

Abstract
In response to different extracellular signals, Rel/NF-κB transcription factors are critical regulators of apoptosis in a variety of cell types. Here we show that in normal B and T cells, expression of the Bcl-2 prosurvival homolog, A1, is rapidly induced in a Rel-dependent manner by mitogens. In B-cell lines derived from c-rel−/− mice, which like primary cells lacking Rel undergo apoptosis in response to antigen receptor ligation, constitutive expression of an A1 transgene inhibits this pathway to cell death. These findings are the first to show that Rel/NF-κB regulates physiologically the expression of a Bcl-2-like protein that is critical for the control of cell survival during lymphocyte activation.
Keywords: Rel/NF-κB, A1, apoptosis, lymphocyte, mitogenesis
Multicellular organisms eliminate unwanted cells that arise during tissue formation and homeostasis, as a result of viral infection or radiation-induced damage, by activating a form of physiological cell death termed apoptosis (Raff 1992; Cory 1995). Although diverse signals trigger cell death, intracellular pathways transmitting this information converge on an evolutionarily conserved biochemical pathway that initiates a common set of events including plasma membrane blebbing, chromatin condensation, and in most cases DNA fragmentation (Wyllie et al. 1980). The executioners of apoptosis are a set of aspartate-specific cysteine proteases termed caspases that serve to degrade critical cellular substrates (Thornberry and Lazebnik 1998). Although the regulatory machinery governing the activation of caspases is less well understood, a key role is played by proteins of the Bcl-2 family, members of which either inhibit or promote cell death. Prosurvival homologs include Bcl-2, BclxL, Bclw, Mcl1, and A1 (also known as Bfl-1; Choi et al. 1995), whereas others such as Bax, Bak, and Bok, and the more distantly related BH3-only containing proteins Bad, Bik, Bid, Hrk, Bim, and Blk are potent activators of apoptosis (Adams and Cory 1998). The mechanism by which prosurvival Bcl2-like proteins function is best understood in the nematode Caenorhabditis elegans in which programmed cell death during development is controlled by the interplay of four proteins: a Bcl-2 homolog CED-9; Egl-1, a BH-3-containing proapoptotic member of the Bcl-2 family; an ATP-binding protein CED4; and the caspase CED-3. CED9 acts by restraining the activity of CED4, an adaptor protein, which in turn is required for the activation of CED3 (Hengartner and Horvitz 1994). Cell death appears to be initiated when Egl-1 antagonizes the activity of CED-9 (Conradt and Horvitz 1998). Recent evidence indicates that prosurvival Bcl-2-like proteins in mammalian cells function in a similar fashion, inhibiting the activation of initiator caspases such as casase-9 by binding to the mammalian homolog of CED4, Apaf1 (Adams and Cory 1998).
Although many components of the cell death machinery preexist in a dormant state (Raff 1992), there is also evidence that for a number of death stimuli, de novo gene expression is necessary for cell survival or apoptosis (Schwartz and Osborne 1993). Consistent with this observation are recent findings that show that nuclear factor-κB (NF-κB)-like transcription factors are essential regulators of apoptosis (Baeuerle and Baltimore 1996). Rel/NF-κB regulates gene expression by binding to decameric sequences (κB motifs) located within the promoters and enhancers of many viral and cellular genes (Baeuerle and Henkel 1994; Baldwin 1996). These proteins are homo- and heterodimers comprised of related subunits that share a conserved amino-terminal motif of ∼300 amino acids termed the Rel homology domain (RHD) that encompasses sequences important for DNA binding, protein dimerization, and nuclear localization (Baeuerle and Henkel 1994). In mammals, there are five distinct subunits. NF-κB1(p50) and NF-κB2(p52) only consist of the Rel homology domain and lack intrinsic transcriptional transactivating properties, whereas Rel, RelA (p65), and RelB have distinct carboxy-terminal transactivation domains (Baeuerle and Henkel 1994). Before stimulation, the major proportion of Rel/NF-κB in most cell types is sequestered in the cytoplasm in an inactive form through an association with regulatory IκB proteins (Finco and Baldwin 1995; Verma et al. 1995). A broad range of agents promote nuclear translocation of cytoplasmic Rel/NF-κB complexes by a mechanism that involves the activation of an IκB kinase complex (for review, see Gerondakis et al. 1998). This phosphorylates specific amino-terminal serine residues within the various IκB isoforms (Brown et al. 1995; DiDonato et al. 1996), thereby targeting IκB for ubiquitin-dependent proteosome-mediated degradation (Finco and Baldwin 1995; Verma et al. 1995).
Various Rel/NF-κB proteins can inhibit or promote apoptosis in a cell-type and stimulus-dependent manner (Sonenshein 1997). Expression of a trans-dominant IκB-α mutant markedly increases death in certain cell lines treated with tumor necrosis factor-α (TNF-α), ionizing radiation, or daunorubicin (Van Antwerp et al. 1996; Wang et al. 1996). An examination of mutant mice lacking specific Rel-related proteins revealed RelA-protected fibroblasts and macrophages against TNF-α-induced apoptosis (Beg and Baltimore 1996), whereas NF-κB1 promotes the survival of quiescent and lipopolysaccharide-activated B cells (Grumont et al. 1998). Yet, despite enforced Rel expression overcoming TNF-α-induced apoptosis in HeLa cells coexpressing the trans-dominant IκB-α mutant (Liu et al. 1996), overexpression of Rel promotes apoptosis in chicken bone marrow cells (Abbadie et al. 1993), and high levels of c-rel mRNA are detected in cells undergoing programmed cell death within the developing chick embryo (Abbadie et al. 1993).
Previously, we have shown that in mitogen-activated primary B cells, Rel is critical for cell cycle progression through G1 and preventing apoptosis (Grumont et al. 1998). Expression of a bcl-2 transgene in primary c-rel−/− and nfkb1−/− B cells inhibited mitogen-induced cell death but failed to promote cell division, raising the possibility that Rel regulated two distinct groups of genes, one crucial for cell cycle progression and the other for apoptosis. To investigate this question, the expression of various prosurvival genes was examined in mitogen stimulated c-rel−/− B cells. Here we show that expression of the Bcl-2 homolog A1, normally rapidly and markedly induced in activated B and T cells, is only weakly up-regulated in mitogen treated c-rel−/− lymphocytes. This Rel-dependent increase in A1 expression was attributable to transcriptional induction mediated through a κB element in the A1 promoter that specifically bound Rel containing complexes. Using B-cell lines derived from c-rel−/− mice, we show that A1 expression inhibits antigen receptor ligation-induced cell death.
Results
Mitogen-induced expression of the Bcl-2 homolog A1 is markedly reduced in primary c-rel−/− B and T cells
To determine whether the survival of mitogen activated B cells involves the transcriptional regulation of a bcl-2-like gene by Rel or NF-κB1, the expression of a number of genes encoding antiapoptotic and proapoptotic Bcl-2 family members was examined in resting and mitogen-activated splenocytes derived from normal, c-rel−/− and nfkb1−/− mice. These experiments showed that expression of bcl-2, bcl-w, bcl-xL, mcl-1, bim, bax, and bad was normal in c-rel−/− and nfkb1−/− lymphocytes in response to a range of mitogens (results not shown). In contrast, A1, a prosurvival bcl-2-like gene previously shown to be induced in macrophages (Lin et al. 1993) and endothelial cells (Karsan et al. 1996) by various stimuli, whereas strongly up-regulated in normal and nfkb1−/− B and T cells, was barely induced in Rel-deficient lymphocytes (Fig. 1). This defect was observed for a range of T-cell (concanavalin A, anti-CD3/anti-CD28 antibodies, PMA; Fig. 1, lanes 2–4) and B-cell [lipopolysaccharide (LPS), anti-RP antibodies, and anti-IgM antibodies; lanes 5–7] mitogens. The impaired expression of A1 does not reflect a universal block in the induction of early response genes in Rel-deficient lymphocytes, as c-myc mRNA is up-regulated normally in mitogen-treated c-rel−/− B cells (Grumont et al. 1998).
Figure 1.
