

Abstract
Bcl-2-family proteins are key regulators of the apoptotic response. Here, we demonstrate that the pro-survival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-κB. We show that bfl-1 gene expression is dependent on NF-κB activity and that it can substitute for NF-κB to suppress TNFα-induced apoptosis. bfl-1 promoter analysis identified an NF-κB site responsible for its Rel/NF-κB-dependent induction. The expression of bfl-1 in immune tissues supports the protective role of NF-κB in the immune system. The activation of Bfl-1 may be the means by which NF-κB functions in oncogenesis and promotes cell resistance to anti-cancer therapy.
Keywords: Rel, NF-κB, Bfl-1, A1, TNFα, apoptosis
Binding of the proinflammatory cytokine tumor necrosis factor α (TNFα) to its receptor triggers competing signaling pathways that determine whether a cell lives or dies. Whereas one pathway is conducive to cell death, the other leads to activation of Rel/NF-κB transcription factors and the coincident inhibition of apoptosis (for review, see Nagata 1997). Accumulating evidence supports a proactive role for NF-κB in the inhibition of cell death induced by TNFα and other death-causing agents (for review, see Van Antwerp et al. 1998). Whereas the activation of NF-κB blocks cell killing, its inhibition enhances the cytotoxicity of TNFα and promotes apoptosis in various cell systems, demonstrating the need for NF-κB function for cell survival (Beg et al. 1995; Beg and Baltimore 1996; White et al. 1995; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996;Wu et al. 1996; Cai et al. 1997; Zong et al. 1997). The protective effect of NF-κB is dependent on RNA and protein synthesis, suggesting that it regulates the expression of genes that confer resistance to death-inducing signals (for review, see Nagata, 1997). The finding that an intact transactivation domain is required for Rel/NF-κB factors to block cell death agrees with this hypothesis (Zong et al. 1998).
Proteins of the Bcl-2 family play critical roles in determining cell fate in the apoptotic pathway. Although some members antagonize cell death, others exhibit a proapoptotic activity (for reviews, see Reed 1996; Adams and Cory 1998). By subtractive cDNA cloning, we identified the prosurvival Bcl-2-homolog Bfl-1/A1 as a direct transcriptional target of NF-κB. We show that ectopically expressed Rel proteins and stimuli that activate endogenous NF-κB factors up-regulate bfl-1 gene expression and that this is inhibited by a dominant IκBαΔN transgene. Expression of Bfl-1 alone conferred resistance to TNFα cytotoxicity, indicating that it can substitute for NF-κB to suppress apoptosis. bfl-1 promoter analysis identified a consensus NF-κB site responsible for its Rel-dependent induction. Together, these results demonstrate that NF-κB directly activates Bfl-1/A1 to inhibit programmed cell death. The preferential expression of bfl-1 in immune tissues supports the protective role of NF-κB in the immune system.
Results and Discussion
We used a PCR-selected subtractive cDNA cloning approach to identify anti-apoptotic genes under Rel/NF-κB control. The HeLa-derived HtTA–CCR43 cell line, which conditionally expresses c-Rel under the control of a tetracycline-regulated promoter, is resistant to TNFα-induced cell death upon induction of c-Rel (Bash et al. 1997; Zong et al. 1998). mRNA from HtTA–CCR43 cells conditionally expressing c-Rel was reverse transcribed and subjected to subtraction and PCR amplification. A subtracted cDNA fragment of ∼700 bp was found to be identical to bfl-1, a member of the Bcl-2 family of apoptosis inhibitors. Bfl-1 was originally isolated from fetal liver and from cytokine-treated endothelial cells (Choi et al. 1995; Karsan et al. 1996) and shares 72% amino acid identity with its mouse homolog A1 (Lin et al. 1993).
Northern blot analysis during a time course of c-Rel induction confirmed the up-regulation of bfl-1 transcripts in HtTA–CCR43 cells, with kinetics that paralleled those of c-rel and of the Rel/NF-κB target gene iκbα (Fig. 1a). Induction of the transactivation-competent RelA subunit of NF-κB also led to a sharp increase in bfl-1 mRNA levels in the tetracycline-regulated HtTA–RelA cell line (Fig. 1b, lanes 4–6; Zong et al. 1998). In contrast no expression was detected in response to the p50/NF-κB1 protein, which lacks a defined transcription activation domain (Fig. 1b, lanes 1–3). Thus, bfl-1 gene expression was specifically up-regulated upon ectopic expression of transcriptionally active Rel/NF-κB subunits.
