

Abstract
The Caenorhabditis elegans HSN motor neurons permit genetic analysis of neuronal development at single-cell resolution. The egl-5 Hox gene, which patterns the posterior of the embryo, is required for both early (embryonic) and late (larval) development of the HSN. Here we show that ham-2 encodes a zinc finger protein that acts downstream of egl-5 to direct HSN cell migration, an early differentiation event. We also demonstrate that the EGL-43 zinc finger protein, also required for HSN migration, is expressed in the HSN specifically during its migration. In an egl-5 mutant background, the HSN still expresses EGL-43, but expression is no longer down-regulated at the end of the cell’s migration. Finally, we find a new role in early HSN differentiation for UNC-86, a POU homeodomain transcription factor shown previously to act downstream of egl-5 in the regulation of late HSN differentiation. In an unc-86; ham-2 double mutant the HSNs are defective in EGL-43 down-regulation, an egl-5-like phenotype that is absent in either single mutant. Thus, in the HSN, a Hox gene, egl-5, regulates cell fate by activating the transcription of genes encoding the transcription factors HAM-2 and UNC-86 that in turn individually control some differentiation events and combinatorially affect others.
Keywords: ham-2, egl-5, unc-86, Hox, neuronal migration, zinc finger
Nervous systems are made of many different types of neurons that differ in position, morphology, connectivity, and neurotransmitter expression. This diversity is generated through transcription factors that direct each neuron’s program of differentiation. Transcription factors required for neuronal development have been identified in vertebrates (Bang and Goulding 1996; DeLapeyriere and Henderson 1997) as well as in Caenorhabditis elegans (Sengupta and Bargmann 1996; Ruvkun 1997), but ordering these transcription factors into regulatory pathways and defining the aspects of cell fate that they control has been difficult.
The establishment of an anteroposterior pattern in the embryo is an early step in the differentiation of neurons, ensuring that neurons in each part of the nervous system will adopt the proper fates. This anteroposterior patterning is established by the regional expression of each member of a cluster of homeobox genes known as Hox genes (Lewis 1978; McGinnis et al. 1984). Hox genes pattern structures as diverse as the segments of Drosophila embryos and the segments of the mammalian hindbrain (Lewis 1978; Keynes and Krumlauf 1994). Mutations in C. elegans and mouse Hox genes both cause transformations in cell fates and defects in neuronal migrations (Salser and Kenyon 1994; Studer et al. 1996).
Although the means by which an initial anteroposterior morphogenetic gradient generates the normal pattern of Hox gene expression have been described in exquisite detail in Drosophila (for review, see St Johnston and Nüsslein-Volhard 1992), how Hox gene products and other transcription factors work together subsequently to specify cellular identity is much less understood. The mechanisms by which the Hox transcription factors activate downstream genes and the identities of these target genes are still being elucidated (for review, see Graba et al. 1997). Some researchers have used the DNA-binding property of Hox homeodomains to identify candidate downstream genes that might determine cell fates. Hox proteins bind the promoters of genes encoding adhesion molecules such as connectin and NCAM, for example, but whether Hox proteins activate these genes directly in vivo has not been fully established (Gould et al. 1990; Gould and White 1992; Edelman and Jones 1998).
Genetic analysis offers another approach to understanding how Hox transcription factors influence cell differentiation. For example, several long-distance cell migrations in C. elegans are under the control of Hox genes. The Hox genes lin-39 and mab-5 have been shown to function cell-autonomously in guiding the migrations of the QR and QL neuroblasts, respectively (Kenyon 1986; Clark et al. 1993). The anteriorly migrating QR neuroblast can be redirected posteriorly by pulses of mab-5 administered by heat shock during its migration (Salser and Kenyon 1992; Harris et al. 1996). The fact that QR responds quickly to these mab-5 pulses suggests that the regulation of cell migrations by these Hox genes is relatively direct and not through a multi-tiered transcriptional cascade. Despite the Hox genes’ importance in directing Q-neuroblast migrations, all known mutations that affect the directional guidance of the Q-cell migrations appear to act upstream of mab-5 (Harris et al. 1996).
Another Hox gene, egl-5, is required for proper migration of the hermaphrodite-specific neurons, or HSNs (Desai et al. 1988). egl-5 is a key regulator of HSN cell identity because egl-5 mutant HSNs are defective both in embryonic differentiation steps, such as cell death and cell migration, as well as in larval differentiation steps, such as neurotransmitter expression (Desai et al. 1988). In this paper, we construct a pathway of transcription factors that act along with egl-5 in directing these steps in HSN differentiation. We show that a zinc finger protein encoded by the gene ham-2 acts downstream of egl-5 to promote HSN migration. ham-2 is the first cell-migration gene found to act downstream of a Hox gene. In contrast, we find that another zinc finger protein required for HSN migration, EGL-43, is activated in the HSN in a Hox-independent manner. The gene unc-86, which encodes a POU homeodomain protein that promotes larval HSN fates (Desai et al. 1988; Finney et al. 1988), has been shown to act downstream of egl-5 (Baumeister et al. 1996). We find here that unc-86 also plays a redundant role with ham-2 in EGL-43 down-regulation in the embryo. Finally, we extend previous results and find that egl-5 is required for proper execution of the sex-specific cell death decision of the HSN, and that this function of egl-5 occurs independently of the ham-2, egl-43, and unc-86 transcription factors.
Results
ham-2 encodes a zinc finger protein required for migration of the HSN motor neurons
The HSNs, a bilateral pair of serotonergic motor neurons, are born in the tail of the embryo and migrate anteriorly to flank the gonad primordium near the middle of the body. The HSNs undergo apoptosis in males but survive in hermaphrodites. Later during larval development the HSNs terminally differentiate. They extend axons that innervate the egg-laying muscles and express the neurotransmitter serotonin (Desai et al. 1988; Garriga et al. 1993a). The HSNs are required for normal egg laying. Laser-operated animals lacking HSNs and mutant animals with defects in HSN development or function are egg-laying defective, or Egl (Trent et al. 1983; Desai et al. 1988). Systematic screens for C. elegans mutants with defective HSNs have identified many genes required for the development of these neurons (Trent et al. 1983; Desai et al. 1988; Desai and Horvitz 1989; Garriga and Stern 1994; Dianne Parry, P.D. Baum, and G. Garriga, unpubl.).
Two ham-2 mutants, n1332 and mu1, were isolated from mutator strains with high rates of transposable element activity. Both mutants have HSN migration defects, but lack obvious pleiotropies (Fig. 1). The original ham-2 mutant, n1332, was reported to have defects in HSN serotonin expression (Desai et al. 1988), but we find 90% of the HSNs of n1332 and mu1 hermaphrodites express detectable levels of serotonin, a percentage much higher than that described originally for n1332 hermaphrodites. Because low-penetrance defects in HSN serotonin expression are displayed by many HSN migration mutants, the ham-2 HSN serotonin defects may result from abnormal cell-body positioning (Garriga et al. 1993a). Two EMS-induced ham-2 mutations, gm16, and gm48, in addition to causing HSN migration defects, also cause first larval stage arrest with a Pun (pharynx unattached to the nose) phenotype (Fig. 2). Larval arrest is complete for gm16 animals, but only partially penetrant for gm48 animals. gm48 hermaphrodites also display the weakest HSN migration defect (Fig. 1).
Figure 1.

HSN migration and cell-death defects of transcription factor mutants. The HSN normally migrates from the tail to the side of the gonad primordium in embryos. This figure shows the final positions of HSNs in L1 larvae from various strains, as scored by Nomarski optics. At the top, the positions of landmark hypodermal cells (eye-shaped symbols) and the gonad (gray oval) are depicted. The arrow indicates the HSN migration route. The area of each circle in the chart is proportional to the percentage of HSNs in that position along the anteroposterior axis of the worm (see key at right). In some strains, some HSNs could not be found in a percentage of the sides scored; these are represented at the right of the figure in the not found column. HSNs that could not be found along the migratory route either failed to migrate from their birthplace in the tail, where we cannot distinguish them from other neurons of the lumbar ganglia, or were missing. (n) The number of HSNs scored. We scored approximately equal numbers of left and right HSNs for each strain. The ced-3 mutation was used to reveal HSNs missing because of programmed cell deaths. The distribution of the HSNs in ced-1 and ced-3 mutants was the same as in wild type and is not shown. The percentage of HSNs that could not be found in egl-5, egl-5; ced-3, and ced-1; egl-5 mutants are indicated. The fractions above the circles for ced-1; egl-5 indicate the number of cell corpses/total number of cell corpses and surviving HSNs at each position. No cell corpses were seen along the HSN migratory route in ced-1 mutants. The unc-86 alleles e1416, n306, n843, and n844 had effects similar to unc-86(n946) on HSN cell-body position, both as single mutants and as double-mutant combinations with the ham-2(mu1) mutation (not shown). 12% of the HSNs in egl-43(n997) mutants migrated out of the tail between V5 (the fourth marker cell from the right) and P11/12 (the most right marker cell) (n = 50). 4% of the HSNs in egl-43(n997); egl-5(n945) migrated out of the tail between P9/10 (the third marker cell from the right) and P11/12 (n = 50). None of the HSNs in egl-43(n997) unc-4(e120); ham-2(n1332) migrated out of the tail (n = 40).