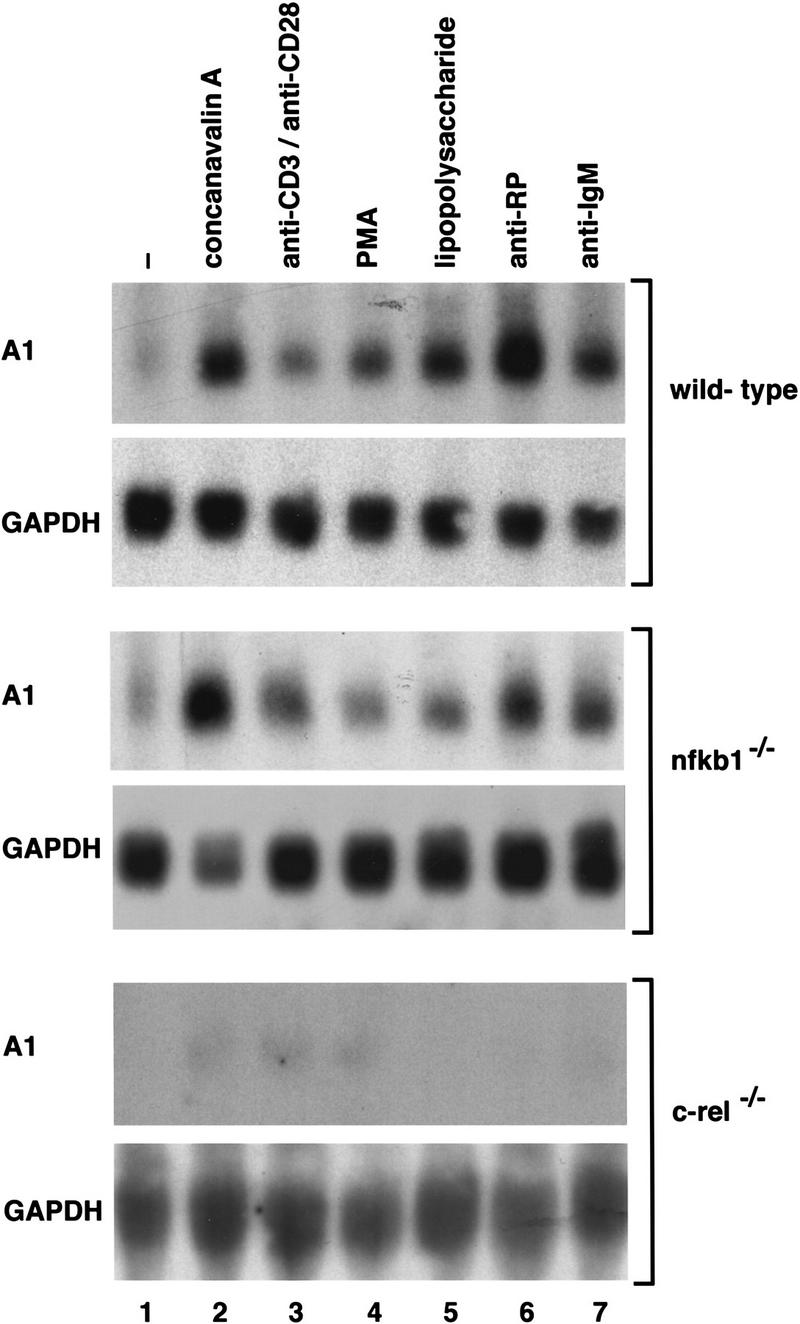
The mitogen-induced expression of A1 mRNA is markedly impaired in c-rel−/− lymphocytes. Ten-microgram samples of total RNA isolated from untreated or mitogen stimulated wild-type, nfkb1−/−, or c-rel−/− splenocytes activated in culture for 2 hr were analyzed by Northern blot hybridization. Filters were sequentially hybridized with murine A1 and rat GAPDH cDNA probes and exposed to autoradiography for between 24 and 48 hr. The stimuli were untreated cells (lane 1); concanavalin A (lane 2); anti-CD3 plus anti-CD28 antibodies (lane 3); phorbol ester (lane 4); lipopolysaccharide (lane 5); anti-RP antibodies (lane 6); anti-IgM antibodies (lane 7).
The promoter in the murine A1 gene contains a κB element that is required for Rel-dependent transcription
To determine whether the Rel-dependent induction of A1 mRNA in lymphocytes was attributable to increased transcription, we first isolated and characterized the promoter region of the mouse A1 gene. The genomic sequence encompassing the initiation codon for the A1 protein (+150), the start site of transcription (Lin et al. 1993), and 2010 nucleotides of additional 5′ flanking sequence is shown in Figure 2. Although scrutiny of the nucleotide sequence upstream of the transcription start site revealed a number of putative binding sites for ubiquitous and tissue-restricted transcription factors, of most interest was the presence of a single κB-like motif, 5′-AGGGGATCCC-3′ located between −488 and −479. To establish whether this κB-like element was involved in Rel-dependent A1 transcription, restriction fragments extending upstream of the 5′-untranslated region (+129) and encompassing the putative κB element (Fig. 3A) were inserted 5′ of the CAT gene in the promoterless reporter plasmid (Lucknow and Schutz 1987; here designated as pCAT) and transiently transfected into the Jurkat T-cell line with or without a c-rel expression vector. The plasmid A1 L-CAT, containing 2010 nucleotides of sequence flanking the transcription start site exhibited significantly higher promoter activity than the parental vector (Fig. 3B, lanes 1,3) and was up-regulated upon cotransfection with c-rel (Fig. 3B, lanes 3,4). Basal promoter activity of A1 S-CAT, in which nucleotides −2010 to −659 were deleted was equivalent to A1 L-CAT (cf. lane 3 with lane 5) and was up-regulated to the same extent by c-rel (Fig. 3B, lanes 4,6). As the Rel-responsive region (−659 to +129) encompassed the putative κB element, its role in Rel-dependent transcription was tested directly by changing the A1 κB site to a version (5′-AGG TCATAAC-3′) unable to bind Rel/NF-κB proteins (Grumont et al. 1993). The plasmid A1κBm L–CAT, containing the mutant κB site within the context of the full-length promoter, while retaining normal basal promoter activity, was not up-regulated by Rel (cf. lane 7 with lane 8), indicating that the unique κB element within the A1 promoter is necessary and sufficient for Rel-dependent A1 transcription.
Figure 2.

Nucleotide sequence of the 5′ flanking region of the murine A1 gene. The DNA-binding motif for Rel/NF-κB located at residues −488 to −479 is boxed. The italicized sequence represents A1 transcript, with nucleotides +1 to +149 (regular type) corresponding to 5′ untranslated sequence and +150 to +155 (boldface type) representing coding sequence (GenBank accession no. 515003). The A1 transcription and translation initiation sites, indicated by the raised and inverted arrows, respectively, were determined by Lin et al. (1993).
Figure 3.

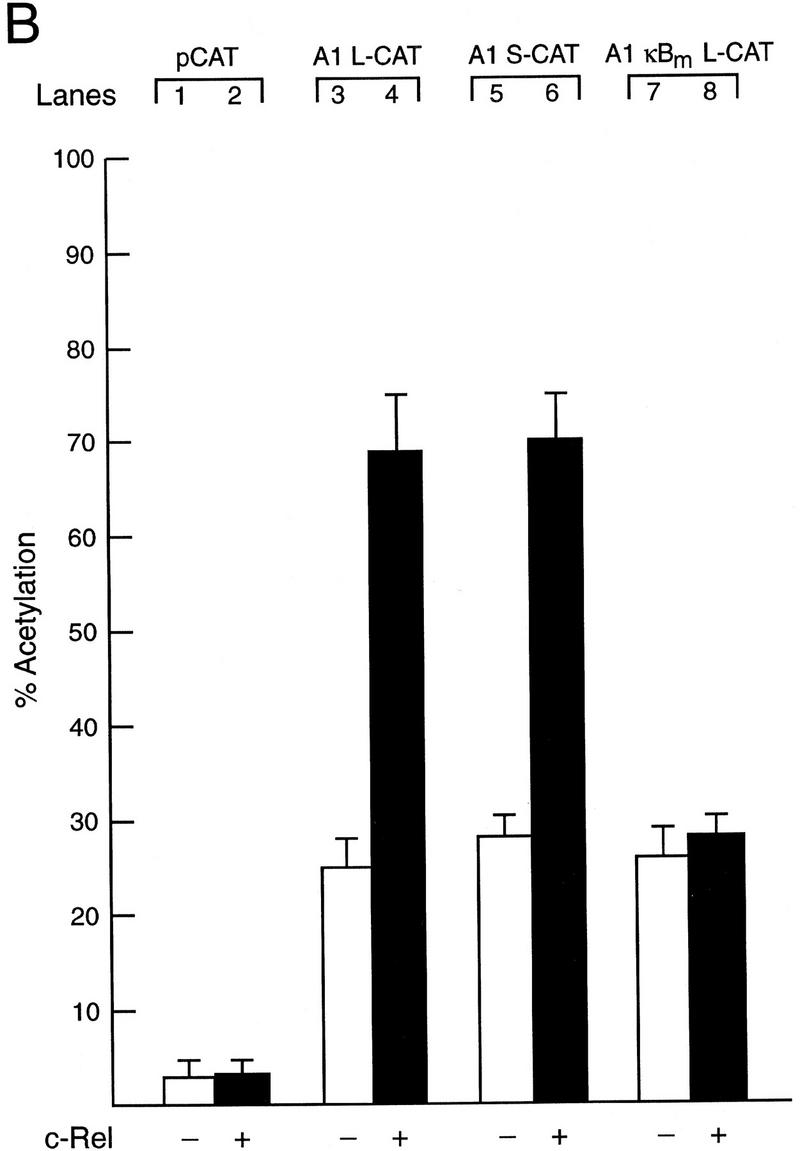
Functional analysis of the murine A1 promoter. (A) Schematic diagram of the A1 5′ flanking region and the CAT reporter plasmids. The numbers in parenthesis indicate the positions of restriction enzyme sites introduced into the murine A1 5′ flanking region by in vitro mutagenesis according to the numbering of the sequence outlined in Fig. 2. The open box represents the putative NF-κB-binding site (5′-AGGGGATCCC-3′: −488 to −479), whereas the corresponding symbol with a cross represents the mutated motif (5′-AGGTCATAAC-3′). The wavy arrow denotes the transcription initiation site and the straight arrow denotes the murine A1 initiation codon. The CAT gene is depicted as a closed box. Plasmid nomencature is indicated to the right of each construct. (B) Mutation of the NF-κB-binding motif abolishes Rel-dependent transcription in T cells. Jurkat cells were transfected transiently with 2 μg of the reporter plasmids pCAT (lanes 1,2), A1 L-CAT (lanes 3,4), A1 S-CAT (lanes 5,6) or A1κBm L-CAT (lanes 7,8) plus 10 μg of the expression plasmid DAMP56 containing no insert (lanes 1,3,5,7) or the c-rel expression plasmid pDAMP56c-rel (lanes 2,4,6,8). Chloramphenicol acetylation for transfections with the pDAMP56 or pDAMP56c-rel are indicated by open and closed bars, respectively. These results represent the mean percentage of chloramphenicol acetylation ± s.d. obtained from five separate sets of transient transfections.