Figure 1.

bfl-1 gene expression is activated by c-Rel and RelA but not by p50/NF-κB1. (a) Expression of bfl-1 transcripts in HtTA–CCR43 cells maintained in the presence (lane 1) or absence of tetracycline for 12, 24, 36, or 48 hr to induce c-rel expression (lanes 2–5). The blot was successively hybridized to 32P-labeled probes for bfl-1, iκbα, c-rel, and gapdh. (b) bfl-1 gene expression in HtTA–p50 and HtTA–RelA cells maintained in the presence (lanes 1,4) or absence of tetracycline for 24 or 48 hr to induce the expression of p50 or relA (lanes 2,3,5,6). The blot was hybridized to bfl-1 and actin probes.
Next, we verified the ability of endogenous NF-κB factors to activate bfl-1 gene expression in response to various stimuli and in different cells. TNFα strongly induced bfl-1 transcripts in human HT1080 fibrosarcoma cells (Fig. 2a, lanes 1,2). bfl-1 mRNA levels were also strongly elevated in human Jurkat T-cells stimulated with phorbol 12-myristate 13-acetate (PMA) plus ionomycin (lanes 3,4). Likewise, the treatment of mouse 70Z/3 pre-B cells with bacterial lipopolysaccharides (LPS) promoted the accumulation of bfl-1/a1 transcripts (lanes 5,6). Consistent with the nuclear NF-κB activity found in mouse WEHI-231 B cells (Liou et al. 1994; Miyamoto et al. 1994), basal bfl-1/a1 expression was observed in these cells (lanes 7,9,11). bfl-1/a1 mRNAs were further induced by different NF-κB-inducing agents, including TNFα, LPS, or PMA (lanes 8,10,12).
Figure 2.


The expression of bfl-1 is dependent on endogenous Rel/NF-κB activity. (a) The activation of NF-κB induces bfl-1 gene expression. Human HT1080 cells untreated or treated with TNFα (lanes 1,2), human Jurkat T cells treated with DMSO as a control (lane 3) or with PMA plus ionomycin (lane 4), mouse 70Z/3 pre-B cells untreated or treated with LPS (lanes 5,6), mouse WEHI-231 B cells untreated or treated with TNFα (lanes 7,8), LPS (lanes 9,10), or PMA (lanes 11,12). Total RNA (20 μg) was hybridized to bfl-1 and actin probes. (b) The induction of bfl-1 gene expression is repressed by a dominant IκBαΔN inhibitor. Jurkat T cells expressing wild-type IκBα or an IκBαΔN transgene were treated with DMSO alone (lanes 1,3) or with PMA plus ionomycin (lanes 2,4). Total RNA (20 μg) was hybridized to 32P-labeled probes for bfl-1, il-8, and 28S rRNA.
The activity of NF-κB is controlled by its association with IκB factors in the cytoplasm. In contrast to wild-type IκBα, which undergoes proteasome-mediated degradation in response to stimuli, an amino-terminally deleted IκBα transgene is resistant to signal-induced degradation and acts as a constitutive repressor of NF-κB in Jurkat–IκBαΔN cells (Chu et al. 1997). We investigated whether NF-κB was necessary for the stimuli-induced activation of bfl-1 by characterizing the effects of wild-type IκBα and a trans-dominant IκBαΔN inhibitor on bfl-1 gene expression. The levels of 28S rRNA and of mRNA for the NF-κB-regulated gene interleukin-8 (IL-8) were monitored as controls. Similar to the induction of bfl-1 in the parental Jurkat T-cell line (Fig. 2a, lanes 3,4), bfl-1 transcripts were sharply elevated by PMA plus ionomycin treatment of T cells expressing a wild-type IκBα transgene (Fig. 2b, lanes 1,2). In contrast, the induction of bfl-1 was reduced by 83% and that of IL-8 was decreased by 65% in cells expressing IκBαΔN (lanes 3,4). This indicated that nuclear NF-κB activity is important for bfl-1 gene expression.
