, PERP could serve as a cell death receptor, albeit a different type with four membrane-spanning domains, to receive either autocrine or paracrine signals. Alternatively, a sequence similarity between PERP and the calcium channel γ-subunit (L.D. Attardi and T. Jacks, unpubl.) suggests that it could have channel or pore activity, perhaps allowing some crucial molecule important for activating apoptosis to pass through. However it exerts its function, PERP might ultimately act by affecting regulators of the apoptotic machinery, such as Bax or Bcl-2, or by directly activating apoptotic effectors like caspases. PERP and PMP-22/gas3, both inducers of cell death, may thus constitute a family of cell death proteins that act through a novel mechanism.
The cell death activated by p53 in response to hyperproliferation is greatly stimulated by cellular stresses such as DNA damage and hypoxia (Lowe et al. 1993; Graeber et al. 1996). Moreover, activation of p53-dependent apoptosis by stress is thought to be a mechanism by which many chemotherapeutic agents act on human tumors (Lowe 1995). Therefore, understanding the p53-mediated apoptosis pathway is crucial not only for understanding its mode of tumor suppression but also for devising new cancer therapies aimed at reconstituting the apoptosis response in the many tumors that lack p53. Finally, the map location of PERP is consistent with it being a tumor suppressor. PERP has been assigned to human chromosome 6q24, a region implicated in the development of many kinds of cancer. For example, loss of heterozygosity within this region is associated with ovarian, breast, and cervical cancers, as well as melanoma (Millikin et al. 1991; Foulkes et al. 1993; Noviello et al. 1996; Mazurenko et al. 1999). PERP, as an apoptosis-specific target gene of p53, might be the target of mutation in such cancers. Future experimentation will unravel the role of PERP both in the p53 apoptotic pathway and in tumor suppression.
Materials and methods
Preparation of G1-arrested RNA populations
Wild-type and p53−/− MEFs were isolated as described previously (see Attardi et al. 1996). To prepare the G1-arrested RNA population for the subtractive hybridization, wild-type MEFs (passage 4) were synchronized by first growing to confluence in DMEM plus 10% FCS for 3 days and then in DMEM plus 0.1% FCS for 4 days. To stimulate re-entry into the cell cycle, the cells were reseeded into DMEM plus 10% FCS at 1.0 × 106 cells per 10-cm dish. After 6 hr, 0.2 μg/ml doxorubicin (Sigma) was added to induce G1 arrest. After 12 hr of doxorubicin treatment, cells were collected by trypsinization and frozen as a pellet for subsequent RNA preparation. Dishes of both treated and untreated wild-type and p53−/− MEFs were collected after 18 hr of doxorubicin treatment for FACS analysis to verify a p53-dependent G1 arrest, as described previously (Brugarolas et al. 1995).
Preparation of apoptotic RNA populations
To obtain E1A-expressing MEFs, both wild-type and p53−/− MEFs were retrovirally infected as described (McCurrach et al. 1997). To generate apoptotic E1A–p53+/+ and doxorubicin-treated E1A–p53−/− cells for RNA preparation, cells were plated at ∼2.5 × 106 cells per 10-cm dish. In addition, to measure apoptosis concurrently, cells of each genotype were plated into wells of a 24-well tissue culture dish at 2.5 × 104 per well. All cells were treated with 0.2 μg/ml doxorubicin. For RNA samples, cells were collected after 17–18 hr of doxorubicin treatment, a time at which 23% or 31% of E1A–p53+/+ cells (two independent experiments) and 0%–1% of E1A–p53−/− cells had undergone apoptosis. Apoptosis levels were measured by collecting both floating and adherent cells from individual 24 wells and determining the percentage of dead cells by trypan blue staining as described (McCurrach et al. 1997). The percentages of apoptotic cells were independently confirmed by staining the cells with DAPI and counting the percentages showing typical apoptotic nuclear morphology (Attardi et al. 1996).