Mitogenic stimulation induces nuclear Rel complexes that bind the κB motif in the A1 promoter
The finding that the A1 κB element was necessary for Rel-dependent induction of A1 transcription prompted an analysis of the nuclear protein-binding properties of this site in resting and mitogen-activated B cells from normal and c-rel−/− mice using electrophoretic mobility shift assays (Fig. 4). Although a single major nuclear complex was detected in both resting wild-type and c-rel−/− B cells (Fig. 4A, lanes 1,3; denoted C2), a novel complex (denoted C1) was up-regulated within 2 hr in anti-IgM antibody-stimulated c-rel+/+ (lane 2), but not c-rel−/− (lane 4) B cells. The A1κB-binding specificity of both complexes in normal (Fig. 4A, lanes 5,6,9,10) and c-rel−/− (lanes 7,8,11,12) B cells was demonstrated by competition with excess nonlabeled A1κB (lanes 5–8) or mutant A1κB (lanes 9–12) probes.
Figure 4.


Analysis of Rel/NF-κB complexes in resting and mitogen-stimulated B cells. Nuclear extracts (1–2 μg) isolated from purified normal and c-rel−/− splenic B cells stimulated with anti-IgM for 2 hr, then incubated with 32P-radiolabeled A1κB probe were resolved on 5% nondenaturing polyacrylamide gels and exposed to autoradiography for 6–24 hr at −70°C. (A) A κB-binding complex rapidly induced by mitogen is absent in c-rel−/− B cells. Nuclear extracts from resting (lanes 1,3,5,7,9,11) and anti-IgM-stimulated (lanes 2,4,6,8,10,12) normal (lanes 1,2,5,6,9,10), and c-rel−/− (lanes 3,4,7,8,11,12) B cells were preincubated in the absence (lanes 1–4) or presence of a 50-fold molar excess of unlabeled A1κB (lanes 5–8) or A1κBm (lanes 9–12) probe before adding radiolabeled A1κB. The inducible slow mobility and constitutive fast mobility complexes are designated C1 and C2, respectively. (B) The inducible C1 complex is a Rel-containing heterodimer. Nuclear extracts from resting (lanes 1–4) and anti-IgM-stimulated (lanes 5–8) wild-type splenic B cells were incubated with preimmune (lanes 1,5) or NF-κB1 (lanes 2,6), Rel (lanes 3,7), and RelA (lanes 4,8) -specific sera before adding the radiolabeled A1κB probe.
The composition of the A1κB-binding complexes in normal B cells was examined by supershift analysis using antibodies specific for the different Rel/NF-κB subunits (Fig. 4B). In anti-IgM-stimulated B cells, C1 is supershifted with antibodies specific for NF-κB1 (lane 6) and Rel (lane 7), but not RelA (lane 8), indicating that this complex is mainly comprised of NF-κB1/Rel heterodimers. Although the mobility of C2 in both resting (lanes 1–4) and stimulated (lanes 5–8) B cells is consistent with this complex being NF-κB1 homodimers, a finding confirmed by shift–Western blotting (R. Grumont, results not shown), it was only weakly supershifted by NF-κB1-specific sera. This anomaly has been observed previously when using this antiserum (Grigoriadis et al. 1996). Although the major anti-IgM-induced nuclear complex in normal B cells binding A1κB is an NF-κB1/Rel heterodimer, in nfkb1−/− B cells, Rel homodimers appear to be the induced nuclear complex that binds this sequence (results not shown), confirming the functional redundancy of the Rel heterodimer partner in A1 gene transcription.
Rescue of Rel-deficient B cells from anti-IgMdependent apoptosis by inducible expression of Rel correlates with up-regulation of A1 expression
To ascertain whether the Rel-dependent induction of A1 in lymphocytes plays a role in protecting cells from mitogen-induced apoptosis, surface IgM-positive B cell lines were derived from tumors arising in control or Rel-deficient mice that expressed a c-myc transgene under the transcriptional control of the immunoglobulin IgH enhancer element (Eμ-myc; Adams et al. 1985). When c-rel+/+ (404.4 and 417.1) and c-rel−/− (B1.1 and B78) Eμ-myc B cell lines were stimulated in culture with anti-IgM antibodies for 72 hr (Fig. 5), the c-rel−/− Eμ-myc B cell lines (lanes 8,11), like primary c-rel B cells underwent apoptosis (91% and 74% death in B1.1 and B78 cells, respectively). In contrast, survival of control c-rel+/+ B cell lines was comparable in untreated and anti-IgM antibody-stimulated cultures (cf. lanes 1,2,4,5). Interestingly, although anti-RP activation increased cell death in primary c-rel−/− B cells (Grumont et al. 1998), the c-rel−/− Eμ-myc cell lines were insensitive to anti-RP antibody-induced apoptosis (Fig. 5, lanes 9,12). Because similar results were obtained with a number of independent c-rel+/+ and c-rel−/− Eμ-myc B cell lines, only data for the 417.1 (c-rel+/+) and B1.1 (c-rel−/−) cell lines are shown for subsequent experiments.
Figure 5.
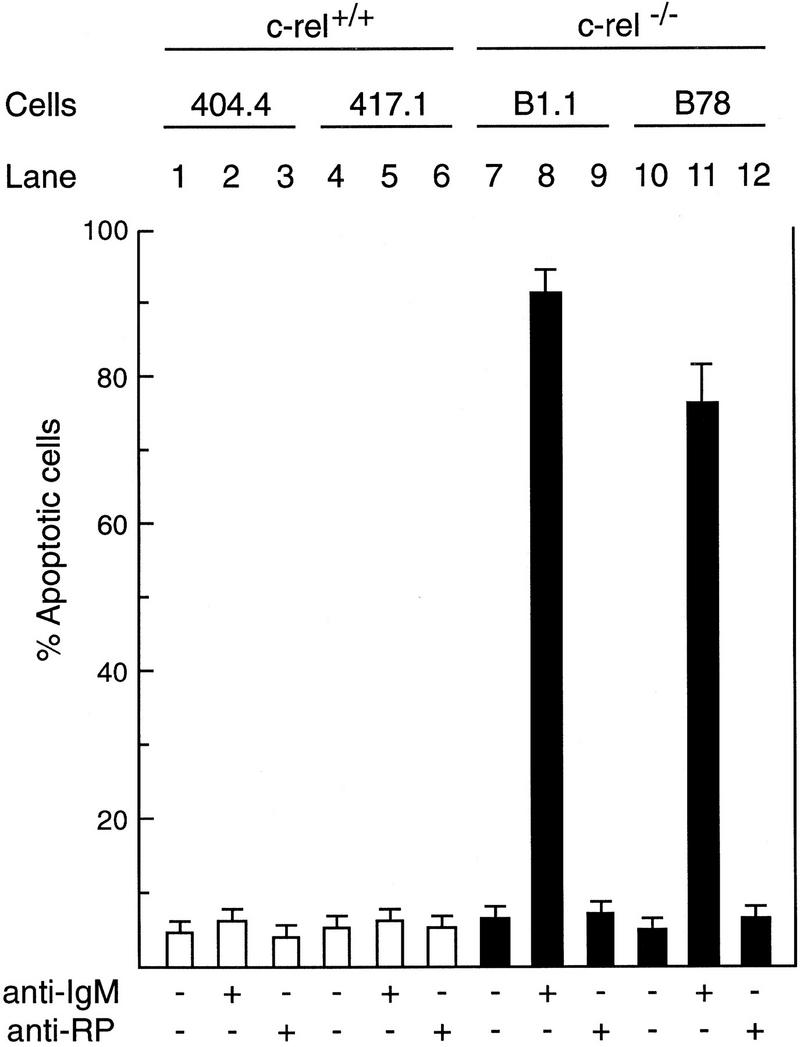
Engaging the antigen receptor of immortalized surface IgM-positive c-rel−/− B-cell lines induces apoptosis. Cell lines derived from tumors arising independently in c-rel+/+ Eμ-myc (404.4, lanes 1–3; 417.1, lanes 4–6), and c-rel−/− Eμ-myc (B1.1, lanes 7–9; B78, lanes 10–12) mice were either untreated (lanes 1,4,7,10) or stimulated in culture for 72 hr with mitogenic antibodies raised against IgM (lanes 2,5,8,11) or the B-cell-specific cell surface protein RP (lanes 3,6,9,12). The frequency of apoptotic cells in each culture is shown for the 72-hr time point and has been expressed as a proportion of the total cell number determined by flow cytometric analysis of fixed cells stained with propidium iodide. More than 99% of each cell line were viable at the start of the experiment. These results represent the mean ± s.d. from six independent experiments.