Similar to c-rel, relA, and their target iκbα, bfl-1 was found to be abundantly expressed in discrete immune tissues. A Northern blot survey showed high levels of bfl-1 transcripts in human spleen, lymph nodes, peripheral blood leukocytes and bone marrow (Fig. 3, lanes 6,7,9,10). Little expression was seen in thymus, fetal liver, ovary, and small intestine, whereas none was detected in prostate, testis, or colon (lanes 1–5,8,11). These results suggest a physiological role for bfl-1 in promoting the survival of defined cell lineages in the immune system.
Figure 3.

bfl-1 is highly expressed in human immune tissues and correlates with endogenous Rel/NF-κB activity. Multiple tissue Northern blots (Clontech) human II (lanes 1–5) and human immune system II (lanes 6–11) were successively hybridized to 32P-labeled probes for bfl-1, iκbα, c-rel, relA, and actin.
In agreement with the anti-apoptotic activity of NF-κB toward TNFα, the transient cotransfection of a CMV-bfl-1 expression vector with a CMV–β-galactosidase reporter plasmid significantly suppressed the TNFα-induced killing of human HeLa cells in the presence of the protein synthesis inhibitor cycloheximide (CHX; Fig. 4a). Quantitation of cell survival showed that the transient expression of bfl-1 increased the viability of HeLa cells 8.5-fold in comparison to cells transfected with the control pCMV plasmid (Fig. 4b). Cell protection was also observed in the human HtTA-1 and HT1080 cell lines (Fig. 4b). Moreover, Bfl-1 also suppressed TNFα-induced apoptosis under conditions in which endogenous NF-κB activity was inhibited by a serine-to-alanine mutant of IκBα that is resistant to signal-induced degradation (IκBαM; Van Antwerp et al. 1996). Whereas HeLa cells transfected with IκBαM were sensitized to TNFα alone, the cotransfection of Bfl-1 rescued the cells from cytolysis (Fig. 4c). These results indicated that Bfl-1 can substitute for NF-κB to block TNFα-induced cytolysis. These data agree with the protective effect of Bfl-1 and its homolog A1 toward various inducers of cell death (D’Sa-Eipper et al. 1996; Karsan et al. 1996; Lin et al. 1996). Together, our findings support a role for Bfl-1 as a survival factor in the NF-κB-signaling pathway that confers resistance to TNFα-induced apoptosis.
Figure 4.
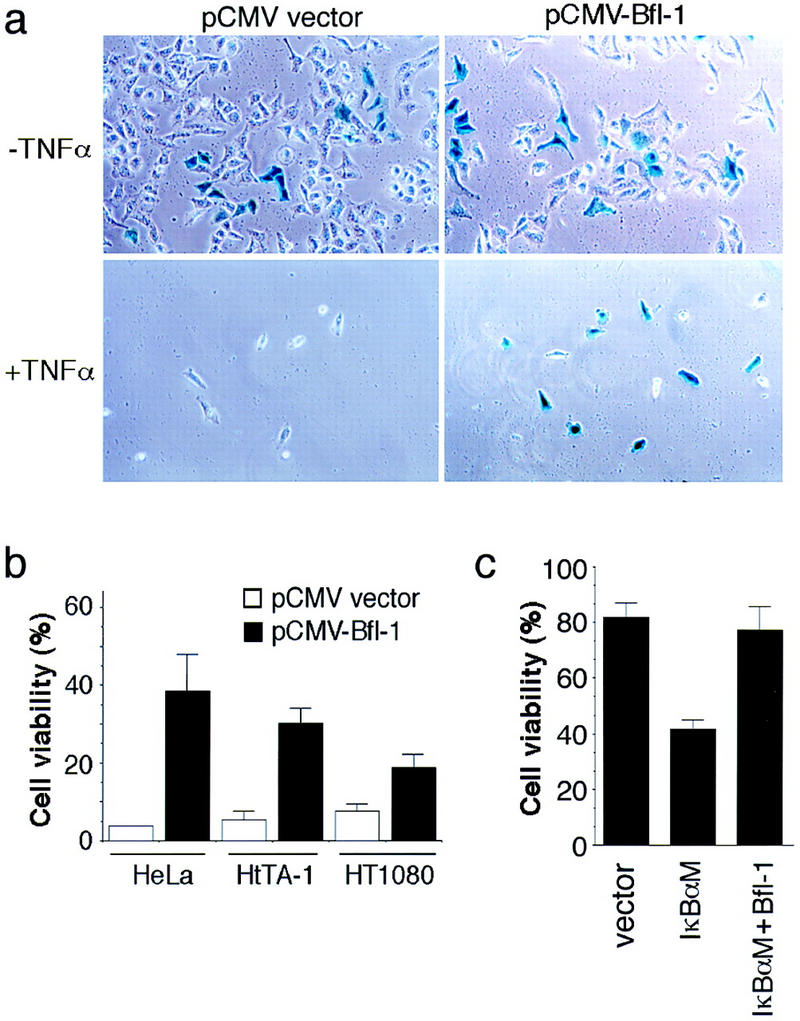
Bfl-1 suppresses TNFα-induced cell death. (a) HeLa cells were cotransfected with pCMV–β-gal, together with an empty pCMV vector or pCMV–bfl-1. The cells were treated with CHX alone or together with TNFα for 16 hr, stained with X-gal, and photographed. (b) Quantitation of cell survival upon expression of bfl-1. The viability of HeLa, HtTA-1, and HT1080 cells transfected as described in a represents the ratio of cells expressing β–gal in wells treated with TNFα plus CHX over that in wells treated with CHX alone. Cells from a minimum of 10 randomly chosen fields were