RNA preparation
Total RNA was prepared from MEFs pelleted and frozen in liquid nitrogen using Ultraspec RNA (Biotecx), according to the manufacturer's instructions. Poly(A)+ RNA was prepared using Poly(A)Quik mRNA Isolation Kit (Stratagene), also according to the manufacturer's directions.
Subtractive hybridization protocol
The subtractive hybridization protocol was performed using the PCR-Select cDNA subtraction kit (Clontech), according to the manufacturer's instructions. Tester cDNA was prepared from ∼2 μg of poly(A)+ RNA derived from doxorubicin-treated E1A–p53+/+ MEFs. Driver cDNA was made from ∼2 μg each of doxorubicin-treated p53+/+ MEF and doxorubicin-treated E1A–p53−/− MEF poly(A)+ RNA mixed together. At the end of the protocol, the PCR products were cloned into the pT7-Blue vector (Novagen). Colonies were subjected to differential screening analysis (Clontech) to eliminate false positives, as described by the manufacturer. Positive clones were analyzed further by sequencing and Northern blotting.
Northern blot analysis
Northern blotting was performed using standard methods. Prehybridization and hybridization were performed using ExpressHyb hybridization solution (Clontech). Probes were prepared using a Prime-It II Random Primer Labeling Kit (Stratagene). To strip blots for reprobing, they were incubated 2 × 15 min in boiling water. cDNAs corresponding to Bax, p21, GAPDH, KILLER/DR5, mdm-2, cyclin G, and cyclin A2 were used as probes (Macleod et al. 1995; Hurford et al. 1997; El-Deiry et al. 1998). EST cDNAs corresponding to IGF–BP3 (IMAGE no. 555520) and PMP-22 (IMAGE no. 1480748) were used as probes.
Time course Northerns
MEFs (1 × 106–3 × 106) of the desired genotype were plated per 10-cm dish for each time point. For the UV assays, apoptosis was induced by removing the medium from the cells and directly exposing the cells (without the lid) to 20 J/m2 UV light using a Stratalinker UV cross-linker (Stratagene). For the TNF-α assays, cells were treated with 20 ng/ml TNF-α (Boehringer Mannheim). At the indicated time points, cells were collected and pellets were frozen for subsequent RNA preparation. At each time point, cell death was measured by trypan blue exclusion.
Luciferase assays
Luciferase assays were performed using the Dual Luciferase System (Promega). MEFs were plated 12 hr before transfection at 2.5 × 104 cells per well of a 24-well plate. A hundred and seventy nanograms of the pPERPluc1 reporter or a control vector was transfected into p53−/− MEFs, wild-type MEFs, or p53−/− MEFs cotransfected with 170 ng of a human p53 expression vector (hp53ΔEGFP; K.Tsai, unpubl.). Transfections were performed using Fugene 6 (Boehringer Mannheim) according to manufacturer's directions. Twenty microliters of lysate was used per luciferase assay. The pRLSV40 vector encoding Renilla luciferase was cotransfected in all conditions at a ratio of 1:50 relative to the reporter, as a control for transfection efficiency. Background luminescence readings in nontransfected control cells were subtracted from all reporter luciferase readings, and data were then normalized to the Renilla luciferase levels.
The pPERPluc1 reporter was constructed by site-directed mutagenesis of the initial ATG of the PERP coding region (to create an NcoI site) using the Quick Change Site Directed mutagenesis kit (Stratagene), followed by joining of the 4-kb PERP promoter region to the initial ATG of luciferase in the pGL3Basic vector (Promega). The PG13–Luc and MG15–Luc vectors were gifts of Wafik El-Deiry.