Figure 2.
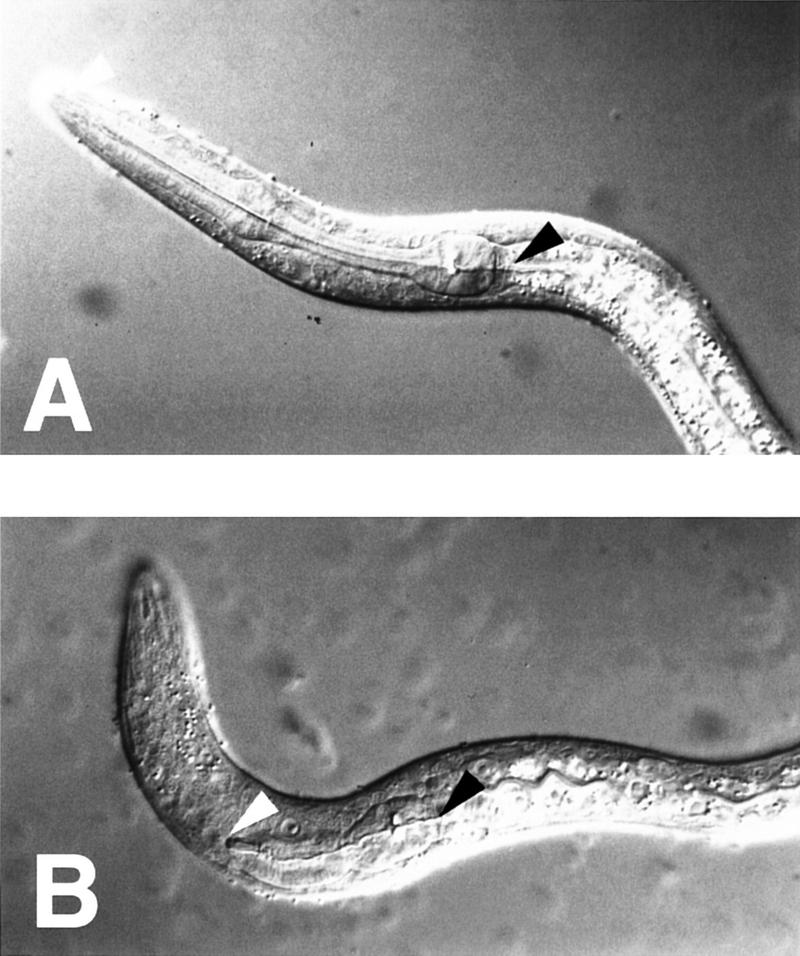
The Pun phenotype. Nomarski photomicrographs of L1 (first stage) larvae. The anterior (white arrowhead) and posterior (black arrowhead) ends of the pharynx are indicated. (A) wild type; (B) ham-2(gm16) mutant.
ham-2 was cloned (see Materials and Methods) and found to encode a protein with zinc finger motifs (Fig. 3). A subclone containing the ham-2 gene rescued the Ham and Pun defects of ham-2 mutants. The alleles n1332 and mu1 are transposon insertions in the first intron of the gene. The alleles gm16 and gm48 are missense mutations. gm16 changes a conserved amino acid in the first zinc finger. This mutation is predicted to alter or destroy the DNA-binding specificity of the protein (Berg and Shi 1996). gm48 alters a conserved histidine that is predicted to coordinate zinc in the first zinc finger. Taken together, the phenotypic and molecular analyses indicate that gm48 reduces ham-2 function, and gm16 may eliminate it. In contrast, n1332 and mu1 appear to be regulatory mutations that reduce severely or eliminate ham-2 function in the HSNs, but not in other cells. Antibody-staining experiments with anti-HAM-2 antiserum are consistent with this hypothesis (see below).
Figure 3.

Two ham-2 transcripts encode zinc finger proteins. (A) Sequence and predicted product of two ham-2 cDNA products are shown. The longer cDNA is trans-spliced to SL1 or SL2 (italics). A shorter transcript is trans-spliced to SL2 at the beginning of the second exon (nucleotide 161). (&) The beginning of each exon. Cysteines and histidines predicted to form zinc fingers are in bold. An acidic region (amino acids 248–263) or a proline-rich region (amino acids 344–364) could potentially act as activation domains. In ham-2(gm16), nucleotide 89 is changed from c to t (underlined). This mutation changes amino acid 30 from S to F. The amino acid in this position of the finger motif is predicted to determine DNA-binding specificity (Berg and Shi 1996). In ham-2(gm48), nucleotide 115 is changed from c to t (underlined). This mutation changes amino acid 39, a histidine predicted to coordinate the zinc atom in the first zinc finger, to a tyrosine. (B) Transposon insertions in the first intron of ham-2. Only a portion of the 2260 nucleotide first intron is shown. Nucleotides are numbered starting with the first nucleotide of the intron. The ham-2(n1332) and ham-2(mu1) transposon insertion sites are indicated.
HAM-2 and EGL-43 proteins are expressed in the HSN nucleus during migration
Like ham-2, the egl-43 gene encodes a zinc finger protein required for HSN migration (Garriga et al. 1993b). Polyclonal antisera raised against the HAM-2 and EGL-43 proteins were used to stain wild-type embryos (Figs. 4 and 5). Experiments to confirm that the staining observed with these antisera reflected the distribution of HAM-2 and EGL-43 are described in Materials and Methods. HAM-2 and EGL-43 proteins localized to cell nuclei, consistent with their proposed function as transcription factors. The HSNs were identified in these embryos by double labeling with an anti-UNC-86 antiserum (Finney and Ruvkun 1990). Both HAM-2 and EGL-43 are expressed in the HSN before and during the cell’s migration from the tail to the gonad primordium of the embryo (Figs. 4 and 5, A–F). Whereas EGL-43 expression is down-regulated after HSN migration, HAM-2 expression continues in larval HSNs (not shown). Both transcription factors are expressed in other cells besides the HSN. In cases in which these cells have been identified, as a group they share no obvious developmental or functional characteristics with the HSN.
Figure 4.