counted. The average survival from three independent experiments is shown. (c) HeLa cells were cotransfected with pCMV-β-gal and an empty pCMV vector or pCMV–IκBαM, alone or together with pCMV-bfl-1. The cells were treated with TNFα alone for 16 hr and stained with X-gal. Cell survival represents the ratio of cells expressing β-gal in wells treated with TNFα over that in wells left untreated. The average survival from three experiments is shown.
Immunofluorescence studies localized Bfl-1 to the cytoplasm (data not shown). This is compatible with the subcellular localization of its homolog Bcl-2 (Krajewski et al. 1993). Consistent with this observation, the inhibitory activity of Bfl-1 toward TNFα-induced cell death was similar to that of Bcl-2 in HeLa cells. Whereas Bfl-1 enabled ∼45%–50% of the cells to survive treatment with TNFα plus CHX, Bcl-2 enabled ∼50%–60% of the cells to escape cytolysis in transient transfection assays (data not shown). However, further studies are needed to determine whether Bfl-1 utilizes the same mechanisms as Bcl-2 to inhibit cell death.
Analysis of the bfl-1 promoter region identified sequence elements responsible for its Rel-dependent induction. Nested PCR amplification of adaptor-ligated human genomic DNA libraries generated products of ∼1.4, 1.3, 0.4, and 0.2 kb (GenomeWalker-kit, Clontech). All shared a common 3′ end derived from the 5′ end of the bfl-1 cDNA and extended 5′ into adjacent genomic sequences. The purified products were directionally cloned into a promoterless reporter plasmid for analysis (Fig. 5a). The detailed characterization of the promoter will be described elsewhere (L.C. Edelstein and C. Gélinas, in prep.).
Figure 5.

The human bfl-1 promoter contains a consensus NF-κB site responsible for its Rel-dependent induction. (a) Schematic representation of CAT reporter gene constructs driven by various regions of the human bfl-1 promoter. (b) c-Rel-dependent transactivation of the bfl-1 promoter. HtTA-1 cells were cotransfected with bfl-1–CAT reporter plasmids together with pCMV–c-rel or an empty pCMV vector as a control. The IL6κBCAT plasmid containing three NF-κB DNA sites derived from the IL6 promoter was used as a positive control. The average CAT activity from three independent experiments is shown. (c) TNFα-inducible activation of the bfl-1 promoter. HtTA-1 cells were transfected with −1374/+81 bfl-1–CAT or the control IL6κBCAT reporter plasmid. Where indicated, cells were stimulated with TNFα for 6 hr prior to harvest. The average fold activation from three independent experiments is shown. (d) κB site-dependent activation of the bfl-1 promoter. Cells were cotransfected with wild-type −1374/+81 bfl-1–CAT or the mutant −1374/+81mκB–CAT reporter plasmid, with a mutated NF-κB site at position −833 (GTTTATTTACC), together with pCMV–c-rel or an empty CMV vector as a control. IL6κBCAT was used as a control.