To demonstrate that anti-IgM signaling was still Rel-dependent in the c-rel−/− B cell lines, we used a chimeric Rel/ER protein comprising a mutant version of the hormone-binding domain of the human estrogen receptor responsive to 4-hydroxy-tamoxifen (4-HT), fused to the carboxyl terminus of full-length mouse Rel. Equivalent strategies have been used to successfully express both v-rel (Boehmelt et al. 1992, 1995; Capobianco and Gilmore 1993) and chicken c-rel (Zurovec et al. 1998) in a hormone-inducible manner in various cell types. The c-rel/ER chimeric cDNA was introduced into control (417.1) and Rel-deficient (B1.1) cells using a puromycin-expressing replication defective retrovirus, and infected cells selected for drug resistance. Because previous studies had shown overexpression of v-rel (Schwartz and Witte 1988) and c-rel (R. Grumont, unpubl.) in lymphoid cell lines can be cytotoxic, we first established the highest concentration of 4-HT tolerated by either c-rel+/+ or c-rel−/− B-cell lines infected with the c-rel/ER virus. Western blots of total cell extracts from infected 417.1 and B1.1 clones (Fig. 6A) using anti-human estrogen receptor (ER) antisera detected a specific protein of ∼104 kD, the expected size of the Rel/ER polypeptide, in cell lines infected with the c-rel/ER (lanes 3,4) but not control virus (lanes 1,2). The nuclear induction of Rel/ER in response to 4-HT was demonstrated in Western blots of nuclear extracts isolated from the c-rel/ER virus-infected cell lines (Fig. 6A, lanes 7,8) and gel shift analysis (Fig. 6B) confirmed that a nuclear complex specifically induced in these B1.1 cells by 4-HT, and which bound to the A1 κB site (lane 2), contained Rel (lane 3). Whereas B1.1 cells expressing Rel/ER were sensitive to anti-IgM-induced apoptosis in the absence of 4-HT (Fig. 6C, lane 7), the nuclear induction of Rel/ER conferred an equivalent level of resistance to anti-IgM-induced death as was observed in control cells (Fig. 6C, lanes 4,8). Most convincingly, A1 gene expression in B1.1 cells containing the c-rel/ER provirus was induced specifically by 4-HT (Fig. 6D, lanes 3,4). Collectively these findings show that Rel/ER can prevent antigen receptor-mediated apoptosis in c-rel−/− B-cell lines and that cell survival correlates with the 4-HT-dependent induction of A1 expression.
Figure 6.


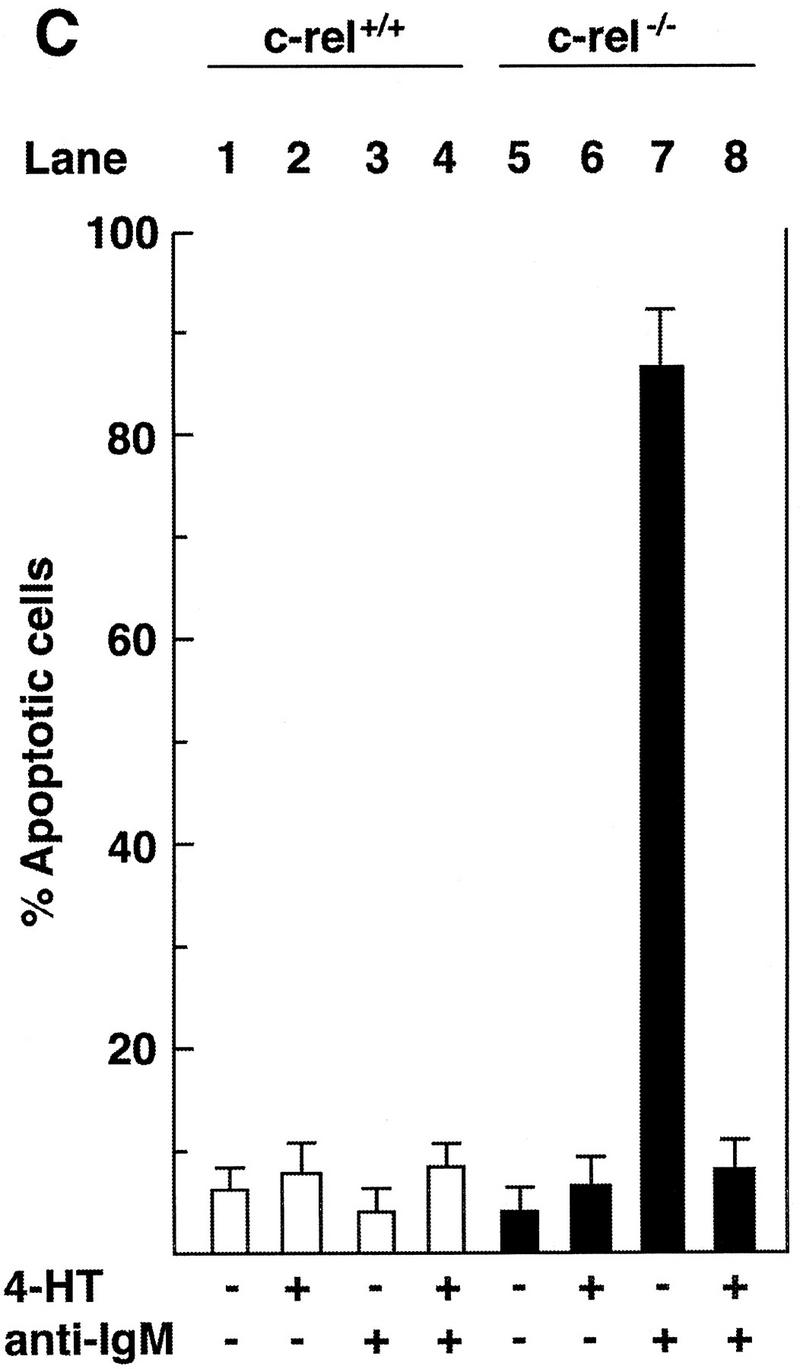
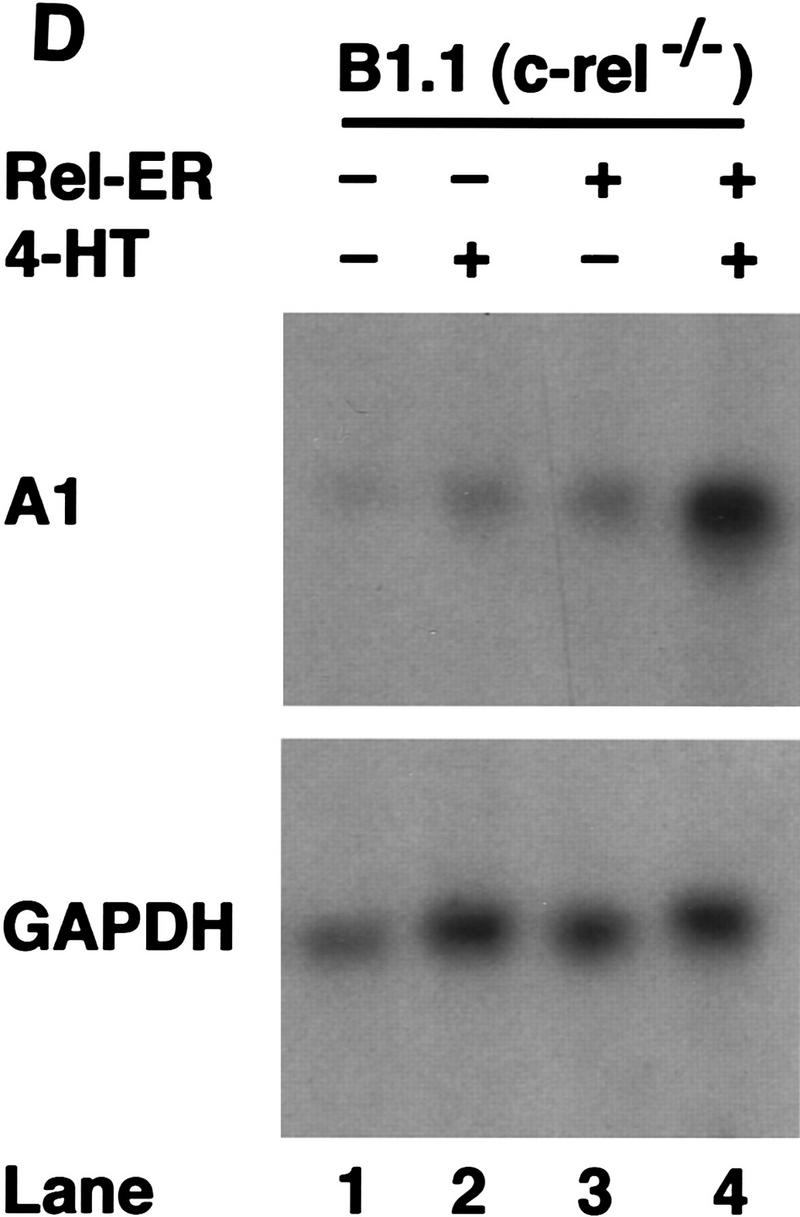
A hormone-regulated Rel protein rescues c-rel−/− B cells from anti-IgM-induced apoptosis. (A) Expression of the Rel/ER protein. Whole cell (lanes 1–4) or nuclear (lanes 5–8) extracts from 417.1 (c-rel+/+; lanes 1,3,5,7) and B1.1(c-rel−/− : lanes 2,4,6,8) cells infected with a control retrovirus (lanes 1,2) or a retrovirus expressing c-rel/ER (lanes 3–8) that had been untreated (lanes 1–6) or stimulated with 50 nm 4-HT (lanes 7,8) were resolved by SDS-PAGE and subjected to Western blot analysis using antibodies raised against the human estrogen receptor. (B) The 4-HT inducible κB-binding nuclear complex in c-rel−/− B cells expressing Rel/ER contains a Rel protein. Nuclear extracts isolated from B1.1 cells infected with the c-rel/ER retrovirus that were untreated (lane 1) or 4-HT activated (lanes 2,3) were preincubated on ice without (lanes 1,2) or with Rel-specific sera for 30 min before adding 1 μg of poly [d(I-C)] and 32P-labeled A1 κB probe and incubating for a further 20 min. The resultant DNA-binding complexes were resolved on 5% nondenaturing polyacrylamide gels and exposed to autoradiography for 6 hr at −70°C. The 4-HT-inducible DNA-binding complex is indicated by an arrow. (C) Inducible expression of Rel/ER protects c-rel−/− B cells from anti-IgM-induced apoptosis. The 417.1 (c-rel+/+; lanes 1–4) and B1.1 (c-rel−/−; lanes 5–8) cell lines infected with the HEDME2SV40puroc-rel/ER retrovirus were either untreated (lanes 1,2,5,6) or stimulated with anti-IgM (lanes 3,4,7,8) for 72 hr in the absence (lanes 1,3,5,7) or presence (lanes 2,4,6,8) of 4-HT. At 24-hr intervals, samples of cells from all cultures were fixed, stained with PI, and analyzed by flow cytometry. The frequency of apoptotic cells, shown for the 72-hr time point is expressed as a proportion of the total cell number. More than 99% of each cell line were viable at the start of the experiment. These results represent the mean ± s.d. from five independent experiments. (D) A1 transcript levels increase in 4-HT-treated c-rel−/− B cells expressing Rel/ER. Ten-microgram samples of total RNA isolated from untreated (lanes 1,3) or 4-HT-stimulated (lanes 2,4) c-rel−/− B1.1 cells infected with the control retrovirus (lanes 1,2) or the c-rel/ER virus (lanes 3,4) were analyzed by Northern blot hybridization. Filters were sequentially hybridized with 32P-radiolabeled murine A1 and rat GAPDH cDNA probes, then exposed for autoradiography at −70°C for 24 hr.