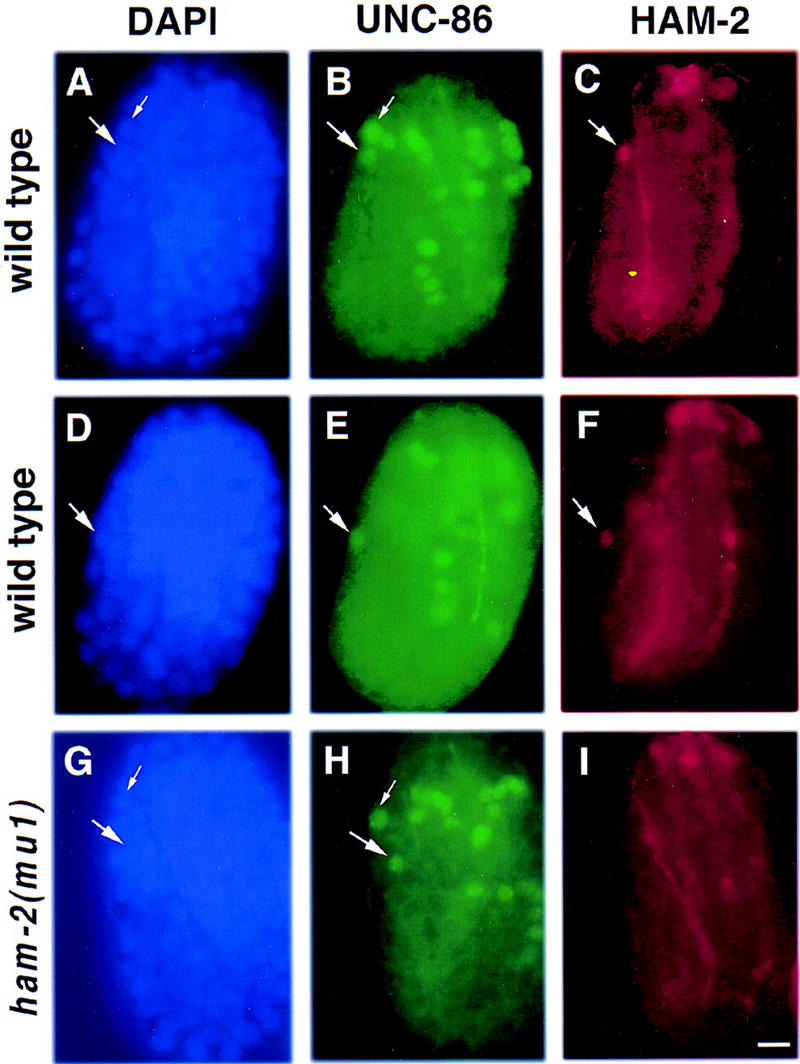
HAM-2 protein is expressed in the HSN nucleus during migration. Fluorescence photomicrographs of wild-type (A–F) and ham-2(mu1) mutant (G–I) embryos that have been stained with DAPI (blue), an UNC-86 antiserum (green) and a HAM-2 antiserum (red). Each panel presents a left lateral view of the embryo, with the developing tail to the left, the head to the right and the end of the tail and the top of the head both oriented upwards. The right sides are out of focus. (A–C) A wild-type embryo at ∼410 min after first cleavage, in which the HSN has not migrated out of the tail. (A) DAPI staining. The nuclei of the HSN (large arrow) and the ALN/PLM precursor (small arrow) are shown. (B) Anti-UNC-86 staining. Both the HSN (large arrow) and the ALN/PLM precursor (small arrow) express UNC-86. (C) Anti-HAM-2 staining. The HSN nucleus (large arrow) expresses HAM-2; several HAM-2 expressing cells in the head are out of focus. (D–F) A wild-type embryo at ∼430 min after first cleavage. (D) DAPI staining reveals the position of the HSN nucleus (arrow). The ALN/PLM precursor nucleus is out of the plane of focus. (E) Anti-UNC-86 staining. Shortly after commencing its anteriorly directed migration, the HSN begins expressing UNC-86 (arrow; Finney and Ruvkun 1990). (F) Anti-HAM-2 staining. During its migration, the HSN also expresses HAM-2 (arrow). (G–I) A ham-2(mu1) mutant embryo at ∼430 min after first cleavage. (G) DAPI staining reveals the positions of the HSN (large arrow) and the ALN/PLM precursor (small arrow) nuclei. (H) Anti-UNC-86 staining. The HSN (large arrow) and ALN/PLM precursor (small arrow) show normal expression of UNC-86. (I) Anti-HAM-2 staining. Although expression of HAM-2 in the head (mostly out of focus) and in the hypodermis (at an earlier stage not shown here) are normal, the HSN does not express HAM-2 in ham-2(mu1) embryos. The HSNs also lack HAM-2 expression in ham2(n1332) animals (not shown). Scale bar, 5 μm.
Figure 5.
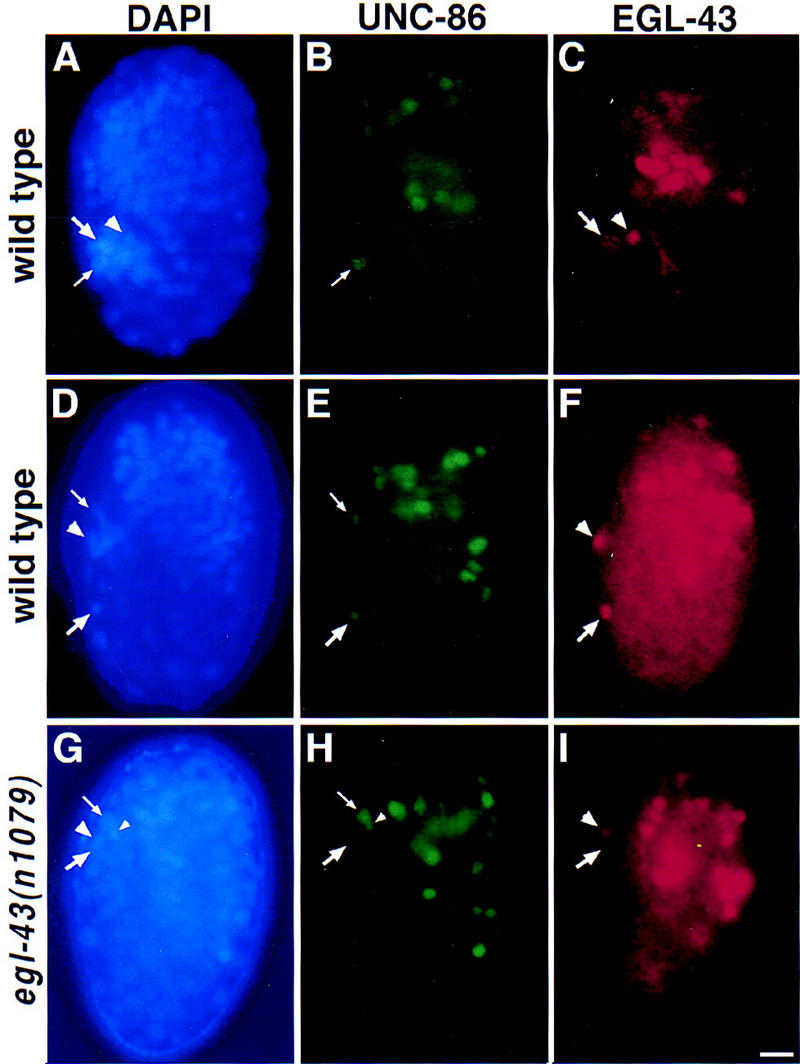
EGL-43 protein is expressed in the HSN nucleus during migration. Fluorescence photomicrographs of wild-type (A–F) and egl-43(n1079) mutant (G–I) embryos that have been stained with DAPI (blue), an UNC-86 antiserum (green), and an EGL-43 antiserum (red). Each panel presents a left lateral view of the embryo, with the developing tail to the left, the head to the right and the end of the tail and the top of the head both oriented upwards. The right sides are out of focus. (A–C) A wild-type embryo at ∼400 min after first cleavage. (A) DAPI staining was used to visualize cell nuclei. The HSN/PHB precursor (large arrow), PHA sensory neuron (arrowhead) and ALN/PLM precursor (small arrow) nuclei are located in the tail. (B) Anti-UNC-86 staining. At this time, only the ALN/PLM precursor nucleus (small arrow) expresses UNC-86 in the tail (Finney and Ruvkun 1990). (C) Anti-EGL-43 staining. At this time, only the HSN/PHB precursor (large arrow), which will divide to generate an HSN and PHB neuron, and the PHA sensory neuron (arrowhead) express EGL-43 in the tail. (D–F) Wild-type embryo at ∼430 min after first cleavage. (D) DAPI staining reveals the positions of the HSN nucleus (large arrow) which has migrated out of the tail, as well as a phasmid neuron nucleus (arrowhead) and an ALN/PLM precursor nucleus (small arrow) in the tail. (E) Anti-UNC-86 staining. Shortly after commencing its anteriorly directed migration, the HSN begins expressing UNC-86 (large arrow; Finney and Ruvkun 1990). (F) Anti-EGL-43 staining. During its migration, the HSN also expresses EGL-43-(large arrow). One of the EGL-43-expressing phasmid neuron nuclei (arrowhead) is also visible. (G–I) egl-43(n1079) mutant embryo at ∼430 min after first cleavage. (G) DAPI staining of this egl-43(n1079) mutant embryo shows the position of the HSN nucleus in the tail (large arrow). The phasmid neuron (large arrowhead), ALN/PLM precursor (small arrow) and the sister cell of the ALN/PLM precursor (small arrowhead) nuclei are indicated. (H) UNC-86 expression. The HSN neuron expressing UNC-86 (large arrow) has not migrated from its birthplace in the tail. The nuclei of the ALN/PLM precursor (small arrow) and its sister cell (small arrow) are indicated. (I) Anti-EGL-43 staining. One of the phasmid neurons (large arrowhead) but not the HSN neuron (large arrow) is expressing EGL-43. Scale bar, 5 μm.