bfl-1 promoter activity was assayed by transient transfection of HeLa cells in the presence or absence of a CMV-c-rel vector. An IL6κBCAT reporter plasmid containing three κB DNA sites served as a positive control. As shown in Figure 5b, all four clones showed minimal basal activity on their own. The cotransfection of pCMV–c-rel enhanced expression from the −1374/+81 and −1240/+81 promoter constructs by 12- and 9-fold, respectively. In contrast, the activity of the −367/+81 and −129/+81 constructs was only marginally increased by c-rel. Our mapping of a consensus NF-κB DNA site at position −833 relative to the transcription start site of bfl-1 agreed with these results (GGGGATTTACC; Fig. 5a). Consistent with these findings, cell treatment with a physiological inducer of NF-κB also activated the bfl-1 promoter. As shown in Figure 5c, TNFα stimulated CAT expression from the bfl-1 promoter, similar to its effect on the control IL6κBCAT reporter plasmid. Inactivation of the consensus NF-κB motif in the context of the bfl-1 promoter region provided direct evidence that bfl-1 is under Rel/NF-κB control (GTTTATTTACC; −1374/+81mκB). Mutation of this NF-κB site decreased gene expression significantly in the presence of c-Rel (Fig. 5d). Together, these findings demonstrate that bfl-1 is a prosurvival gene under direct Rel/NF-κB control.
Prosurvival members of the Bcl-2 family have been shown to block apoptosis in lymphoid cells under conditions in which NF-κB activity was inhibited (Wu et al. 1996; for review, see Sonenshein 1997). This raised the possibility that some members of the Bcl-2 family may lie downstream of NF-κB in the survival cascade. Our demonstration that bfl-1/a1 is a transcriptional target of NF-κB provides the first direct evidence that a Bcl-2-family member is controlled by NF-κB proteins. These findings are consistent with previous reports indicating that bfl-1/a1 gene expression is induced by proinflammatory cytokines in endothelial, leukemic, and hemopoietic cells (Moreb and Schweder 1997; Lin et al. 1993; Karsan et al. 1996b).
Our data also show clearly that bfl-1 can suppress TNFα-induced cytolysis. The anti-apoptotic activity of Bfl-1 in this context agrees with work indicating that Bfl-1/A1 can confer resistance to a variety of death inducers in different cells (D’Sa-Eipper et al. 1996; Karsan et al. 1996a; Lin et al. 1996). Thus, Bfl-1 may be viewed as an important player in the survival pathway. It remains possible that bfl-1 may act in combination with other anti-apoptotic genes to block cell death efficiently in response to different stimuli and in different cells. For example, whereas NF-κB was recently implicated in inducing expression of the death inhibitors c-IAP1, c-IAP2, and the TRAF1 (TNFR-associated factor 1) and TRAF2 factors, all four proteins must act in combination to efficiently block TNF-induced apoptosis in cells where NF-κB is inactive (Chu et al. 1997; Wang et al. 1998; You et al. 1997). The presence of an NF-κB site in the promoter region of the zinc finger protein A20 suggests that it may also be under NF-κB control (Krikos et al. 1992), although A20 failed to rescue RelA−/− cells from TNFα-induced cytolysis (Beg and Baltimore 1996). Similarly, the immediate-early response gene IEX-1L was shown to be involved in NF-κB-mediated cell survival, but its mechanism of action remains to be clarified (Wu et al. 1998). Preliminary data from our laboratory agree with these reports and suggest that other anti-apoptotic factors are also regulated by Rel/NF-κB (C. Chen and C. Gélinas, in prep.). It will thus be important to evaluate how their activities are coordinated by the NF-κB-signaling pathway and to determine whether they function individually or cooperatively in response to different stimuli and in different cellular environments.
The coinciding expression of bfl-1 and c-rel in the white pulp of the spleen, the germinal centers of lymphatic tissues, and inflammatory cells (Carrasco et al. 1994; Jung-ha et al. 1998) supports a model whereby Bfl-1 may be a critical factor for carrying out the protective role of Rel/NF-κB in the immune system and during the inflammatory response. Bfl-1 was shown previously to cooperate with the adenovirus E1A protein in inducing cell transformation and to be overexpressed in certain cancers (Choi et al. 1995; D’Sa-Eipper et al. 1996). Although the participation of Bfl-1 in oncogenesis is still a topic of controversy (Jung-ha et al. 1998), the activation of Bfl-1 by NF-κB may also be a means by which NF-κB functions in oncogenesis and promotes the resistance of tumor cells to anti-cancer therapy.