Enforced expression of A1 inhibits anti-IgM-induced apoptosis in Rel-deficient B cells
To assess the role of A1 in preventing enhanced death of c-rel−/− B cells in response to anti-IgM activation, 417.1 (c-rel+/+) or B1.1 (c-rel−/−) cells stabily expressing amino terminally Flag-tagged A1 or an inactivating mutant, A1G87E, were generated. Cells expressing comparable levels of either A1 or A1G87E (Fig. 7A) were then treated with anti-IgM antibody and cell viability assessed after 72 hr (Fig. 7B). Expression of wild-type or mutant A1 did not significantly alter the survival of untreated or anti-IgM-activated 417.1 cells (lanes 1–5). In contrast, A1, but not mutant A1 significantly reduced anti-IgM-induced apoptosis in B1.1 cells (Fig. 7B, cf. lane 8 with lane 10). Consistent with the protective effect of Bcl-2 in primary c-rel−/− B cells (Grumont et al. 1998), high level expression of Bcl-2 in B1.1 cells also inhibited anti-IgM-induced apoptosis (26% apoptosis after 72 hr; results not shown). These results show that expression of the prosurvival protein A1 can protect B cells against anti-IgM induced cell death.
Figure 7.

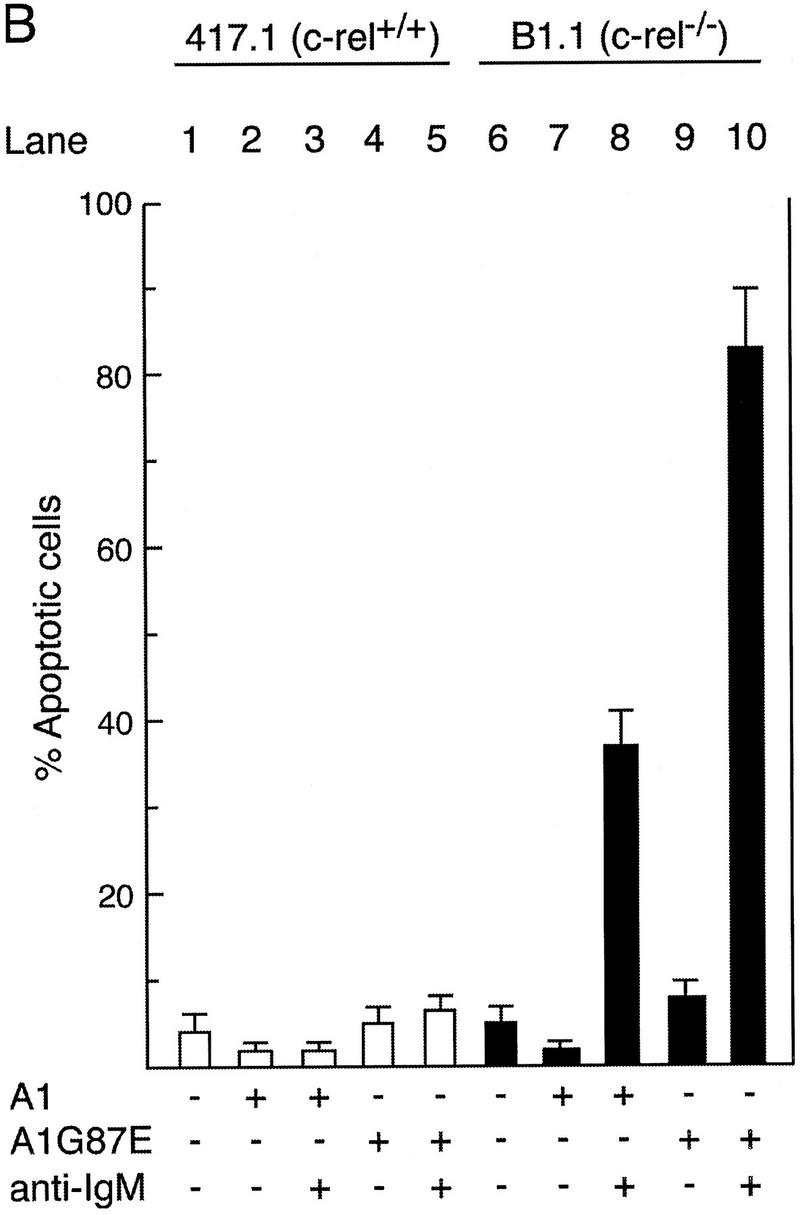
Constitutive expression of A1 protects c-rel−/− B cells from anti-IgM-induced apoptosis. (A) Expression of wild-type and G87E mutant A1 proteins in Eμ-myc B cell lines. c-rel+/+ (417.1) and c-rel−/− (B1.1) Eμ-myc B cell lines stabily transfected with a vector control (solid line) Flag-A1 (amino-terminal Flag-tagged A1; open dark line) or Flag-A1G87E (amino-terminal Flag-tagged A1 mutant G87E; open light line) were fixed, permeabilized, and stained with an anti-Flag monoclonal antibody and analyzed by flow cytometry as described (Huang et al. 1997a). (B) Enforced expression of A1 protects c-rel−/− B-cell lines from BCR ligation-induced apoptosis. Parental Eμ-myc cells (lanes 1,6) and stabily transfected clones expressing Flag-tagged A1 (lanes 2,3,7,8) or Flag-tagged A1 G87E (lanes 4,5,9,10) were either untreated (lanes 1,2,4,6,7,9) or stimulated with anti-IgM (lanes 3,5,8,10) for 72 hr. The frequency of apoptotic cells expressed as a proportion of the total cell number in each culture was determined by flow cytometric analysis of fixed cells stained with PI. More than 99% of each cell line were viable at the start of the experiment. These results represent the mean ± s.d. from six independent experiments.
Discussion
The Rel /NF-κB family of transcription factors have been shown to regulate apoptosis in response to a variety of cytotoxic signals and agents (Sonenshein 1997). Here we demonstrate that in activated B and T cells, Rel induces directly the transcription of the bcl-2 homolog A1 and that expression of this prosurvival gene is required to prevent apoptosis that occurs during the engagement of the B-cell antigen receptor (BCR).
A1 represents a subclass of Bcl-2-like proteins induced by Rel/NF-κB
Previous studies have implicated genes encoding several different types of anti-apoptotic proteins as transcriptional targets of Rel/NF-κB. These include cellular inhibitor of apoptosis proteins (c-IAP1: Wang et al. 1998; c-IAP2: Chu et al. 1997, Wang et al. 1998; xIAP: Stehlik et al. 1998; and ch-IAP1: You et al. 1997), TNF receptor-associated factors (TRAF1, TRAF2; Wang et al. 1998), and a novel protein IEX-1L (Wu et al. 1998), all of which participate in protecting cells against TNF-α-induced cell death. Here we show that A1, a bcl-2 homolog up-regulated in primary lymphocytes by different mitogens, represents a novel class of Rel/NF-κB-regulated prosurvival genes. A1 mRNA is also rapidly induced in macrophages stimulated with granulocyte–macrophage colony-stimulating factor (GM-CSF) or LPS (Lin et al. 1993) and human umbilical vein endothelial cells treated with phorbol ester, TNF-α, or IL-1β (Karsan et al. 1996). Although it remains to be determined whether these diverse stimuli induce A1 expression in these cells in a Rel/NF-κB-dependent manner, it is highly probable that given each of these agents is known to activate these transcription factors (Baeuerle and Henkel 1994). The induction of A1 by cytokines may also confer a protective role on cells during inflammatory responses, particularly as the resistance of endothelial cells to the effects of TNF-α (Pohlman and Harlan 1989) is consistent with A1 serving such a function.