Cloning PERP full-length cDNA
The full-length PERP cDNA was isolated by 5′ and 3′ RACE using the Marathon cDNA Amplification Kit (Clontech). The sequence was analyzed by MOTIF (http://www.motif.genome.ad.jp) and Pfam (http://www.sanger.ac.uk/Software/Pfam/). Topology was examined with the TmPred program (http://www.ch.embnet.org/software/TMPRED_form.html). The similarity to the calcium channel was noted with the SSEARCH program (http://genome.eerie.fr/bin/ssearch-guess.cgi). Alignment of PERP with PMP-22 family members was performed with the DNAStar program Megalign. The human chromosomal map position of PERP in the human genome was based on the Sanger Centre estimate (http://www.sanger.ac.uk/HGP/Chr6).
Mammalian expression studies
The PERP ORF was amplified by PCR using primers containing Sal1 sites and was cloned into the KA mammalian expression vector (a pcDNA3 derivative encoding the HA tag; R. Shaw, pers. comm.). The resulting vector is KA–PERP–HA, encoding a form of PERP with the HA tag at the carboxyl terminus. pCEP4-hp53 and CMV–β-galactosidase (for reference, see Attardi et al. 1996) have been described previously, and pcDNA-3hu Bcl-2 was the gift of G. Nunez, University of Michigan, Ann Arbor.
MEFs grown on polylysine-coated glass coverslips were transfected with expression constructs using Fugene 6 as described above. Transfected cells were immunostained 24–48 hr after transfection, as described previously (Attardi et al. 1996). To detect PERP–HA, cells were stained with a monoclonal anti-HA antibody (1:1000; Boehringer Mannheim). For the staining of the Golgi apparatus, anti-β-COP (1:100; Affinity Bioreagents, Inc.) was used. To detect mitochondria, Mito-Tracker (Molecular Probes) was used according to the manufacturer's instructions. To detect p53, human p53 monoclonal antibody 1801 (1:50; Oncogene Science) was used. To detect β-galactosidase, a rabbit anti-β-galactosidase antibody was used (1:50, 5′–3′). FITC-coupled anti-mouse antibodies and rhodamine-coupled anti-rabbit antibodies (Cappel, 1:1000) were used to visualize monoclonal and polyclonal antibodies, respectively. DAPI (1 μg/ml) was used to stain nuclei. Cell death was assessed by examining the morphology of positively staining cells.
Retroviral infections
Wild-type and p53−/− MEFs were infected with wild-type or mutant E2F-1ΔC (Δ amino acids 410–437) retroviruses, as described previously (McCurrach et al. 1997), using pMIG–E2F-1 and pMIG–E2F-1ΔC constructs (K. Tsai, unpubl.). Forty-eight hours after the infections were initiated, MEFs were collected for RNA preparation as described above.
Acknowledgments
We thank Ken Tsai, Reuben Shaw, Karen Cichowski, Karlyne Reilly, and Raluca Verona for critical reading of the manuscript. We thank Dan Chasman for help with sequence analysis programs and Matt Petitt for assistance with graphics. We are grateful to Melissa Rolls and Jamie White for expertise and reagents to examine PERP subcellular localization. We thank Annemieke deVries for assistance with the UV-induced apoptosis assays. We appreciate the advice of Andy Samuelson and Marisol Soengas on retroviral infections. We are grateful to Wafik El-Deiry for the KILLER/DR5 probe and the PG13–Luc and MG15–Luc plasmids, to Gigi Lozano for the mdm-2 probe, to Jackie Lees for the cyclin A2 and cyclin G probes, and to Gabriel Nunez for the Bcl-2 expression plasmid. This work has been supported by funding from the American Cancer Society, the Bunting Institute at Radcliffe College, and Merck to L.D.A. S.W.L. is a Rita Allen Foundation Scholar. T.J. is an Associate Investigator at the Howard Hughes Medical Institute. This work has been supported by grant CA13106 to S.W.L., and Howard Hughes Medical Institute and NCI funding to T.J.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tjacks@mit.edu; FAX (617) 253-9863.