Materials and methods
Cells and endogenous NF-κB activation
Parental HtTA-1 cells and the HtTA-1-derived HtTA–CCR43, HtTA–RelA, and HtTA–p50 cell clones that expressed c-rel, relA, and p50, respectively, under tetracycline-regulated control have been described (Gossen and Bujard 1992; Bash et al. 1997; Zong et al. 1998). Human HeLa cervical carcinoma cells, HT1080 fibrosarcoma cells and Jurkat T-lymphocytic leukemia cells, and mouse 70Z/3 pre-B cells and WEHI-231 mature B-cells were obtained from ATCC. Jurkat–IκBαwt and Jurkat–IκBαΔN T cells were a gift of D.W. Ballard (Chu et al. 1997). Endogenous NF-κB activity was induced by treatment with TNFα (Sigma; 10 ng/ml) for 2 hr (WEHI-231) or 3 hr (HT1080), with PMA (50 ng per ml) plus ionomycin (1 μm in DMSO) for 2 hr or DMSO alone as a control (0.05%), with LPS (10 μg/ml) for 4 hr, or with PMA (100 nm) for 2 hr.
Subtractive hybridization and cloning of a bfl-1 cDNA
Poly(A)+ RNA was isolated from HtTA–CCR43 cells using a QuickPrep RNA purification kit (Pharmacia). Purified mRNA (2 μg) was reverse transcribed and subjected to subtractive hybridization with a PCR-Select cDNA subtraction kit (Clontech). Subtracted cDNA fragments were cloned in the pCRII vector (Invitrogen) and sequenced (Sequenase 2.0; U.S. Biochemical). A full-length HA-tagged bfl-1 cDNA clone was obtained by RT–PCR of total RNA from human HeLa cells in two successive rounds of amplification. The 5′ primer used in the first round was GCGTTCCAGATTACGCTAGCTTGATGACAGACTGTGAATTTGGA, and the 3′ primer was CTGCTTAAGAGCTCTCAACATGATTGCTTCAGG. In the second round, the 5′ primer containing an HA tag was GGATCCGCCATGGCATACCCATATGATGTTCCAGATTACGCT. The 3′ bfl-1-specific primer was identical to that used in the first round. The amplification product was cloned in a pCMV vector for transient transfection assays (pcDNA3; Invitrogen). The bfl-1 cDNA sequence was confirmed with a T7 sequencing kit (Pharmacia).
Northern blot analysis
Total RNA (20 μg) extracted with RNAzol B (TEL-TEST) was fractionated in a 1% agarose–formaldehyde gel and transferred onto a Hybond-NX membrane (Amersham). The membrane was baked for 10 min at 80°C under vacuum and UV cross-linked with a Stratalinker (Stratagene). Multiple tissue Northern blots were purchased from Clontech (human, human II, human immune system II). Probes were generated by random priming with Klenow polymerase in the presence of [32P]dCTP and [32P] dGTP (Feinberg and Vogelstein 1983). Membranes were hybridized in 5× SSC (0.75 m NaCl, 75 mm Na citrate at pH 7.0), 5× Denhardt’s solution, 0.5% SDS, and 100 μg/ml sheared salmon sperm DNA at 65°C overnight. Membranes were washed twice in 2× SSC, 0.1% SDS, and twice in 1× SSC, 0.1% SDS, at 65°C, followed by autoradiography.
TNFα-induced apoptosis
Cell resistance to TNFα-induced apoptosis was assayed as described (White et al. 1992). Cells (3 × 106) were incubated with pCMV–β-gal (5μg), together with pCMV–bfl-1 (15 μg) or an empty pCMV vector as a control, and electroporated at 220 V, 960 μF using a Bio-Rad Gene Pulser. The cells were then distributed equally into two 35-mm wells. After 24 hr, the cells were treated with CHX alone (30 μg/ml) or together with TNFα (10 ng/ml) for 16 hr. The cells were fixed and stained with X-gal and photographed at a magnification of 200x. In assays of cell death performed in the absence of CHX, HeLa cells were coelectroporated with pCMV–β-gal (3 μg), an empty CMV vector, or pCMV–IκBαM (12 μg) to constitutively repress NF-κB, alone or together with pCMV–bfl-1 (6 μg). Cells were then distributed equally into two 35-mm wells and treated 24 hr later with TNFα (10 ng/ml) for 16 hr.