Although we clearly demonstrate that A1 is a critical Rel-regulated target gene required to prevent antigen receptor ligation-induced death in B cells, the experimental evidence indicates that there are likely to be other genes regulated by Rel that participate in this anti-apoptotic process. First, in contrast to expression of the Rel/ER protein, enforced expression of A1 in the c-rel−/− B-cell lines only provided partial protection against anti-IgM antibody-induced apoptosis. This difference is unlikely to simply reflect the level of A1 expression as A1 mRNA levels were higher in transfected c-rel−/− B cells compared with Rel/ER-induced expression in the same cells (results not shown). Second, Bcl-2 was still not fully protective, although its activity is more potent than A1 in a variety of cell types (Lin et al. 1996; D. Huang, unpubl.). The conclusion that Rel regulates multiple prosurvival genes necessary for complete protection against BCR ligation-induced apoptosis is consistent with the complete inhibition of TNF-α-induced apoptosis in a fibrosarcoma cell line requiring the induction of TRAF1, TRAF2, c-IAP1, and c-IAP2 by NF-κB (Wang et al. 1998). Similarly, another NF-κB-regulated gene, IEX-1L, is alone not fully protective against TNF-α-induced apoptosis (Wu et al. 1998). Using Rel/NF-κB transcription factors to induce the expression of several genes that inhibit cell death may represent a mechanism whereby a cell can attempt to block apoptosis at multiple points in the signaling pathways used to initiate apoptosis.
Other Rel-regulated prosurvival genes involved in preventing apoptosis during B-cell activation remain to be identified. One potential candidate, c-myc, shown to be a transcriptional target of Rel/NF-κB crucial in preventing anti-IgM-induced apoptosis in the immature B cell line W231 (Sonenshein 1997) does not appear to be implicated in this survival process in mature B cells. The induction of c-myc in primary c-rel−/− B cells by anti-IgM treatment is normal (Grumont et al. 1998), and here we demonstrate that despite the constitutive expression of a c-myc transgene in immortalized c-rel−/− B cell lines, anti-IgM still induced apoptosis in these cells.
A1-dependent survival of lymphocytes during mitogenesis
Lymphocytes are prone to apoptosis throughout most stages of their maturation (Strasser 1995). In the B-cell lineage, ∼75% of pre-B cells produced in the bone marrow undergo apoptosis as a direct result of nonproductive immunoglobulin gene rearrangements preventing these cells from receiving a BCR-mediated survival signal. Immature B cells that exit the bone marrow also require positive selection, as only 5% survive and differentiate into mature long-lived peripheral B cells (Osmond 1993). Recently it was shown that when immature B cells emerging from the bone marrow become long-lived peripheral B cells, there is a corresponding 10-fold increase in A1 mRNA expression (Tomayko and Cancro 1998). As this coincides with an increase in nuclear Rel that accompanies pre-B to B-cell differentiation (Grumont and Gerondakis 1994), it is tempting to speculate that Rel is responsible for increased constitutive A1 expression and in turn, that A1 plays a central role in survival during this phase of B-cell differentiation. Consistent with Rel-regulating A1 expression in resting B cells, is the reduction in constitutive A1 mRNA levels in c-rel−/− lymphocytes (see Fig. 1).
Although the constitutive increase in A1 expression may be critical for the survival of mature B cells, it does not explain why resting lymphocytes up-regulate A1 in response to mitogenic activation. One plausible explanation is that expression of a prosurvival gene like A1 increases the likelihood of a quiescent lymphocyte successfully exiting the resting state to undergo cell division. The transition from G0 to G1 is accompanied by extensive changes in the expression of many genes including those encoding growth factors, growth factor receptors, and cell cycle regulators (Herschman 1991), all of which must be coordinated precisely to ensure DNA replication and mitosis to occur correctly. Because the exit from G0 in a resting lymphocyte population is asynchronous (Pike et al. 1987), presumably reflecting differences in the expression of key proteins critical in this early stage of division, A1 may help to protect cells receiving a mitogenic signal that are not yet ready to enter the cell cycle. Consistent with this notion is the high level of death in cultures of normal B and T cells during the first 24 hr of mitogenic stimulation (Strasser 1995; Grumont et al. 1998). It is noteworthy that to date there has been no documentation of the rapid induction of Bcl-2-like prosurvival genes in growth factor-stimulated quiescent fibroblasts, which in contrast to lymphocytes exit G0 and enter S phase synchronously (Riddle et al. 1979).
Our study also establishes that the mechanism by which Rel promotes G1 progression and limits cell death during mitogenesis appears to involve the regulation of two distinct groups of genes, one crucial for cell cycle progression and the other set essential for regulating apoptosis. Despite enforced expression of the c-myc transgene abrogating the Rel-dependent cell cycle block in c-rel−/− Eμ-myc B-cell lines, these cells remain sensitive to apoptosis induced by BCR engagement. Because anti-IgM antibody treatment does not induce cell cycle arrest in c-rel−/− Eμ-myc B cells (R. Grumont, unpubl.), Rel-dependent survival of B cells in response to BCR activation appears to be genetically distinct from the cell cycle regulation.
The molecular mechanism by which A1 protects B cells from BCR-mediated cell death remains to be determined. Because proapoptotic and anti-apoptotic Bcl-2-like proteins form heterodimers and seemingly titrate each others function (Adams and Cory 1998), the induction of survival promoting family members may serve to bind proapoptotic relatives, thereby preventing the displacement of CED-4-like proteins and inhibiting the activation of initiator caspases. Although A1 specifically binds to some proapoptotic relatives, such as Bak and Bik, it does not antagonize the activity of all (Holmgreen and S. Cory, in prep.), suggesting additional complexity in its regulation. Despite Bcl-2 also restraining cell cycle entry (Huang et al. 1997b), a property that accommodates a model in which resting cells are protected from the action of mitogens until they are ready to divide, A1 is unlikely to function in such a manner, as it lacks a BH4 domain that is critical in Bcl-2 for inhibition of cell cycle entry but dispensable for anti-apoptotic function (Huang et al. 1997b; Adams and Cory 1998). Prosurvival proteins, such as Bcl-2 and A1, might also function by regulating activators of caspase 9, such as Apaf-1 and cytochrome c. Recent evidence indicates that in mammalian cells, prosurvival Bcl-2-like proteins bind to and sequester mammalian Apaf1 (for review, see Adams and Cory 1998; Green and Reed 1998). Alternatively, as Bcl-2 is found in the mitochondrial membrane, preservation of the integrity of this organelle by Bcl-2 or A1 could inhibit caspase-9 activation indirectly by eliminating the availability of cytochrome c (Green and Reed 1998). Ultimately, the identification of other Rel/NF-κB-regulated genes that together with A1 prevent apoptosis in various cell types should offer important insight into the mechanisms by which these transcription factors regulate this physiological process in response to various extracellular signals.
Materials and methods
Genomic clones, plasmid constructs, and retroviral expression vector
Phage genomic clones encoding the mouse A1 gene were isolated from a 129/Sv liver genomic library (Stratagene, San Diego, CA). The nucleotide sequence of the flanking region upstream of translation initiation site was determined using automated sequencing. Plasmids A1 L-CAT and A1 S-CAT consisted of the 2.14-kb HindIII–XbaI and 787-bp HindIII–XbaI genomic fragments, respectively, from the 5′ flanking sequence of the murine A1 gene inserted upstream of the CAT gene in the promoterless reporter plasmid pBLCAT3 (Luckow and Schutz 1987). A1 κBm L-CAT is a derivative of A1 L-CAT in which the NF-κB-binding site (5′-AGGGGATCCC-3′) at position −488 to −479 was altered by in vitro mutagenesis (Ho et al. 1989) to 5′-AGGTCATAAC-3′. The mouse A1 and A1 G87E mutant cDNAs, cloned into the puromycin resistance plasmid pEF FlagA pGKpuro (Huang et al. 1997a) were a kind gift of D. Huang (Walter and Eliza Hall Institute). The c-rel/ER chimeric cDNA consisted of the mutant form of the hormone binding domain of the human estrogen receptor (amino acid residues 282–595; a kind gift of P. Boulet, Walter and Eliza Hall Institute) that had been engineered to be nonresponsive to β-estradiol but retains responsiveness to 4-HT fused in-frame to the 3′ end of the mouse Rel cDNA (amino acid residues 1–588; Grumont and Gerondakis 1990). This cDNA was then subcloned into the retroviral vector pHEDME2SV40puro. Precise details of these constructs are available on request.
Mice
The generation of the c-rel−/− (Kontgen et al. 1995), nfkb1−/− (Sha et al. 1995), and Eμ-myc transgenic (Adams et al. 1985) mice have been described previously. All mouse strains had been backcrossed for >nine generations with C57BL/6 mice before being used in this study. c-rel−/− Eμ-myc mice were generated by intercrossing the two parental strains, with inheritance of the mutant c-rel allele and the Eμ-myc transgene determined by PCR analysis of tail biopsy DNA samples using specific oligonucleotide primers (details of PCR genotyping available on demand).