Hypodermal nuclei also express HAM-2 briefly. We examined embryos double stained with a mouse anti-HAM-2 antiserum and a rabbit antiserum recognizing the hypodermal transcription factor LIN-26. Initially, hypodermal cells expressed both proteins, but HAM-2 expression ceased when embryos reached comma stage, whereas LIN-26 expression persisted (Labouesse et al. 1996; data not shown). Because lin-26 mutants arrest as embryos, and ham-2(gm16) mutants arrest as L1 larvae, we tested whether lin-26 might regulate ham-2 hypodermal expression. We found that HAM-2 staining in the hypodermal cells was not affected in lin-26 null mutants (M. Labouesse, pers. comm.), suggesting that ham-2 does not function downstream of lin-26. The pattern of anti-HAM-2 staining was similar to the expression of GFP from a ham-2–gfp transgene.
Regulatory mutations in ham-2 and egl-43 block protein expression in the HSN
The ham-2 alleles n1332 and mu1, which contain transposon insertions in the first intron of the gene (Fig. 3), and the egl-43 alleles, n997, and n1079, which contain an identical 790-bp deletion 3′ to the coding region (Materials and Methods), are likely to be regulatory mutations. Consistent with this hypothesis, staining of these mutants showed reduced protein expression in the HSN, with protein levels in most other cells unaffected. Staining of n1332 and mu1 mutants with an anti-HAM-2 antiserum showed no detectable HAM-2 expression in the HSNs during their migrations, despite normal expression in some other neurons and in the hypodermis (Fig. 4G–I; Table 1; data not shown). Faint staining was seen occasionally in the postmigratory HSNs of older ham-2(mu1) animals (not shown). Likewise, staining of egl-43(n1079) animals with an anti-EGL-43 antiserum revealed that most cells still expressed normal protein levels, but that the HSNs expressed no detectable EGL-43 (Fig. 5G–I; data not shown). HSN expression of UNC-86 was normal in the ham-2 and egl-43 mutants (Figs. 4 and 5, H). We also found that ham-2 mutants had normal HSN EGL-43 expression, and egl-43 mutants had normal HSN HAM-2 expression, suggesting that these genes act in parallel to regulate HSN migration (Table 1). The egl-43 deletion and the ham-2 transposon insertions likely disrupt enhancers required for HSN expression, but neither the egl-43 3′ region nor the ham-2 first intron are sufficient to drive HSN expression in GFP reporter constructs (data not shown).
Table 1.
Expression of HSN transcription factors in transcription factor mutant backgrounds
|
Strain
|
L.G.
|
Encodes
|
EGL-43 expression
|
HAM-2 expression
|
UNC-86 expression
|
|---|---|---|---|---|---|
| egl-5(n945) | III | Hox | persists in HSNs | absent in HSNs | absent in HSNs |
| egl-43(n1079) | II | Zn finger | absent in HSNs, ADEs and phasmid neurons | w.t. | w.t. |
| ham-2(n1332) | X | Zn finger | w.t. | absent HSNs | w.t. |
| unc-86(n946) | III | POU | w.t. | w.t. | absent in all UNC-86-expressing cells |
L1 larvae carrying loss-of-function mutations in genes encoding HSN transcription factors were stained with antisera to EGL-43, HAM-2 (Materials and Methods),and UNC-86 proteins (Finney and Ruvkun 1990). Over 50 animals of each mutant background were stained with each antiserum. (L.G.) Linkage group for each gene. The cloning of egl-5, egl-43, and unc-86 were described in Wang et al. (1993), Garriga et al. (1993b), and Finney and Ruvkun (1990), respectively. The absence of UNC-86 expression in the HSNs of egl-5 mutants was first noted in Baumeister et al. (1996).
In addition to eliminating EGL-43 expression in the HSN, the egl-43(n1079) mutation also affects EGL-43 expression in the ADE neuron and in the phasmid neurons PHA and PHB. Reduced EGL-43 expression in the phasmid neurons correlates with the phasmid dye-loading defect also observed in egl-43 mutants (Garriga et al. 1993b). Because the HSNs and PHBs are sister cells, current C. elegans mosaic analysis techniques cannot distinguish between egl-43 acting in the HSN or the PHB to promote HSN migration (Herman 1989; Sulston et al. 1983). It is likely, however, that egl-43 acts cell-autonomously to promote HSN migration, because in individual animals the severity of the HSN migration defects does not correlate with the presence or absence of EGL-43 in the phasmid neurons (Table 2). We have been unable to detect any defects in the development of the ADE neurons of egl-43 mutants (data not shown).
Table 2.
EGL-43 expression in the phasmid neurons does not affect the extent of HSN migration in egl-43 mutants
|
No. of phasmid neurons expressing EGL-43
|
HSN position
|
|||
|---|---|---|---|---|
|
>20%
|
1–20%
|
0%
|
n
|
|
| 2 | 0 | 66 | 33 | 6 |
| 1 | 15 | 58 | 27 | 26 |
| 0 | 13 | 56 | 31 | 16 |
egl-43(n1079) L1 larvae were double strained with an antiUNC-86 antiserum to detect the HSNs (Finney and Ruvkun 1990) and an anti-EGL-43 antiserum to detect the phasmid neurons. HSN positions were measured relative to the phasmid neurons and gonad, which mark the beginning (0%) and end (100%) of the HSN migration route, respectively. Animals were then scored for expression of EGL-43 in both, one; or neither of the PHA and PHB phasmid neurons on that side of the animal.
egl-5 regulates ham-2 and egl-43 expression in the HSN
In most HSN migration mutants, the HSNs either fail to migrate or migrate partially out of the tail (Desai et al. 1988; Forrester and Garriga 1997; Garriga and Stern 1994). Because HSN migration mutants all have a similar phenotype, it has been impossible to perform genetic epistasis experiments to order them into a pathway. However, as some of the genes required for HSN migration have now been cloned, antibody staining experiments make it possible to order these genes based on molecular epistasis (Table 1). We found that HSN expression of HAM-2 and EGL-43 was normal in most HSN migration mutant backgrounds. However, we found striking effects on the expression of HAM-2 and EGL-43 in egl-5 mutants. egl-5 encodes an Abdominal-B homolog that plays a critical role in specifying most steps in HSN differentiation, including cell migration, cell death, and neurotransmitter expression (Desai et al. 1988; Wang et al. 1993). First, egl-5 mutants showed no HSN expression of HAM-2, suggesting that egl-5 acts upstream of ham-2. Consistent with this hypothesis, egl-5; ham-2 double mutants did not display a more severe HSN migration defect than egl-5 single mutants (not shown). Second, egl-5 mutants failed to down-regulate HSN expression of EGL-43 normally after HSN migration was complete (Fig. 6). This deregulation of egl-43 expression is probably not a result of the egl-5 migration defect, because EGL-43 is turned off at the normal time in other mutants with misplaced HSNs (data not shown). The lineal sisters of the HSNs, the PHB sensory neurons, also express EGL-43 from when they are born during embryogenesis, but they continue to express EGL-43 though adulthood. Thus, one interpretation of the continued EGL-43 expression is that egl-5 mutant HSNs are transformed partially into their sister cell. To test this hypothesis, we determined whether egl-5 HSNs express an srb-6–gfp transgene. srb-6 encodes a seven-transmembrane molecule that may function as chemoreceptor for the detection of volatile repellents (Troemel et al. 1995). The phasmid neurons PHA and PHB express srb-6–gfp, whereas the HSNs do not. Consistent with the hypothesis that HSNs are transformed partially into PHBs, egl-5 HSNs often express srb-6–gfp (Fig. 6).
Figure 6.