Cloning of the human bfl-1 promoter, transient CAT assays, and mutagenesis
The human bfl-1 promoter region was isolated by nested PCR amplification with a GenomeWalker-PromoterFinder kit (Clontech) and cloned in a promoterless vector expressing a CAT reporter gene (pCAT-basic; Promega). bfl-1 promoter activity was analyzed by transient transfection of HtTA-1 cells with bfl-1–CAT reporter plasmids (3 μg) in the presence of a CMV–c-rel expression vector (1 μg; Xu et al. 1993) or an empty pCMV vector as a control. Where indicated, cells were treated with TNFα (10 ng/ml) for 6 hr before harvest. An IL6κBCAT reporter plasmid containing three NF-κB DNA sites from the IL6 promoter was used as a positive control (Xu et al. 1993). The −1374/+81 bfl-1 promoter region cloned in pAlter-1 was subjected to site-directed mutagenesis to inactivate the consensus NF-κB motif at position −833 (GTTTATTTACC, −1374/+81mκB; Altered Sites Mutagenesis System, Promega). Mutation of the consensus NF-κB site was confirmed by sequencing.
Acknowledgments
We are very grateful to C. Labrie for allowing C.C. to clone HA-bfl-1 in his laboratory, to D.W. Ballard for Jurkat–IκBαwt and Jurkat–IκBαΔN cells, and to H. Bujard for the gift of HtTA-1 cells. We thank A. Rabson, B. Rayet, A. Shatkin, and E. White for helpful comments on the manuscript. This work was supported by grants from the National Institutes of Health (NIH CA54999), The Council for Tobacco Research USA (4175), and by the New Jersey Commission on Science and Technology. L.C.E. is supported by NIH Biotechnology pre-doctoral training grant GM08339. C.C. is a postdoctoral fellow of the New Jersey Commission on Cancer Research and The Foundation of UMDNJ.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gelinas@mbcl.rutgers.edu; FAX (732) 235-5289.
References
- Adams JM, Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Bash J, Zong W-X, Gélinas C. c-Rel arrests the proliferation of HeLa cells and affects critical regulators of the G1/S-phase transition. Mol Cell Biol. 1997;17:6526–6536. doi: 10.1128/mcb.17.11.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-κB in preventing TNFα-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes & Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- Cai Z, Korner M, Tarantino N, Chouaib S. IκBα overexpression in human breast carcinoma MCF7 cells inhibits nuclear factor-κB activation but not tumor necrosis factor α-induced apoptosis. J Biol Chem. 1997;272:96–101. doi: 10.1074/jbc.272.1.96. [DOI] [PubMed] [Google Scholar]
- Carrasco D, Weih F, Bravo R. Developmental expression of the mouse c-rel proto-oncogene in hematopoietic organs. Development. 1994;120:2991–3004. doi: 10.1242/dev.120.10.2991. [DOI] [PubMed] [Google Scholar]
- Choi SS, Park I-C, Yun JW, Sung YC, Hong SI, Shin H-S. A novel Bcl-2 related gene, Bfl-1, is overexpressed in stomach cancer and preferentially expressed in bone marrow. Oncogene. 1995;11:1693–1698. [PubMed] [Google Scholar]
- Chu Z-L, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-κB control. Proc Natl Acad Sci. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Sa-Eipper C, Subramanian T, Chinnadurai G. Bfl-1, a Bcl-2 homologue, suppresses p53-induced apoptosis and exhibits potent cooperative transforming activity. Cancer Res. 1996;56:3879–3882. [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung-ha H, Kim D, Lee S-B, Hone S-I, Park S-Y, Huh J, Kim C-W, Kim S-S, Lee Y, Choi SS, Shin H-S. Expression of Bfl-1 in normal and tumor tissues: Bfl-1 overexpression in cancer is attributable to its preferential expression in infiltrating inflammatory cells. Hum Pathol. 1998;29:723–728. doi: 10.1016/s0046-8177(98)90282-9. [DOI] [PubMed] [Google Scholar]
- Karsan A, Yee E, Harlan JM. Endothelial cell death induced by tumor necrosis factor α is inhibited by the Bcl-2 family member, A1. J Biol Chem. 1996a;271:27201–27204. doi: 10.1074/jbc.271.44.27201. [DOI] [PubMed] [Google Scholar]