Purification of primary B lymphocytes
Small resting B lymphocytes were purified from the spleens of 6- to 8-week-old wild-type, nfkb1−/−, and c-rel−/− mice by negative sorting on a FACS II or FACStar Plus cell sorter (Becton Dickinson, San Jose, CA) as described previously (Grumont et al. 1998). The purity of all sorted B cells was verified by staining cells with a phycoerthyrin-labeled anti-B220 antibody (CALTAG Labs., San Francisco, CA) and ranged between 97% and 99% purity.
Generation of Eμ-myc B-cell lines
Eμ-myc and c-rel−/− Eμ-myc mice older than 6 wk were monitored daily and mice that appeared ill or exhibited enlarged lymph nodes indicative of lymphomas were sacrificed. Single cell suspensions prepared from bone marrrow, spleen, thymus, and peripheral lymph nodes were serially diluted in DMEM/10% FCS/50 μm 2-mercaptoethanol, and passaged every 2 or 3 days for a period of 6 wk, at which time ∼60% of tumors had adapted to growth in culture. Surface IgM-positive B-cell tumor lines identified by immunofluorescence staining and flow cytometry were cloned using the cell deposition unit of a FACStar Plus (Becton Dickinson). A more detailed characterization of the c-rel−/−Eμ-myc B-cell lines will be described elsewhere.
Cell culture, retroviral production, and infection
The parental c-rel+/+Eμ-myc (404.4 and 417.1) and c-rel−/− Eμ-myc (B1.1 and B78) B-cell lines were routinely passaged in DMEM/10% FCS/50 μm 2-mercaptoethanol, whereas those expressing 4-HT inducible Rel/ER were grown in media lacking phenol red. Retroviral stocks were obtained by Superfect (Quiagen, Hilden, Germany)-mediated transfection of the pHEDME2SV40puro and pHEDME2SV40puroc-rel/ER vectors into BOS23 cells as described (Pear et al. 1993). B-cell lines 417.1 (c-rel+/+) and B1.1(c-rel−/−) were infected by incubating 106 cells for 2 hr with 0.25 ml (∼106 virus/ml) of viral stock in 1 ml of phenol red-free media containing 5 μg/ml of polybrene. Cells were washed once, resuspended in 2 ml of complete media (phenol red free) and incubated for 48 hr before adding puromycin (3.5 μg/ml; Sigma). After 14 days of selection, drug-resistant cells were cloned in 96-well plates using the cell deposition unit of a FACStar Plus. Constitutive and 4-HT inducible (a concentration of 50 nm was used) nuclear expression of Rel/ER was confirmed by Western blotting (see below).
Lymphocyte activation in tissue culture
All cell lines and primary cells were cultured in the high glucose version of DMEM supplemented with 13 mm folic acid, 250 mm l-asparagine, 50 μm 2-mercaptoethanol, and 10% fetal bovine serum. Spleen cells were cultured at an initial concentration of 1 × 106 cells/ml. FACS-purified B cells were cultured at an initial concentration of 3 × 105 cells/ml. B lymphocytes were stimulated with LPS (DIFCO) at a concentration of 20 μg/ml, affinity-purified goat anti-mouse IgM (Fab′)2 fragments (Jackson Immuno Research, Bar Harbor, ME) at 20 μg/ml and 10 μg/ml for primary cells and cell lines, respectively, or rat anti-mouse RP monoclonal antibody at 1 μg/ml (Miyake et al. 1994). T lymphocytes were stimulated with concanavalin A (Pharmacia) at 2 μg/ml, PMA (Sigma) at 2 ng/ml, or by culturing in T75 flasks (Falcon) coated with monoclonal hamster antibodies specific for mouse CD3 and mouse CD28 as described (Grumont et al. 1998).
Cell viability assays
Cell viability was determined by flow cytometric analysis of cellular DNA content. In brief, cells were first fixed (>8 hr at 4°C) in 70% ethanol, treated with 0.5 μg/ml DNase-free RNase A (Boehringer Mannheim, Indianapolis, IN) for 20 min at room temperature, and finally stained with 69 μm PI in 0.1 m sodium citrate (pH 7.4) for 30 min at 4°C. Flow cytometric analysis (10,000 cells/sample) were performed on a FACScan at low flow rate (100–300 cells/second).
Transfections and CAT assays
The mouse B cell lines 417.1 and B1.1 were each transfected by electroporation (350 V, 960 μF; Bio-Rad Gene Pulser) with 10 μg of the plasmids pEFPGKpuro (Huang et al. 1997a), pEFPGKpuro Flag A1 or pEFPGKpuroFlag A1G87E and selected by growth in puromycin (3 μg/ml). Drug-resistant cell lines were cloned using the cell deposition unit of a FACStar Plus. Clones expressing high levels of A1 or A1G87E were identified by immunofluorescence staining of fixed permeablized cells followed by flow cytometric analysis as described (Huang et al. 1997a). Jurkat T cells were transfected transiently using Superfect (Qiagen) as specified by the manufacturer. Briefly, 20 μl of Superfect reagent was added to DNA samples diluted in serum and antibiotic-free DMEM to a final volume of 150 μl, vortexed and incubated for 10 min at room temperature. The DNA suspension was added dropwise to each of 2 × 106 cells that had been seeded into six-well plates (Costar) at a final density of 4 × 105/ml. After 48 hr, cells were harvested and CAT assays performed on cell extracts. Equimolar amounts (1–2 μg) of each CAT reporter plasmid were used together with a threefold molar excess of pDAMP56 or pDAMP56c-rel (Grumont et al. 1993). Approximately 48 hr after transfection, cells were harvested and extracts prepared as described (Grumont et al. 1993). Extracts were standardized for protein content and CAT assays performed with 15 μg of protein. Reaction products were subjected to thin layer chromatography and the fraction of acetylated chloramphenicol determined by PhosphorImager (Molecular Dynamics) analysis. Transfections were performed five times, with a maximum variance of ∼15% observed between replicate experiments.
Northern blot analysis
Total RNA isolated from cells using RNAgents (Promega) was fractionated on 1% formaldehyde–agarose gels and transferred onto Hybond C membranes (Amersham). Filters were baked, prehybridized in 50% formamide, 5× SSC, 0.02% Ficoll, 0.02% wt/vol polyvinylpyrrolidone, 0.02% wt/vol albumin, 500 μg/ml of denatured hering sperm DNA, and then hybridized for 18 hr at 42°C with probes at a concentration of 2 × 106 cpm/ml. Filters were washed in 0.2× SSC, 0.1% SDS at 65°C and exposed to autoradiography at −70°C. For successive hybridizations, filters were first treated by boiling in 10 mm EDTA, 0.1% SDS to remove bound probe. The probes used were a 0.6-kb BamHI–XbaI murine A1 cDNA (Lin et al. 1993) and a 1.1-kb PstI rat GAPDH cDNA (Piechaczyk et al. 1984) insert that had been radiolabeled by random primer extension with [α-32P]dATP to specific activities ranging between 5 × 108 and 109 cpm/μg.
Subcellular fractionation and immunoblotting
Approximately 106 cells resuspended in lysis buffer [50 mm Tris-HCl (pH 7.4), 50 mm NaCl, 3 mm MgCl2, 5% (wt/vol) glycerol, 0.5% (vol/vol) NP-40, 5 μg/ml each leupeptin, aprotinin, and 0.5 mm PMSF] were subjected to Dounce homogenization. The nuclear fraction recovered by centrifugation (1000g for 5 min at 4°C) was washed once in ice-cold wash buffer [10 mm Tris-HCl (pH 7.4), 15 mm KCl, 1.5 mm MgCl2, 0.5 mm PMSF, and 5% glycerol], pelleted, and lysed in 0.5% SDS. For immunoblotting, total cell extracts (2 × 105 cells) or nuclear pellets (106 cells) boiled in 0.5% SDS, were electrophoresed on 8% SDS–polyacrylamide gels and transferred to nitrocellulose membranes as described (Grumont and Gerondakis 1994). Filters were incubated with an affinity-purified rabbit anti-human estrogen receptor specific polyclonal immunoglobulin (cat sc-543, Santa Cruz Biotechnology, Santa Cruz, CA) and bound antibody revealed by horseradish peroxidase-conjugated goat anti-rabbit Ig (Silenus) and enhanced chemoluminescence (Amersham).
EMSA assays
The A1 κB probe was prepared by end-labeling the double-stranded oligonucleotide 5′-CTGCTGTTTCAGGGGATCCCAGGTTTCGTC-3′ from the murine A1 promoter and EMSA reactions performed as previously described (Grumont and Gerondakis 1994). For competition analysis, a 50-fold excess of unlabeled A1 κB or mutant A1 κB (5′-CTGCTGTTTCAGGTCATAACAGGTTTCGTC-3′) competitor DNA was added to the reaction at room temperature for 15 min before the addition of radiolabeled probe. For supershift analysis, antibodies that specifically recognize NF-κB1, RelA, and Rel (Grumont and Gerondakis 1994) were incubated for 30 min before the addition of the radiolabeled probe. All EMSA reactions were then incubated for 20 min at room temperature, 2 μl of Ficoll dye added and the reactions fractionated on 5% nondenaturing polyacrylamide gels. Gels were then dried and exposed to autoradiography at −70°C.