References
- Attardi LD, Lowe SW, Brugarolas J, Jacks T. Transcriptional activation by p53, but not induction of the p21 gene, is essential for oncogene-mediated apoptosis. EMBO J. 1996;15:3693–3701. [PMC free article] [PubMed] [Google Scholar]
- Attardi LD, Jacks T. The role of p53 in tumour suppression lessons from mouse models. Cell Mol Life Sci. 1999;55:48–63. doi: 10.1007/s000180050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. p14ARF links the tumour suppressors RB and p53. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- Brancolini C, Marzinotto S, Edomi P, Agostoni E, Fiorentini C, Muller HW, Schneider C. Rho-dependent regulation of cell spreading by the tetraspan membrane protein Gas 3/PMP22. Mol Biol Cell. 1999;10:2441–2459. doi: 10.1091/mbc.10.7.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GH. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377:646–649. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- Caelles C, Helmberg A, Karin M. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature. 1994;370:220–223. doi: 10.1038/370220a0. [DOI] [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes & Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge S, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- El-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol. 1998;8:345–357. doi: 10.1006/scbi.1998.0097. [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler K, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Fabretti E, Edomi P, Brancolini C, Schneider C. Apoptotic phenotype induced by overexpression of wild-type gas3/PMP22: Its relation to the demyelinating peripheral neuropathy CMT1A. Genes & Dev. 1995;9:1846–1856. doi: 10.1101/gad.9.15.1846. [DOI] [PubMed] [Google Scholar]
- Fearnhead HO, Rodriguez J, Govek EE, Guo W, Kobayashi R, Hannon G, Lazebnik YA. Oncogene-dependent apoptosis is mediated by caspase-9. Proc Natl Acad Sci. 1998;95:13664–13669. doi: 10.1073/pnas.95.23.13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes WD, Ragoussis J, Stamp GW, Allan GJ, Trowsdale J. Frequent loss of heterozygosity on chromosome 6 in human ovarian carcinoma. Br J Cancer. 1993;67:551–559. doi: 10.1038/bjc.1993.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes & Dev. 1999;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia mediated selection of cells with diminished apoptotic potential in solid tumors. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes & Dev. 1995;9:2170–2183. doi: 10.1101/gad.9.17.2170. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Hurford RK, Cobrinik D, Lee M, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes & Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- Juin P, Hueber AO, Littlewood T, Evan G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome C release. Genes & Dev. 1999;13:1367–1381. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan M, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Lanni JS, Lowe SW, Licitra EJ, Liu JO, Jacks T. p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc Natl Acad Sci. 1997;94:9679–9683. doi: 10.1073/pnas.94.18.9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Lowe SW. Cancer therapy and p53. Curr Opin Oncol. 1995;7:547–553. doi: 10.1097/00001622-199511000-00013. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus-5 E1A and accompanies apoptosis. Genes & Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- Lowe SM, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- Macleod K, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21, during cell growth, differentiation and DNA damage. Genes & Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JFJ, Nelson CE, Kim DH. Germline p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- Mazurenko N, Attaleb M, Gritsko T, Semjonova L, Pavlova L, Sakharova O, Kisseljov F. High resolution mapping of chromosome 6 deletions in cervical cancer. Oncol Rep. 1999;6:859–863. doi: 10.3892/or.6.4.859. [DOI] [PubMed] [Google Scholar]
- McCurrach ME, Connor TMF, Knudson CM, Korsmeyer SJ, Lowe SW. Bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci. 1997;94:2345–2349. doi: 10.1073/pnas.94.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millikin D, Meese E, Vogelstein B, Witkowski C, Trent J. Loss of heterozygosity for loci on the long arm of chromosome 6 in human malignant melanoma. Cancer Res. 1991;51:5449–5453. [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Murphy M, Hinman A, Levine AJ. Wild-type p53 negatively regulates the expression of a microtubule-associated protein. Genes & Dev. 1996;10:2971–2980. doi: 10.1101/gad.10.23.2971. [DOI] [PubMed] [Google Scholar]