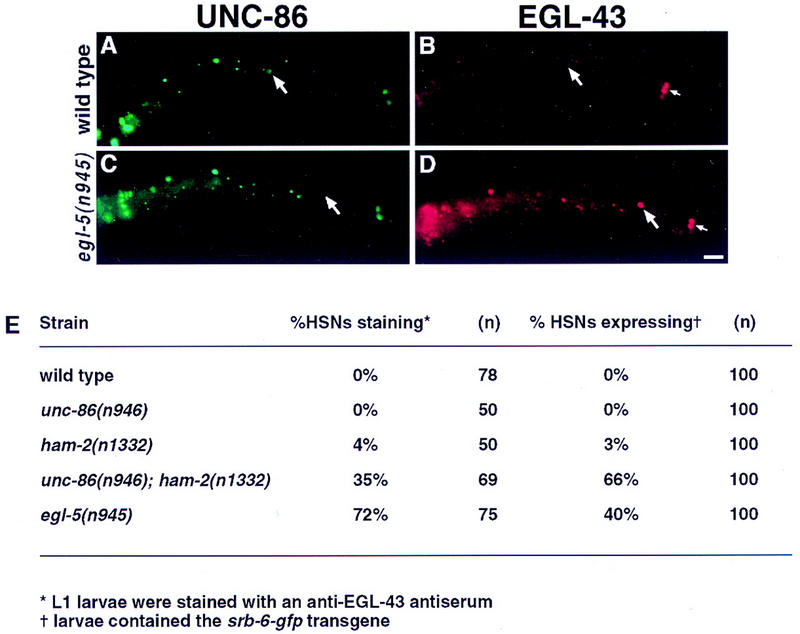
Some HSN migration mutants fail to down-regulate EGL-43 and repress srb-6. (A–D) Fluorescence photomicrographs of wild-type (A,B) and egl-5(n945) mutant (C,D) L1 larvae that have been fixed and stained with UNC-86 and EGL-43 antisera. All four panels present left lateral views of the animals with anterior to the left. The right sides are out of focus. (A) In wild-type animals, the HSN neuron is located in the midbody and expresses UNC-86 (arrow). (B) EGL-43 expression in the same larva as in A. The PHA and PHB phasmid neuron nuclei (small arrow) express EGL-43 but the HSN neuron (arrow) does not. (C) In egl-5(n945) L1 larvae, posteriorly displaced HSNs (arrow) often fail to express UNC-86 (Baumeister et al. 1996). (D) EGL-43 expression in the same larva pictured in C. In contrast to wild-type, the posteriorly displaced HSN does express EGL-43 (arrow). The phasmid neurons also express EGL-43 (small arrow). Scale bar, 15 μm. (E) Table quantifying persistence of EGL-43 expression and presence of srb-6–gfp in the HSNs of transcription factor mutants.
unc-86; ham-2 double mutants phenocopy egl-5 mutants in their failure to down-regulate HSN EGL-43 expression
Expression of the UNC-86 POU homeodomain transcription factor in the HSN requires egl-5 (Baumeister et al. 1996), similar to the egl-5 requirement for HAM-2 expression in the HSN. UNC-86 expression in the HSN begins when the neuron initiates its migration, and we found low penetrance HSN migration defects in unc-86 mutants (Fig. 1). In contrast to the weak early HSN migration defect, unc-86 mutants display severe defects in HSN traits expressed later during larval development: hood formation (a morphological marker of neuronal maturation) and serotonin expression fail to occur (Desai et al. 1988). From the analysis of single mutants, it appeared that ham-2 and unc-86 play distinct roles in early and late HSN development, respectively. Nonetheless, because UNC-86 expression in the HSN begins early, it remained possible that unc-86 played a redundant role in early HSN development that was masked by ham-2 function. To test this possibility, we examined the HSN phenotype of an unc-86; ham-2 double mutant. We did not uncover an additional role for unc-86 in HSN migration: unc-86; ham-2 double mutants had no more severe an HSN migration defect than ham-2 mutants (Fig. 1). To our surprise, we found that the unc-86; ham-2 double mutant displayed synthetic phenotypes not seen in either single mutant alone: EGL-43 expression was often not properly down-regulated and srb-6–gfp was expressed ectopically in the HSNs of unc-86; ham-2 mutants (Fig. 6). As in wild-type animals, the HSNs of unc-86 or ham-2 single mutants down-regulated EGL-43 expression after they finished migrating and did not express srb-6–gfp (Fig. 6).
egl-5 regulates the HSN’s decision to live or die
In addition to their other differentiation defects, the HSNs of egl-5 mutants also display defects in sex-specific programmed cell death. During male embryonic development, the HSNs normally die (Sulston and Horvitz 1977). Desai et al. (1988) reported that HSNs sometimes survived in egl-5 males. In addition, they found that egl-5 was able to suppress the inappropriate cell deaths of HSNs in hermaphrodites carrying dominant alleles of egl-1. The gene egl-1 encodes a negative regulator of the Bcl-2 homolog CED-9 that is necessary for all programmed cell deaths in C. elegans, suggesting that dominant egl-1 alleles are regulatory mutations that cause the HSNs to die in hermaphrodites, the male HSN fate (Trent et al. 1983; Conradt and Horvitz 1998).
We identified a new cell-death phenotype for egl-5 mutants: the HSNs occasionally died in egl-5 hermaphrodites (Fig. 1). When we scored egl-5 mutants by Nomarski optics, only 76% of the HSNs were located along their migratory route. The missing HSNs might have failed to migrate from their birthplace in the tail, where they would be indistinguishable from other neurons of the lumbar ganglia; alternatively, these HSNs might have died. To distinguish between these possibilities, we determined the positions of the HSNs in egl-5; ced-3 hermaphrodites (Fig. 1). The gene ced-3 is necessary for all programmed cell deaths in C. elegans; in ced-3 males; for example, the HSNs of males survive (Ellis and Horvitz 1986). We found that the percentage of HSNs detected along the migratory route increased to 85% in egl-5; ced-3 double-mutant hermaphrodites, consistent with the hypothesis that HSNs undergo apoptosis occasionally in egl-5 hermaphrodites. Presumably, the 15% of HSNs still unaccounted for in these double mutants were hidden among the neurons of the tail. To confirm that HSN apoptosis was taking place, we made use of the ced-1 mutation, which disrupts the cell corpse engulfment process and allows corpses to persist (Hedgecock et al. 1983). When we could not identify an HSN on a given side of a ced-1; egl-5 hermaphrodite, an ectopic cell-death corpse was often present along the HSN’s migratory route on that side (Fig. 1). This phenotype was not observed in ced-1 hermaphrodites (not shown).
Because the ham-2 and unc-86 genes act redundantly downstream of egl-5 to down-regulate EGL-43 HSN expression, it seemed plausible that ham-2 and unc-86 could also coordinately control HSN sex-specific cell death. However, no HSNs were missing in unc-86; ham-2 hermaphrodites—all HSNs were detected along their migratory route (Fig. 1). In addition, no HSNs survived in unc-86; egl-1; ham-2 hermaphrodites (not shown). In contrast to the egl-5 mutants, the HSN cell death decision was implemented properly in unc-86; ham-2 animals.
Discussion
Throughout the animal kingdom, Hox genes play a fundamental role in anteroposterior patterning, but the mechanisms by which they specify cell fates have not been examined in detail at single-cell resolution. Here, we have identified ham-2, a gene that encodes a probable zinc-finger transcription factor required for HSN migration. We have shown that HAM-2 acts downstream of the Hox protein EGL-5, and examined its regulatory interactions with the Hox-dependent transcription factor UNC-86 and the Hox-independent transcription factor EGL-43, two other proteins that also promote HSN differentiation. Until now, it has been difficult to determine unambiguously the order of actions of genes regulating HSN differentiation (Desai et al. 1988). Using analysis of single and multiple mutants, as well as molecular analysis of protein expression in different mutant backgrounds, we have been able to propose a regulatory pathway for HSN development (Fig. 7).
Figure 7.

A transcriptional network regulates HSN development. This figure shows transcription factors expressed in the HSN and required for HSN differentiation. The arrows designate functional relationships determined by genetic criteria; these relationships may be direct or indirect. The zinc finger protein encoded by the gene sem-4, which acts in late HSN differentiation (Basson and Horvitz 1996), is not depicted in this chart because its relationships to egl-5 and unc-86 have not been determined. Consistent with egl-43 and egl-5/ham-2 acting independently to regulate HSN migration, HSN migration in egl-43; egl-5 and egl-43; ham-2 double mutants is more severely defective than in any of the single mutants.
The Abdominal-B homolog egl-5 is a general regulator of HSN identity. Unlike mutations in other transcription factors, which only affect certain aspects of HSN differentiation, egl-5 mutations disrupt HSN sex-specific cell death, migration, and later phenotypes, such as neurotransmitter expression. egl-5 is also required for proper expression of two downstream transcription factors, UNC-86 and HAM-2, in the HSNs, and may act through these genes to implement HSN cell fate. Because HSN expression of EGL-43 and srb-6–gfp in egl-5 mutants resembles the normal expression of these markers in the lineal sisters of the HSNs, the PHB sensory neurons, many of these defects may result from a partial transformation of HSN into PHB. Although egl-5 plays an important role in determining HSN identity, it is not the sole determinant of HSN identity. The various HSN phenotypes of egl-5 null mutants are not fully penetrant, and UNC-86 and HAM-2 are occasionally expressed in HSNs lacking egl-5 function. The HSNs, although misplaced and morphologically abnormal, can still migrate partially in egl-5 null mutants. Furthermore, egl-5 alone cannot confer HSN identity on a given cell: EGL-5 is expressed in other tail neurons that differentiate into non-HSN fates (Wang et al. 1993).