- Karsan A, Yee E, Kaushansky K, Harlan JM. Cloning of a human Bcl-2 homologue: Inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 1996b;87:3089–3096. [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: Residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor α-inducible zinc finger protein, A20, is mediated by κB elements. J Biol Chem. 1992;267:17971–17976. [PubMed] [Google Scholar]
- Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J Immunol. 1993;151:1979–1988. [PubMed] [Google Scholar]
- Lin EY, Orlofsky A, Wang HG, Reed JC, Prystowsky MB. A1, a Bcl-2 family member, prolongs cell survival and permits myeloid differentiation. Blood. 1996;87:983–992. [PubMed] [Google Scholar]
- Liou HC, Sha WC, Scott ML, Baltimore D. Sequential induction of NF-κB/Rel family proteins during B-cell terminal differentiation. Mol Cell Biol. 1994;14:5349–5359. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Schmitt MJ, Verma IM. Qualitative changes in the subunit composition of κB-binding complexes during murine B-cell differentiation. Proc Natl Acad Sci. 1994;91:5056–5060. doi: 10.1073/pnas.91.11.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreb JS, Schweder M. Human A1, a Bcl-2-related gene, is induced in leukemic cells by cytokines as well as differentiating factors. Leukemia. 1997;11:998–1004. doi: 10.1038/sj.leu.2400719. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Reed JC. Mechanisms of Bcl-2 family protein function and dysfunction in health and disease. Behring Inst Mitt. 1996;97:72–100. [PubMed] [Google Scholar]
- Sonenshein GE. Rel/NF-κB transcription factors and the control of apoptosis. Sem Cancer Biol. 1997;8:113–119. doi: 10.1006/scbi.1997.0062. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNFα-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNFα-induced apoptosis by NF-κB. Trends Cell Biol. 1998;8:107–111. doi: 10.1016/s0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- Wang C-Y, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wang C-Y, Mayo MW, Baldwin AS., Jr TNFα and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- White DW, Roy A, Gilmore TD. The v-Rel oncoprotein blocks apoptosis and proteolysis of IκBα in transformed chicken spleen cells. Oncogene. 1995;10:857–868. [PubMed] [Google Scholar]
- White E, Sabbatini P, Debbas M, Wold WSM, Kusher DI, Gooding LR. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor α. Mol Cell Biol. 1992;12:2570–2580. doi: 10.1128/mcb.12.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, FitzGerald MJ, Rothstein TL, Sherr DH, Sonenshein GE. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- Wu MX, Ao Z, Prasad KVS, Wu R, Schlossman SF. IEX-1L, an apoptosis inhibitor involved in NF-κB-mediated cell survival. Science. 1998;281:998–1001. doi: 10.1126/science.281.5379.998. [DOI] [PubMed] [Google Scholar]
- Xu X, Prorock C, Ishikawa H, Maldonado E, Ito Y, Gélinas C. Functional interaction of the v-Rel and c-Rel oncoproteins with the TATA-binding protein and association with transcription factor IIB. Mol Cell Biol. 1993;13:6733–6741. doi: 10.1128/mcb.13.11.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Ku P-T, Hrdlickova R, Bose HR. ch-IAP1, a member of the inhibitor-of-apoptosis protein family, is a mediator of the antiapoptotic activity of the v-Rel oncoprotein. Mol Cell Biol. 1997;17:7328–7341. doi: 10.1128/mcb.17.12.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong W-X, Farrell M, Bash J, Gélinas C. v-Rel prevents apoptosis in transformed lymphoid cells and blocks TNFα-induced cell death. Oncogene. 1997;15:971–980. doi: 10.1038/sj.onc.1201266. [DOI] [PubMed] [Google Scholar]
- Zong W-X, Bash J, Gélinas C. Rel blocks both anti-Fas- and TNFα-induced apoptosis and an intact Rel transactivation domain is essential for this effect. Cell Death Differ. 1998;5:963–972. doi: 10.1038/sj.cdd.4400441. [DOI] [PubMed] [Google Scholar]