Acknowledgments
We thank Professor Anthony De Franco for helpful discussions during the preliminary phase of this work, Dr David Huang for many reagents and technical advice, Dr. Tom Gilmore for advice on Rel/ER fusion proteins, Dr. Bill Sha and Professor David Baltimore for making the nfkb1−/− mice available, Dr. Alan Harris for Eμ-myc transgenic mice, Dr. Kensuke Miyake for the anti-RP antibody, Dr. Frank Battye and colleagues for assistance with cell sorting, Julie Merryfull and Kat Grey for animal husbandry and Drs. Andreas Strasser, David Huang, Paul Ekert, and Liam O’Conner for critical discussions and comments on the manuscript. This work is supported by the National Health and Medical Research Council (Australia), the Anti-Cancer Council of Victoria, a Commonwealth AIDS Research Grant (No. 971274) and the International Association for Cancer Research (St.Andrew, UK).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gerondakis@wehi.edu.au; FAX 61-3-93470852.
References
- Abbadie C, Kabrun N, Bouali F, Smardova J, Stehelin D, Vandenbunder B, Enrietto PJ. High levels of c-rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. Cell. 1993;75:899–912. doi: 10.1016/0092-8674(93)90534-w. [DOI] [PubMed] [Google Scholar]
- Adams JM, Cory S. The bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RB, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. NF-kappa B: Ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappa B in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Boehmelt G, Walker A, Kabrun N, Mellitzer G, Beug H, Zenke M, Enrietto PJ. Hormone-regulated v-rel estrogen receptor fusion protein: Reversible induction of cell transformation and cellular gene expression. EMBO J. 1992;11:4641–4652. doi: 10.1002/j.1460-2075.1992.tb05566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmelt G, Madruga J, Dorfler P, Briegel K, Schwarz H, Enrietto PJ, Zenke M. Dendritic cell progenitor is transformed by a conditional v-Rel estrogen receptor fusion protein v-RelER. Cell. 1995;80:341–352. doi: 10.1016/0092-8674(95)90417-4. [DOI] [PubMed] [Google Scholar]
- Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- Capobianco AJ, Gilmore TD. A conditional mutant of vRel containing sequences from the human estrogen receptor. Virology. 1993;193:160–170. doi: 10.1006/viro.1993.1112. [DOI] [PubMed] [Google Scholar]
- Choi SS, Park IC, Yun JW, Sung YC, Hong SI, Shin HS. A novel Bcl-2 related gene, Bfl-1, is overexpressed in stomach cancer and preferentially expressed in bone marrow. Oncogene. 1995;11:1693–1698. [PubMed] [Google Scholar]
- Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-iap2 is under NF-κB control. Proc Natl Acad Sci. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–529. doi: 10.1016/s0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- Cory S. Regulation of lymphocyte survival by the bcl-2 gene family. Annu Rev Immunol. 1995;13:513–543. doi: 10.1146/annurev.iy.13.040195.002501. [DOI] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finco TS, Baldwin AS. Mechanistic aspects of NF-kappa B regulation: The emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- Gerondakis S, Grumont R, Rourke I, Grossman M. The regulation and roles of Rel/NκB transcription factors during lymphocyte activation. Curr Opin Immunol. 1998;10:353–359. doi: 10.1016/s0952-7915(98)80175-1. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Grigoriadis G, Zhan Y, Grumont RJ, Metcalf D, Handman E, Cheers C, Gerondakis S. The Rel subunit of NF-kappaB-like transcription factors is a positive and negative regulator of macrophage gene expression: Distinct roles for Rel in different macrophage populations. EMBO J. 1996;15:7099–7107. [PMC free article] [PubMed] [Google Scholar]
- Grumont RJ, Gerondakis S. The murine c-rel proto-oncogene encodes two mRNAs the expression of which is modulated by lymphoid stimuli. Oncogene Res. 1990;5:245–254. [PubMed] [Google Scholar]
- ————— The subunit composition of NF-kappa B complexes changes during B-cell development. Cell Growth Differ. 1994;5:1321–1331. [PubMed] [Google Scholar]
- Grumont RJ, Richardson IB, Gaff C, Gerondakis S. rel/NF-kappa B nuclear complexes that bind κB sites in the murine c-rel promoter are required for constitutive c-rel transcription in B-cells. Cell Growth Differ. 1993;4:731–743. [PubMed] [Google Scholar]
- Grumont RJ, Rourke IJ, O’Reilly LA, Strasser A, Miyake K, Sha W, Gerondakis S. B lymphocytes differentially use the Rel and nuclear factor kappaB1 (NF-kappaB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J Exp Med. 1998;187:663–674. doi: 10.1084/jem.187.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev. 1994;4:581–586. doi: 10.1016/0959-437x(94)90076-f. [DOI] [PubMed] [Google Scholar]
- Herschman HR. Primary response genes induced by growth factors and tumor promoters. Annu Rev Biochem. 1991;60:281–319. doi: 10.1146/annurev.bi.60.070191.001433. [DOI] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Huang DC, Cory S, Strasser A. Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene. 1997a;14:405–414. doi: 10.1038/sj.onc.1200848. [DOI] [PubMed] [Google Scholar]
- Huang DC, O’Reilly LA, Strasser A, Cory S. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 1997b;16:4628–4638. doi: 10.1093/emboj/16.15.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsan A, Yee E, Kaushansky K, Harlan JM. Cloning of human Bcl-2 homologue: Inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 1996;87:3089–3096. [PubMed] [Google Scholar]
- Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes & Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J Immunol. 1993;151:1979–1988. [PubMed] [Google Scholar]
- Lin EY, Orlofsky A, Wang HG, Reed JC, Prystowsky MB. A1, a Bcl-2 family member, prolongs cell survival and permits myeloid differentiation. Blood. 1996;87:983–992. [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Luckow B, Schutz G. CAT constructions with multiple unique restriction sites for the functional analysis of eukaryotic promoters and regulatory elements. Nucleic Acids Res. 1987;15:5490. doi: 10.1093/nar/15.13.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, Yamashita Y, Hitoshi Y, Takatsu K, Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: Unresponsiveness of X-linked immunodeficient B cells. J Exp Med. 1994;180:1217–1224. doi: 10.1084/jem.180.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmond DG. The turnover of B-cell populations. Immunol Today. 1993;14:34–37. doi: 10.1016/0167-5699(93)90322-C. [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechaczyk M, Blanchard JM, Marty L, Dani C, Panabieres F, El Sabouty S, Fort P, Jeanteur P. Post-transcriptional regulation of glyceraldehyde-3-phosphate-dehydrogenase gene expression in rat tissues. Nucleic Acids Res. 1984;12:6951–6963. doi: 10.1093/nar/12.18.6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike BL, Alderson MR, Nossal GJ. T-independent activation of single B cells: An orderly analysis of overlapping stages in the activation pathway. Immunol Rev. 1987;99:119–152. doi: 10.1111/j.1600-065x.1987.tb01175.x. [DOI] [PubMed] [Google Scholar]
- Pohlman TH, Harlan JM. Human endothelial cell response to lipopolysaccharide, interleukin-1, and tumor necrosis factor is regulated by protein synthesis. Cell Immunol. 1989;119:41–52. doi: 10.1016/0008-8749(89)90222-0. [DOI] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Riddle VG, Dubrow R, Pardee AB. Changes in the synthesis of actin and other cell proteins after stimulation of serum-arrested cells. Proc Natl Acad Sci. 1979;76:1298–1302. doi: 10.1073/pnas.76.3.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz LM, Osborne BA. Programmed cell death, apoptosis and killer genes. Immunol Today. 1993;14:582–590. doi: 10.1016/0167-5699(93)90197-S. [DOI] [PubMed] [Google Scholar]
- Schwartz RC, Witte ON. A recombinant murine retrovirus expressing v-rel is cytopathic. Virology. 1988;165:182–190. doi: 10.1016/0042-6822(88)90671-x. [DOI] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Sonenshein GE. Rel/NF-kappa B transcription factors and the control of apoptosis. Semin Cancer Biol. 1997;8:113–119. doi: 10.1006/scbi.1997.0062. [DOI] [PubMed] [Google Scholar]
- Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-κB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor a-induced apoptosis. J Exp Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A. Life and death during lymphocyte development and function: Evidence for two distinct killing mechanisms. Curr Opin Immunol. 1995;7:228–234. doi: 10.1016/0952-7915(95)80007-7. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Tomayko MM, Cancro MP. Long-lived B cells are distinguished by elevated expression of A1. J Immunol. 1998;160:107–111. [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: Intimate tales of association and dissociation. Genes & Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Kornuluk RG, Goeddel DV, Baldwin AS., Jr NF-κB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wu MX, Ao Z, Prasad KV, Wu R, Schlossman SF. IEX-1L, an apoptosis inhibitor involved in NF-kappaB-mediated cell survival. Science. 1998;281:998–1001. doi: 10.1126/science.281.5379.998. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JF, Currie AR. Cell death: The significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- You M, Ku P-T, Hrdlickova R, Bose HR., Jr ch-IAP1, a member of the inhibitor-of-apoptosis protein family, is a mediator of the antiapoptotic activity of the v-Rel oncoprotein. Mol Cell Biol. 1997;17:7328–73541. doi: 10.1128/mcb.17.12.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurovec M, Petrenko O, Roll R, Enrietto PJ. A chicken c-rel-estrogen receptor chimeric protein shows conditional nuclear localization, DNA binding, transformation and transcriptional activation. Oncogene. 1998;16:3133–3142. doi: 10.1038/sj.onc.1201860. [DOI] [PubMed] [Google Scholar]