We have shown that ham-2 is required for HSN migration. Our analysis of two regulatory mutations in the ham-2 gene, which cause HSN migration defects and which reduce or eliminate HAM-2 expression in the HSN without affecting other cells, suggests that ham-2 functions in the HSN. By contrast, mutations that alter the ham-2 coding region, and thus presumably disrupt ham-2 function in all the cells in which it is normally expressed, do not cause additional neuronal migration defects, but do cause defects in the attachment of the pharynx to the front of the worm. Based on this additional phenotype, it is possible that adhesion molecules might be among HAM-2’s targets.
ham-2 mutants do not have as severe an HSN migration defect as egl-5 mutants do. Either none of our ham-2 alleles is null, or egl-5 has additional targets that promote HSN migration. Finally, although the ham-2 alleles n1332 and mu1 do not appear to produce defects in later stages of HSN development, such as serotonin expression, we cannot rule out the possibility that these alleles provide some ham-2 function in the HSN during those later stages. It is possible that ham-2 is not completely specific for the early migratory stage of HSN development.
unc-86 has been shown previously to be required for late HSN fates, such as hood formation and serotonin expression (Desai et al. 1988). Although UNC-86 is expressed in the HSN during migration, null unc-86 mutants have weak migration defects, and do not enhance the migration defects of ham-2 mutants. unc-86 plays a second role in early HSN differentiation, but this role is only revealed in a ham-2 double mutant.
egl-43 has been described previously as a transcription factor required for HSN migration (Garriga et al. 1993b). Here we show that the identical 3′ deletion found in the egl-43 alleles n997 and n1079 is likely to be a regulatory mutation that removes EGL-43 from only a few cells: the HSNs, the ADE neurons, and the phasmid neurons. egl-43 appears to act in the HSNs to promote HSN migration and in the phasmid neurons to promote their differentiation. Unlike HAM-2 and UNC-86, the onset of EGL-43 expression in the HSN is independent of egl-5. On the other hand, egl-5 appears to act via ham-2 and unc-86 to down-regulate EGL-43 expression and repress srb-6 expression in the HSN.
We have also demonstrated a role for egl-5 in proper execution of the sex-specific cell-death decision that the HSN makes during embryonic development. egl-5 function is required to prevent both the inappropriate cell death of the HSN in hermaphrodites and the aberrant survival of the HSN in males. Although we have identified roles for unc-86 and ham-2 in mediating many egl-5 functions in the HSN, we were unable to uncover any role for these genes in regulating HSN cell death. One possible mediator of egl-5’s role in the HSN cell-death decision is the gene tra-1, which encodes a zinc-finger protein that acts autonomously as the terminal regulatory gene of the sex-determination pathway and controls the HSN sexual fate in hermaphrodites by preventing its death (Hunter and Wood 1990; Zarkower and Hodgkin 1992). As a regulator of HSN sex-specific survival, EGL-5 could act by altering expression of egl-1 in the HSN, either directly by binding to egl-1 regulatory sequences or indirectly though TRA-1.
Why have Hox targets been difficult to identify and analyze through genetic means? Our results suggest three reasons that Hox target genes may have mutant phenotypes that are unrecognizably different from the mutant phenotypes of the Hox genes themselves. First, Hox target genes may play general roles in development, with their roles downstream of individual Hox genes comprising only a minor part of their function. Specific regulatory mutations that reduced ham-2 function in the HSN were isolated in screens for HSN migration defects. The widespread expression pattern of ham-2, and the lethal phenotype of ham-2(gm16) suggest that ham-2’s role downstream of egl-5 in promoting HSN migration is only one part of its biological function. Second, our results show that Hox target genes may each only carry out a small subset of the roles played by the Hox gene itself, and that these target genes may have redundant roles. Although certain HSN differentiation events regulated by egl-5, such as cell migration and neurotransmitter synthesis, are carried out by individual downstream regulators, others, such as the down-regulation of EGL-43, may be carried out by multiple downstream targets in a redundant fashion. Still other steps in HSN differentiation regulated by egl-5, such as the sex-specific cell-death decision, may be carried out by additional downstream genes that still remain to be identified. Third, Hox genes are not fully responsible for cell fate determination. The HSNs migrate partially even in egl-5 null mutants, suggesting that HSN migration is determined only partly by Hox-directed cell-fate determination and partly regulated by Hox-independent regulators, such as egl-43. Taken together, the complexity of egl-5 function suggests that it will be difficult to identify Hox targets by genetic phenotypes alone.
Often the expression of one or two transcription factors can confer a specific cell fate. For example, expression of both unc-86 and mec-3 is required for mechanosensory neuron cell fate (Way and Chalfie 1989; Xue et al. 1993). Yet a few transcription factors are known that are only responsible for one portion of a neuron’s differentiation program. These transcription factors may act after more general fate determinants in the process of neuronal differentiation (Lewin 1994). For example, the Hox genes mab-5 and lin-39 are not master regulators of Q-neuroblast identity in the way that egl-5 is a master regulator of HSN identity. Mutations in these genes affect the direction and extent of Q-neuroblast migrations, but other aspects of these cells are not affected (Wang et al. 1993). As another example, the ODR-7 transcription factor appears to be required for regulating the odorant specificity of the AWA sensory neuron, but is not required for proper morphological development of that neuron (Sengupta et al. 1994). Here, in the case of the HSN, we have described a collection of transcription factors, unc-86, egl-43, and ham-2, that appear to regulate different subsets of HSN differentiation.
A challenge for the future will be to identify the targets of egl-43 and ham-2 that act directly in HSN migration. The relevant effectors may be represented among the large collection of mutants required for HSN migration (Garriga and Stern 1994).
Materials and methods
C. elegans genetics
Strains were grown at 20°C unless otherwise stated, and were maintained as described by Brenner (1974). In addition to the wild-type strain N2, strains with the following mutations were used in this work: L.G. I: ced-1(e1735) (Hedgecock et al. 1983). L.G. II: egl-27(n170) (Trent et al. 1983); egl-43(n997), egl-43(n1079) (Desai et al. 1988); unc-4(e120) (Brenner 1974). L.G. III: unc-86(e1416), unc-86(n306), unc-86(n843), unc-86(n844), unc-86(n946) (Chalfie et al. 1981; Desai and Horvitz 1989); egl-5(n945) (Desai and Horvitz 1989); gmIs12 (srb-6–gfp) + rol-6 (su1006) (Troemel et al. 1995; N. Hawkins, pers. comm.). L.G. IV: egl-18(n162) (Trent et al. 1983); egl-20(n585) (Trent et al. 1983); ced-3(n717) (Ellis and Horvitz 1986). L.G. V: him-5(e1490) (Hodgkin et al. 1979). L.G. X: lin-32(u282) (Chalfie and Au 1989); unc-20(e112ts) (Brenner 1974); lin-18(e620) (Ferguson and Horvitz 1985); dpy-23(e840) (Hodgkin 1983); ham-2(n1332) (Desai et al. 1988); ham-2(mu1) (Cynthia Kenyon, pers. comm.); ham-2(gm16) and ham-2(gm48) (this study); lon-2(e678) (Brenner 1974).
Isolation of new ham-2 alleles
ham-2(gm16) was isolated in a ham-2 noncomplementation screen. Briefly, unc-20(e112ts) lin-18(e620) hermaphrodites were mutagenized with EMS and crossed to ham-2(n1332) lon-2(e678) males. 16,200 non-Unc cross progeny were screened for Egl defects, yielding one new allele, gm16. ham-2(gm48) was isolated in a direct screen for mutants with misplaced HSNs (Dianne Parry, P.D. Baum, and G. Garriga, unpubl.).