- Naef R, Suter U. Many facets of the peripheral myelin protein PMP22 in myelination and disease. Micr Res Tech. 1999;41:359–371. doi: 10.1002/(SICI)1097-0029(19980601)41:5<359::AID-JEMT3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Noviello C, Coujal F, Theillet C. Loss of heterozygosity on the long arm of chromosome 6 in breast cancer: Possibly four regions of deletion. Clin Cancer Res. 1996;2:1601–1606. [PubMed] [Google Scholar]
- O'Connor L, Strasser A. Fas, p53, and apoptosis. Science. 1999;284:1431b. (in Technical Comments). [Google Scholar]
- Okamoto K, Beach D. Cyclin G is a transcriptional target of the tumor suppressor protein p53. EMBO J. 1994;13:4816–4823. doi: 10.1002/j.1460-2075.1994.tb06807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerated programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Owen-Scaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang W-W, Kruzel E, Radinsky R. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol. 1995;15:3032–3040. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Kaelin W, Jr, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KM, Vousden KH. Characterisation of structural p53 mutants which show selective defects in apoptosis but not cell cycle arrest. Mol Cell Biol. 1998;18:3692–3698. doi: 10.1128/mcb.18.7.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbatini P, Lin J, Levine AJ, White E. Essential role for p53-mediated transcription in E1A-induced apoptosis. Genes & Dev. 1995;9:2184–2192. doi: 10.1101/gad.9.17.2184. [DOI] [PubMed] [Google Scholar]
- Sakamuro D, Sabbatini P, White E, Prendergast GC. The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene. 1997;15:887–898. doi: 10.1038/sj.onc.1201263. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Chancer PF. Myelin genes: Getting the dosage right. Nat Genet. 1995;11:226–228. doi: 10.1038/ng1195-226. [DOI] [PubMed] [Google Scholar]
- Schneider C, King RM, Philipson L. Genes specifically expressed at growth arrest of mammalian cells. Cell. 1988;54:787–793. doi: 10.1016/s0092-8674(88)91065-3. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Tumor surveillance via the ARF–p53 pathway. Genes & Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- Soengas M, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW. Apaf-1 and Caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- Spreyer P, Kuhn G, Hanemann CO, Gillen C, Schaal H, Kuhn R, Lemke G, Muller HW. Axon-regulated expression of a Schwann cell transcript that is homologous to a 'growth arrest-specific' gene. EMBO J. 1991;10:3661–3668. doi: 10.1002/j.1460-2075.1991.tb04933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, Van Dyke T. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21 waf1/cip1. Genes & Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Welcher AA, Suter U, De Leon M, Jackson Snipes G, Shooter EM. A myelin protein is encoded by the homologue of a growth arrest-specific gene. Proc Natl Acad Sci. 1991;88:7195–7199. doi: 10.1073/pnas.88.16.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GS, Burns TF, McDonald ER, III, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, El-Deiry W. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–143. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci. 1994;91:3802–3806. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature. 1997;385:637–640. doi: 10.1038/385637a0. [DOI] [PubMed] [Google Scholar]
- Yonish-Rouach E. Transcriptional activation plays a role in the induction of apoptosis by transiently transfected wild-type p53. Oncogene. 1996;12:2197–2205. [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoidl G, Blass-Kampmann S, D'Urso D, Schmalenbach C, Muller HW. Retroviral-mediated gene transfer of the peripheral myelin protein PMP-22 in Schwann cells: Modulation of cell growth. EMBO J. 1995;14:1122–1128. doi: 10.1002/j.1460-2075.1995.tb07095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoidl G, D'Urso D, Blass-Kampmann S, Schmalenbach C, Kuhn R, Muller HW. Influence of elevated expression of rat wild-type PMP22 and its mutant PMP22Trembler on cell growth of NIH3T3 fibroblasts. Cell Tissue Res. 1997;287:459–470. doi: 10.1007/s004410050770. [DOI] [PubMed] [Google Scholar]