Cloning of ham-2
ham-2 was genetically mapped to the interval between dpy-23 and lon-2 on the X chromosome, and then RFLP mapping was used to locate ham-2 on the physical map. Three RFLPs were identified by probing genomic Southern blots of polymorphic C. elegans strains with cosmid clones from the region: gmP1 (a HinDIII digest of the strain RW7000 probed with the cosmid W01H2), gmP2 (a XhoI digest of the strain N62 with the cosmid F02E8, and muP1 (an EcoRI digest of N62 with the cosmid K02G10; Mary Sym, pers. comm.). Transheterozygous strains were constructed that had a dpy-23(e840) ham-2(n1332) lon-2(e678) X chromosome and either a RW7000 or an N62 X chromosome. Lon non-Dpy recombinants were selected and progeny of these recombinants that were homozygous for the Lon non-Dpy recombinant chromosome were scored for the Ham phenotype and assayed for polymorphisms by Southern blot. Of 19 Lon non-Dpy recombinants, all picked up gmP1. Of 20 Lon non-Dpy recombinants, 4 out of 5 Ham recombinants, and all 15 non-Ham recombinants picked up gmP2. In addition, none of the 5 Ham recombinants, and 4 out of 15 non-Ham recombinants picked up muP1. Taken together, these data placed ham-2 between the gmP2 and muP1 RFLPs.
Two overlapping cosmids from this region, C04F8 and F42G12, rescued the HSN migration defect of ham-2(n1332) mutants. In addition, a Southern blot of ham-2(n1332) and ham-2(mu1) genomic DNA probed with the cosmid C04F8 revealed DNA rearrangements relative to wild type (not shown). Finally, a 7-kb AvrII–KpnI genomic subclone predicted to contain only the ham-2 gene (pGM134) rescued the HSN migration defect of ham-2(n1332) mutants. A GFP construct, pGM138, was constructed by cloning the GFP gene from pPD95.69 (Andrew Fire, pers. comm.) as a BamHI–FspI fragment into pGM134 cut with BamHI and KpnI (blunted).
Analysis of ham-2 transcripts
The ham-2 genomic region had been sequenced by the C. elegans Genome Sequencing Project (cosmid C07A12; accession no.U41542). A cDNA for the long transcript of ham-2, yk82h11, was a gift of Yuji Kohara (National Institute of Genetics, Mishima, Japan). The cDNA was sequenced using the method of Strathmann et al. (1991). We also searched for alternative transcripts using the 5′ RACE technique, as well as RT–PCR with gene-specific primers and primers to the SL-1 and SL-2 splice leaders. The long SL-1 transcript was amplified consistently; the SL-2 transcripts were only detected in a minority of PCR reactions. The genomic organization of the ham-2 gene does not conform to the usual pattern for SL2 trans-splicing of polycistronic mRNAs (Blumenthal 1995).
Allele sequencing
All mutant alleles were sequenced using multiple independent PCR amplifications of genomic DNA. The egl-43 alleles were determined previously to have deletions 3′ to the coding region of the gene based on Southern blotting (Garriga et al. 1993b). Sequencing of the egl-43(n997) and egl-43(n1079) alleles revealed identical 790-bp deletions corresponding to nucleotides 21,803–22,592 of the genomic sequence derived by the C. elegans Genome Sequencing Project from the cosmid R53 (accession no. Z66515).
anti-HAM-2 and anti-EGL-43 antisera
The BamHI–KpnI carboxy-terminal fragment of the yk82h11 ham-2 cDNA was cloned into the pRSET B 6×His vector (Invitrogen). This carboxy-terminal portion was chosen because the amino terminus showed extensive homology to other predicted C. elegans proteins. Antibodies to this carboxy-terminal fusion protein would be expected to react with both the long and short ham-2 gene products. The protein was expressed in E. coli, purified with nickel-agarose resin and injected into mice. Ascites fluid was induced and collected. Antisera from mice were able to recognize specifically bacterially expressed HAM-2 on a Western blot (not shown). Nematodes were stained by the method of Finney and Ruvkun (1990). The staining pattern for anti-HAM–2 antisera is likely to be correct because the expression pattern obtained with the antisera was identical to that seen with a genomic ham-2–GFP construct (pGM138). In addition, the antisera showed an identical staining pattern after affinity purification against bacterial HAM-2 immobilized on nitrocellulose strips as described (Harlow and Lane 1988).
To generate EGL-43 protein, three cDNAs corresponding to different regions of EGL-43 were amplified by RT–PCR and cloned into the PstI site of the pRSET B 6×His vector (Invitrogen); protein was made and antisera raised as described above. Antisera were affinity purified using columns of protein coupled to Reacti-Gel 6× (Pierce). Antisera raised against bacterial fusion proteins containing two distinct nonoverlapping regions of EGL-43 produced identical staining patterns. In addition, animals homozygous for the mnDf24 deficiency, which completely removes the egl-43 gene (Sigurdson et al. 1984), did not show any nuclear staining, suggesting that the staining pattern observed was specific for EGL-43. The genotypes of putative mnDf24 L1 larvae were confirmed by PCR.
EGL-43-expressing cells in embryos and larvae were identified by position, and, where possible, these identifications were confirmed by double-staining experiments using other markers. Besides the HSN/PHB precursor, the HSNs, and the PHA and PHB phasmid neurons (Fig. 4; Tables 1 and 2), EGL-43 was also expressed in the FLPL/R, PVDL/R, ADFL/R, ADLL/R, LUAL/R (only during embryogenesis), ADEL/R, RIGL/R, ASHL/R, ASJL/R, ASIL/R, CEPDL/R, CEPVL/R, OLQDL/R, OLQVL/R, IL1VL/R, IL1DL/R, URADL/R, the T-blast cells, and the somatic gonad precursors Z1 and Z4 as well as some of their descendants.
Acknowledgments
We are grateful to Cynthia Kenyon for the gift of the ham-2(mu1) allele, Yuji Kohara for a cDNA clone, Andy Fire for plasmids, Åshild Vik, Thomas Harbaugh, and Millicent Yee for molecular biology assistance, Gary Ruvkun and Michael Finney for anti-UNC-86 antiserum, Michel Labouesse for anti-LIN-26 antiserum and lin-26 mutants, Nancy Hawkins for the srb-6-gfp transgenic strain, and Wendy Katz and Mary Sym for mapping data. Some strains were provided by the Caenorhabditis Genetics Center, St. Paul, MN. We thank members of the Garriga and Meyer laboratories for advice and assistance. We also thank Michael Levine for critical reading of the manuscript. This work was supported by a National Institutes of Health grant NS32057 to G.G. C.G. and C.A.F. were supported by National Science Foundation fellowships.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL garriga@peregrine.berkley.edu; FAX (510) 642-7000.
References
- Bang AG, Goulding MD. Regulation of vertebrate neural cell fate by transcription factors. Curr Opin in Neurobiol. 1996;6:25–32. doi: 10.1016/s0959-4388(96)80005-5. [DOI] [PubMed] [Google Scholar]
- Basson M, Horvitz HR. The Caenorhabditis elegans gene sem-4 controls neuronal and mesodermal cell development and encodes a zinc finger protein. Genes & Dev. 1996;10:1953–1965. doi: 10.1101/gad.10.15.1953. [DOI] [PubMed] [Google Scholar]
- Baumeister R, Liu Y, Ruvkun G. Lineage-specific regulators couple cell lineage asymmetry to the transcription of the Caenorhabditis elegans POU gene unc-86 during neurogenesis. Genes & Dev. 1996;10:1395–1410. doi: 10.1101/gad.10.11.1395. [DOI] [PubMed] [Google Scholar]
- Berg JM, Shi Y. The galvanization of biology: A growing appreciation for the roles of zinc. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- Blumenthal T. Trans-splicing and polycistronic transcription in Caenorhabditis elegans. Trends Genet. 1995;11:132–136. doi: 10.1016/s0168-9525(00)89026-5. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Au M. Genetic control of differentiation of the Caenorhabditis elegans touch receptor neurons. Science. 1989;243:1027–1033. doi: 10.1126/science.2646709. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Horvitz HR, Sulston JE. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell. 1981;24:59–69. doi: 10.1016/0092-8674(81)90501-8. [DOI] [PubMed] [Google Scholar]
- Clark SG, Chisholm AD, Horvitz HR. Control of cell fates in the central body region of C. elegans by the homeobox gene lin-39. Cell. 1993;74:43–55. doi: 10.1016/0092-8674(93)90293-y. [DOI] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:419–529. doi: 10.1016/s0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- DeLapeyriere O, Henderson CE. Motoneuron differentiation, survival and synaptogenesis. Curr Opin Genet Devel. 1997;7:642–650. doi: 10.1016/s0959-437x(97)80012-3. [DOI] [PubMed] [Google Scholar]
- Desai C, Horvitz HR. Caenorhabditis elegans mutants defective in the functioning of the motor neurons responsible for egg laying. Genetics. 1989;121:703–721. doi: 10.1093/genetics/121.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai C, Garriga G, McIntire SL, Horvitz HR. A genetic pathway for the development of the Caenorhabditis elegans HSN motor neurons. Nature. 1988;336:638–646. doi: 10.1038/336638a0. [DOI] [PubMed] [Google Scholar]
- Edelman GM, Jones FS. Gene regulation of cell adhesion: A key step in neural morphogenesis. Brain Res Rev. 1998;26:337–352. doi: 10.1016/s0165-0173(97)00034-9. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Ferguson EL, Horvitz HR. Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics. 1985;110:17–72. doi: 10.1093/genetics/110.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Finney M, Ruvkun G, Horvitz HR. The C. elegans cell lineage and differentiation gene unc-86 encodes a protein with a homeodomain and extended similarity to transcription factors. Cell. 1988;55:757–769. doi: 10.1016/0092-8674(88)90132-8. [DOI] [PubMed] [Google Scholar]
- Forrester W, Garriga G. Genes necessary for C. elegans cell and growth cone migrations. Development. 1997;124:1831–1843. doi: 10.1242/dev.124.9.1831. [DOI] [PubMed] [Google Scholar]
- Garriga G, Stern MJ. Hams and Egls: Genetic analysis of cell migration in Caenorhabditis elegans. Curr Opin Genet Dev. 1994;4:575–580. doi: 10.1016/0959-437x(94)90075-e. [DOI] [PubMed] [Google Scholar]
- Garriga G, Desai C, Horvitz HR. Cell interactions control the direction of outgrowth, branching and fasciculation of the HSN axons of Caenorhabditis elegans. Development. 1993a;117:1071–1087. doi: 10.1242/dev.117.3.1071. [DOI] [PubMed] [Google Scholar]
- Garriga G, Guenther C, Horvitz HR. Migrations of the Caenorhabditis elegans HSNs are regulated by egl-43, a gene encoding two zinc finger proteins. Genes & Dev. 1993b;7:2097–2109. doi: 10.1101/gad.7.11.2097. [DOI] [PubMed] [Google Scholar]
- Gould AP, White RA. Connectin, a target of homeotic gene control in Drosophila. Development. 1992;116:1163–1174. doi: 10.1242/dev.116.4.1163. [DOI] [PubMed] [Google Scholar]
- Gould AP, Brookman JJ, Strutt DI, White RA. Targets of homeotic gene control in Drosophila. Nature. 1990;348:308–312. doi: 10.1038/348308a0. [DOI] [PubMed] [Google Scholar]
- Graba Y, Aragnol D, Pradel J. Drosophila Hox complex downstream targets and the function of homeotic genes. BioEssays. 1997;19:379–388. doi: 10.1002/bies.950190505. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Harris J, Honigberg L, Robinson N, Kenyon C. Neuronal cell migration in C. elegans: Regulation of Hox gene expression and cell position. Development. 1996;122:3133–3140. doi: 10.1242/dev.122.10.3117. [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Sulston JE, Thomson JN. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science. 1983;220:1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- Herman RK. Mosaic analysis in the nematode Caenorhabditis elegans. J Neurogenet. 1989;5:1–24. doi: 10.3109/01677068909167261. [DOI] [PubMed] [Google Scholar]
- Hodgkin J. X chromosome dosage and gene expression in C. elegans: Two unusual dumpy genes. Mol Gen Genet. 1983;192:452–458. [Google Scholar]
- Hodgkin JA, Horvitz HR, Brenner S. Nondisjunction mutants of the nematode Caenorhabditis elegans. Genetics. 1979;91:67–94. doi: 10.1093/genetics/91.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CP, Wood WB. The tra-1 gene determines sexual phenotype cell-autonomously in C. elegans. Cell. 1990;63:1193–1204. doi: 10.1016/0092-8674(90)90415-b. [DOI] [PubMed] [Google Scholar]
- Kenyon C. A gene involved in the development of the posterior body region of C. elegans. Cell. 1986;46:477–487. doi: 10.1016/0092-8674(86)90668-9. [DOI] [PubMed] [Google Scholar]
- Keynes R, Krumlauf R. Hox genes and regionalization of the nervous system. Annu Rev Neurosci. 1994;17:109–132. doi: 10.1146/annurev.ne.17.030194.000545. [DOI] [PubMed] [Google Scholar]
- Labouesse M, Hartwieg E, Horvitz HR. The Caenorhabditis elegans LIN-26 protein is required to specify and/or maintain all non-neuronal ectodermal cell fates. Development. 1996;122:2823–2833. doi: 10.1242/dev.122.9.2579. [DOI] [PubMed] [Google Scholar]
- Lewin B. On neuronal specificity and the molecular basis of perception. Cell. 1994;79:935–943. doi: 10.1016/0092-8674(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- McGinnis W, Levine MS, Hafen E, Kuroiwa A, Gehring WJ. A conserved DNA sequence in homoeotic genes of the Drosophila Antennapedia and bithorax complexes. Nature. 1984;308:428–433. doi: 10.1038/308428a0. [DOI] [PubMed] [Google Scholar]
- Ruvkun G. Patterning the nervous system. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 543–581. [PubMed] [Google Scholar]
- Salser SJ, Kenyon C. Activation of a C. elegans Antennapedia homologue in migrating cells controls their direction of migration. Nature. 1992;355:255–228. doi: 10.1038/355255a0. [DOI] [PubMed] [Google Scholar]
- Salser SJ, Kenyon C. Patterning C. elegans: Homeotic cluster genes, cell fates and cell migrations. Trends Genet. 1994;10:159–164. doi: 10.1016/0168-9525(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Bargmann CI. Cell fate specification and differentiation in the nervous system of Caenorhabditis elegans. Devel Genet. 1996;18:73–80. doi: 10.1002/(SICI)1520-6408(1996)18:1<73::AID-DVG8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Colbert HA, Bargmann CI. The C. elegans gene odr-7 encodes an olfactory-specific member of the nuclear receptor superfamily. Cell. 1994;79:971–980. doi: 10.1016/0092-8674(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Sigurdson DC, Spanier GJ, Herman RK. Caenorhabditis elegans deficiency mapping. Genetics. 1984;108:331–345. doi: 10.1093/genetics/108.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D, Nusslein-Volhard C. The origin of pattern and polarity in the Drosophila embryo. Cell. 1992;68:201–219. doi: 10.1016/0092-8674(92)90466-p. [DOI] [PubMed] [Google Scholar]
- Strathmann M, Hamilton BA, Mayeda CA, Simon MI, Meyerowitz EM, Palazzolo MJ. Transposon-facilitated DNA sequencing. Proc Natl Acad Sci. 1991;88:1247–1250. doi: 10.1073/pnas.88.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer M, Lumsden A, Ariza-McNaughton L, Bradley A, Krumlauf R. Altered segmental identity and abnormal migration of motor neurons in mice lacking Hoxb-1. Nature. 1996;384:630–634. doi: 10.1038/384630a0. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Trent C, Tsung N, Horvitz H. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Chou JH, Dwyer ND, Colbert HA, Bargmann CI. Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell. 1995;83:207–218. doi: 10.1016/0092-8674(95)90162-0. [DOI] [PubMed] [Google Scholar]
- Wang BB, Muller-Immergluck MM, Austin J, Robinson NT, Chisholm A, Kenyon C. A homeotic gene cluster patterns the anteroposterior body axis of C. elegans. Cell. 1993;74:29–42. doi: 10.1016/0092-8674(93)90292-x. [DOI] [PubMed] [Google Scholar]
- Way JC, Chalfie M. The mec-3 gene of Caenorhabditis elegans requires its own product for maintained expression and is expressed in three neuronal cell types. Genes & Dev. 1989;3:1823–1833. doi: 10.1101/gad.3.12a.1823. [DOI] [PubMed] [Google Scholar]
- Xue D, Tu Y, Chalfie M. Cooperative interactions between the Caenorhabditis elegans homeoproteins UNC-86 and MEC-3. Science. 1993;261:1324–1328. doi: 10.1126/science.8103239. [DOI] [PubMed] [Google Scholar]
- Zarkower D, Hodgkin J. Molecular analysis of the C. elegans sex-determining gene tra-1: A gene encoding two zinc finger proteins. Cell. 1992;70:237–249. doi: 10.1016/0092-8674(92)90099-x. [DOI] [PubMed] [Google Scholar]